")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 13
Disabling the Nuclear Translocalization of RelA/NF-κB by a Small Molecule Inhibits Triple-Negative Breast Cancer Growth
Authors Kanzaki H, Chatterjee A , Hossein Nejad Ariani H , Zhang X, Chung S, Deng N, Ramanujan VK, Cui X, Greene MI, Murali R
Received 11 March 2021
Accepted for publication 13 May 2021
Published 5 July 2021 Volume 2021:13 Pages 419—430
DOI https://doi.org/10.2147/BCTT.S310231
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Hirotaka Kanzaki,1 Avradip Chatterjee,1 Hanieh Hossein Nejad Ariani,1 Xinfeng Zhang,2 Stacey Chung,2 Nan Deng,3,4 V Krishnan Ramanujan,4 Xiaojiang Cui,1,2,4 Mark I Greene,5 Ramachandran Murali1
1Department of Biomedical Sciences, Research Division of Immunology; 2Department of Surgery; 3Biostatistics and Bioinformatics Research Center; 4Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, CA, 90048, USA; 5Department of Pathology and Laboratory of Medicine, University of Pennsylvania, Philadelphia, PA, 19104, USA
Correspondence: Ramachandran Murali Email [email protected]
Introduction: Constitutive activation of NF-κB has been implicated as being contributive to cancer cell growth, drug resistance, and tumor recurrence in many cancers including breast cancer. Activation of NF-κB leads to nuclear translocation of RelA, a critical component of the NF-κB transcription factor complex, which subsequently binds to specific DNA sites and activates a multitude of genes involved in diverse cell functions. Studies show that triple-negative breast cancer (TNBC) cells possess constitutively active NF-κB and concomitantly have higher levels of nuclear localization of RelA than cytoplasmic RelA. This feature is considered to be associated with the response to chemotherapy. However, currently, there is no specific inhibitor to block nuclear translocation of RelA.
Methods: A structure-based approach was used to develop a small-molecule inhibitor of RelA nuclear translocation. The interaction between this molecule and RelA was verified biophysically through isothermal titration calorimetry and microscale thermophoresis. TNBC cell lines MDA-MB-231 and MDA-MB-468 and a human TNBC xenograft model were used to verify in vitro and in vivo efficacy of the small molecule, respectively.
Results: We found that the small molecule, CRL1101, bound specifically to RelA as indicated by the biophysical assays. Further, CRL1101 blocked RelA nuclear translocation in breast cancer cells in vitro, and markedly reduced breast tumor growth in a triple-negative breast cancer xenograft model.
Conclusion: Our study demonstrates that CRL1101 may lead to new NF-κB-targeted therapeutics for TNBC. Further, blocking of nuclear translocation of shuttling transcription factors may be a useful general strategy in cancer drug development.
Keywords: transcription factors, breast cancer, computer aided drug design, nuclear transport, drug-target
Introduction
Triple-negative breast cancer (TNBC), a clinical breast cancer subtype lacking estrogen receptor (ER), progesterone receptor (PR) and overexpression of Her2, is highly proliferative but more sensitive to systemic chemotherapies. However, patient outcomes of TNBC are poor compared to the prognosis of other subtypes of breast cancer. While 93% of Her2+ breast cancer patients remain in remission for 5 years, this is only true for 77% of those individuals with TNBC. Although immunotherapies are showing promise in the treatment of TNBC, chemotherapy remains a standard approach.1 Nonetheless, for 20–30% of patients, there is a high risk for relapse within 3 years even after resection of primary tumors and chemotherapy.2
The NF-κB transcription factor is a central mediator of cell function and fate. Numerous studies have shown that NF-κB plays a critical role in promoting breast tumor growth, progression, and resistance to drug treatment.3 NF-κB can also be activated by multiple stimuli that may sometimes reduce the efficacy of treatments. For example, obesity is considered as a general risk-factor for breast cancer. In fact, for TNBC patients, studies show that adipose tissues elicit inflammatory cytokines such as TNFα in a RelA dependent manner,4,5 and reduce the efficacy of cancer treatment.6,7 In another study, Wee et al.8 showed that resistance to paclitaxel treatment in TNBC is conferred by an overexpression of interleukin-1 receptor associated kinase 1 (IRAK1) in response to NF-κB-mediated cytokine production.
The activation of NF-κB can occur in response to different stimuli such as radiation, chemo-agents, and pro-inflammatory cytokines.9 NF-κB can also be activated at different stages of tumor progression and can facilitate tumor growth. We and others recently observed that resistance to Trastuzumab leads Her2+ breast cancer cells to display the phenotypes of TNBC through the activation of NF-κB.10,11 TNBC cells are known to harbor constitutive activation of NF-κB,12,13 which is implicated in poor prognosis for TNBC patients.
NF-κB is a protein complex consisting of Rel family proteins (RelA, RelB and cRel) located in the cytoplasm, where they reside in a resting state when binding to IκB proteins (IκBα, IκBβ), which masks the nuclear translocation signal in Rel proteins and thus prevents their DNA binding. Upon activation, IκB proteins are degraded through phosphorylation by IκB kinases (IKK). Rel proteins in complex with p50/p52 proteins are then translocated to the nucleus where they bind to DNA.14 NF-κB activation can occur through either canonical or non-canonical signaling. In canonical signaling, RelA is translocated into the nucleus, whereas in non-canonical signaling RelB/cRel are translocated.
Much effort has been directed to develop specific NF-κB inhibitors. NF-κB activation can be blocked by interference at any stage of the NF-κB signaling cascades: upstream regulators, IKK complex, NF-κB nuclear translocation, DNA binding, transactivation, and post-translational modification. However, the only regulated step in the activation process is the phosphorylation of IκB by IKK.15 Given IKK’s central role in signal integration and relay in regulating NF-κB activation, IκB kinases have been studied to develop NF-κB signaling inhibitors. However, decades of work has not yielded a clinically useful IKK inhibitor.
Recently, using TNBC tissues, Kim et al.16 have shown that among Rel proteins, a disproportionate accumulation of RelA in the cancer cell nucleus indicated poor prognosis. These observations, consistent with the established role of NF-κB in TNBC, further suggest that dysregulation of RelA shuttling between cytoplasm and nucleus is essential for TNBC progression. Thus, small molecules that are aimed at sequestering RelA in the cytoplasm and thereby blocking its DNA binding and transactivation may effectively silence anti-apoptotic gene expression.
Umezawa developed an irreversible RelA inhibitor, DHMEQ, that shows anti-cancer activity in leukemias.17 This is the only RelA inhibitor developed thus far. However, this irreversible inhibitor binds to a free cysteine (free-Cys) at position 38 located in the DNA binding domain of RelA.18 Since this free-Cys in the DNA binding domain is conserved among all three REL proteins in the NF-κB complex (Figure S1), we surmised that DHMEQ will be non-specific and likely block cRel, RelB and RelA. Thus, we hypothesized that preventing RelA nuclear translocation by an allosteric inhibitor will be an alternate approach to inhibiting constitutively active NF-κB, rather than using RelA-DNA binding inhibitors or IKK inhibitors. Here, we show that a small molecule designed by a structure-based approach disabled the nuclear translocation of the RelA protein. The inhibitor blocked activation of NF-κB and its target genes, and inhibited xenograft tumor growth. Thus, our strategy to block RelA using an allosteric inhibitor may be relevant for cancer therapy and represents a novel approach to specifically target NF-κB activation.
Materials and Methods
Cell Lines and Reagents
Spontaneously immortalized mouse embryo fibroblasts (MEFs) were obtained from Rodent transgenic core facility at Cedars-Sinai and maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% donor calf serum (Invitrogen) and 100U/mL penicillin and 100 mg/mL streptomycin.
Human triple-negative breast cancer cell lines MDA-MB-231 and MDA-MB-468 were obtained from ATCC. These TNBC cells are highly proliferative, lack the expression of hormone receptors ER (estrogen receptor) and PR (progesterone receptor), and do not overexpress the Her2 receptor. Cells were grown in the Roswell Park Memorial Institute (RPMI)-1640 (Mediatech, Manassas, VA, USA) supplemented with 10% donor calf serum (Invitrogen), 100U/mL penicillin and 100 mg/mL streptomycin (Invitrogen). Both cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. CRL1101 was purchased from Chembridge, Inc. (San Diego, CA).
Bioinformatics Analysis of TCGA Database
The RNA-Seq data of Breast invasive carcinoma (BRCA) was obtained from the Broad Institute GDAC Firebrowse (http://firebrowse.org), which contains The Cancer Genome Atlas (TCGA) data version 2016_01_28. We downloaded the data of mRNAseq preprocess level 3 and used the raw counts of UNC RNASeqV2 in our study. The clinical information and PAM50 subtypes of the TCGA-BRCA dataset were gathered from the released dataset of Netanely et al.19 by Shamir lab. The clinical subtypes were then defined using estrogen receptor (ER), progesterone receptor (PR) and Her2 status. We matched sample IDs from the two resources and obtained 1031 tumor samples and 111 normal samples for our bioinformatics analysis. DESeq220 was applied to normalize the raw counts of RNA-seq data after filtering out low abundance genes, and differential expression analysis was performed. The p-values of multiple tests were adjusted using Benjamini-Hochberg’s method.21
Design and Development of RelA Inhibitor
The compounds have been designed using a stepwise procedure that identifies pseudo-allosteric cavities used to induce allosteric modification (CIAM) as described previously22,23 to disable protein–protein interaction. The procedure has been modified to constrain ligand-induced structural changes in a protein complex; these steps involve identification of rigid cavity, virtual screening using GLIDE (Schrodinger, Inc.) and molecular simulations using the DESMOND function in Schrodinger (San Diego, CA, USA). Briefly, the three-dimensional structures of RelA dimer-DNA complexes (PBD code: 2RAM,24 1NFI25 and 1VKX26) were used as template. Computational analysis was performed at different temperature and simulation times to identify critical sites (S276 and NLS) to disrupt the nuclear localization signaling region (NLS) located between DNA-binding domain and transactivation domain (TAD) at the C-terminus. We have screened chemicals from Chembridge database (> 500,000 compounds) that are filtered for drug-like properties that can disrupt NLS motif structural disposition thereby preventing RelA binding to DNA. CRL1101 was identified as a potential inhibitor for biological characterization. Structure analysis and modeling were performed using software from SBGRID.27 The calculations were performed at Cedars-Sinai high-performance computing center facility.
Expression and Purification of Recombinant RelA
Human RELA encoding amino acid residues 20–321 was cloned into pET-21(a) vector and expressed as a C-terminal His-tag fusion protein in BL21 (DE3) cells. The cells were resuspended in 1x phosphate buffered saline (PBS), sonicated and soluble lysate was passed through a Ni-NTA 5 mL FF column (GE Healthcare) equilibrated in buffer A (20 mM Tris pH 7.4, 500 mM NaCl). The column was washed and eluted with buffer A supplemented with 40 mM and 400 mM Imidazole, respectively. The sample was dialyzed against 20 mM Tris pH 7.4 and put through a SP sepharose 5 mL FF column (GE Healthcare). The protein was eluted using 0 to 300 mM NaCl gradient, dialyzed against PBS + 15 mM β-ME, concentrated and stored at −80 °C.
Isothermal Titration Calorimetry (ITC)
The binding thermodynamics of RelA to the CRL1101 was measured by ITC using a high precision VP-ITC titration calorimetric system (Microcal LLC, Northampton, MA). The calorimetric cell containing RelA at a concentration of 6–10 μM dissolved in 10 mM Tris (pH 8.0), 1mM EDTA was titrated with CRL1101 at a concentration ranging from 400–600 μM. Injection volumes were 10 μL. The heat evolved upon each injection of CRL1101 were obtained from the integral of calorimetric signal. The heat associated with binding of CRL1101 to RelA was obtained by subtracting the heat of dilution from the heat of reaction. The measurements were made at 25°C. Data were analyzed and fitted by using the data analysis software supplied by Microcal (ORIGIN 5.0). The free energy generated by RelA binding to CRL1101 was estimated from ∆G = -RT lnK = ∆H -T∆S.
Ligand Binding by Microscale Thermophoresis
Monolith NT.115 Microscale Thermophoresis (MST) instrument (Nanotemper Technologies) was used for this assay. Monolith protein labeling kit RED-NHS was purchased from Nanotemper Technologies. Briefly, recombinant RelA protein was labeled using RED-NHS 2nd generation labelling kit (NanoTemper) following manufacturer’s instructions. A serial dilution of ligand CRL1101 (0.25mM to 0.00763μM) was prepared and titrated against 0.55 µM labeled RelA. The assay was read in 1% excitation power and 40% of MST power.
Nuclear Localization of RelA in TNBC Cell Lines by Fluorescent Microscopy
Cells were treated with or without 15μM of CRL1101 in 1% dimethyl sulfoxide (DMSO). 45 min after treatment, cells were fixed with 4% formaldehyde for 10 min and sequentially treated with 0.1% Triton X-100 for 10 min. Cells were blocked in 1% bovine serum albumin (BSA) in phosphate buffered saline (PBS) for 10 min. Cells were incubated with anti-RelA(p65) antibody (1:150, Abcam, Cambridge, UK) in PBS with 1% BSA overnight at 4 °C and a secondary antibody (1:1000 anti-rabbit Alexa 488, Invitrogen, Carlsbad, CA, USA) for 1 h in the dark. Analyses were performed using a microscope (Nikon ECLIPSE Ti-U, Nikon, Tokyo, JAPAN). Green signal shows RelA(p65), blue signal shows 4ʹ,6-diamidino-2-phenylindole (DAPI).
Cell Viability Assay
Cell viability was quantitated using a colorimetric MTT assay according to the procedures described previously. Briefly, 100 μL of target cell suspension (1 X 104 cells) were added to each well of 96-well plate, and the plate was incubated for 24 h at 37 °C in a humidified 5% CO2 atmosphere. Following incubation, 10 μM of MTT working solution was added to each well, and the plates were incubated for 3 h at 37 °C. After addition of inhibitor, the absorbance values were measured with a microplate reader at 570 nm. The percentage of survival was calculated using the following formula: survival percentage = (absorbance of CRL1101 treated wells – blank wells/absorbance of untreated wells – blank wells) X 100. The inhibition constant (IC50) of the inhibitor was obtained by regression analysis using Excel.
Western Blot Analysis
Expression and phosphorylation changes of NF-κB related molecules after CRL1101 treatment were examined by Western blot. Western blot analysis was performed to detect NF-κB (p105 and p50), RelA (p65), IκBα, p-IκBα and β-actin protein expression. β-actin was used as a loading control. Time course for 0–60 min after 15μM of CRL1101 treatment was examined.
Anchorage Independent Growth Assay
The effects of CRL1101 on clonogenic survival were analyzed. Base layers consisting of growth medium containing 0.53% low-melting point agarose (Invitrogen) were poured onto 6-well plates and allowed to solidify. Cells were seeded at concentration of 10,000 cells/well in triplicate in top layers consisting of growth medium containing 0.32% agarose. After 24 h, 15 µM of CRL1101 was added. Cells were incubated for 14 days to form visible colonies. The colonies were fixed and stained by 0.05% Crystal violet in 50% methanol.
NF-κB Reporter Assay
MDA-MB-231 cells transfected with the pGL3-luciferase (empty vector control) or pGL3-NF-κB luciferase vector were treated with 10 µM CRL1101 or DMSO vehicle for 24 h the day after transfection. 10 µM CRL1101 was used in this assay instead of 15 µM due to reduced cell viability. Cells were grown in 10% FBS medium. Reporter activity was measured by luciferase assays and was normalized to β-galactosidase activity.
Cell Migration Assay
Cells were seeded in 6-well plates at 1×105 per well and treated with either vehicle or 10 µM CRL1101 (of note, 10 µM was used due to reason stated above). A line was drawn on the underside of the well with a pipette tip. Cell migration was assessed by measuring the distance between wound edges. Migration results are expressed as the average migration distance (μm±SD). The cells were monitored by phase contrast microscopy on an inverted microscope. All the data presented are from at least 3 independent experiments performed in duplicate.
Tumor Growth Study
NCr homozygous athymic (nude) mice (eight weeks-old) were purchased from Charles River Laboratories. An aliquot of 2×106 MDA-MB-231 cells were suspended in 200 mL of PBS and injected subdermally in the right thigh of each animals. When xenograft tumors reached ~200–230 mm3 in volume, animals were regrouped into two treatment groups (n=6): Vehicle control and CRL1101 alone. After tumor cell engraftment when tumor size is palpable, on day 9, mice were treated with CRL1101 (25mg/kg) intraperitoneally every other day. Animals were maintained in accordance with guidelines of the Institutional Animal Care and Use Committee (IACUC) of Cedars-Sinai Medical Center. Tumor growth was monitored three times weekly for four weeks. Tumor volume was calculated by the formula: π/6 x (larger diameter) x (smaller diameter)2. Mice body weight during the treatment was measured every 2 days.
Statistical Analysis
All experiments were repeated independently a minimum of three times and expressed as mean values with 95% confidence. The activity of CRL1101 was analyzed using cell viability assay and assessed by paired t-test. The other results were assessed by one-way ANOVA following by Scheffe’s F-test. A value of p < 0.05 was considered to indicate statistical significance.
Results
RelA is Overexpressed in TNBC Compared to Other Breast Cancer Subtypes
NF-κB is constitutively activated in TNBC and plays an important role in cancer cell survival and resistance.12,28 The activation of NF-κB is also critical for the function of breast cancer stem cells (BCSC)29 that promotes tumor re-emergence and metastasis. In either neoadjuvant or adjuvant settings, certain chemotherapeutic agents activate NF-κB. Indeed, our TCGA data analysis show that the expression of RelA is significantly higher in TNBC than in normal breast tissue and other breast cancer subtypes (Figure 1A and B). The results are consistent with observation made by others using breast cancer tissues. Jones et al. observed nuclear NF-κB in TNBC tissues by immunohistochemistry (IHC) in response to chemotherapy correlated with high-grade tumor.30 Similarly, Wang et al.31 showed overexpression of RelA is associated with TNBC by interfering with celecoxib mediated apoptosis. These studies show overexpression of RelA, as measured by mRNA levels and constitutive expression of nuclear RelA, promotes cancer growth and progression in TNBC. Furthermore, using TNBC tissues, Kim et al.16 have shown that among Rel proteins, an accumulation of nuclear RelA in the cancer cells indicated poor prognosis. Hence, we hypothesize that blocking RelA transported from cytoplasm into nucleus will limit tumor growth.
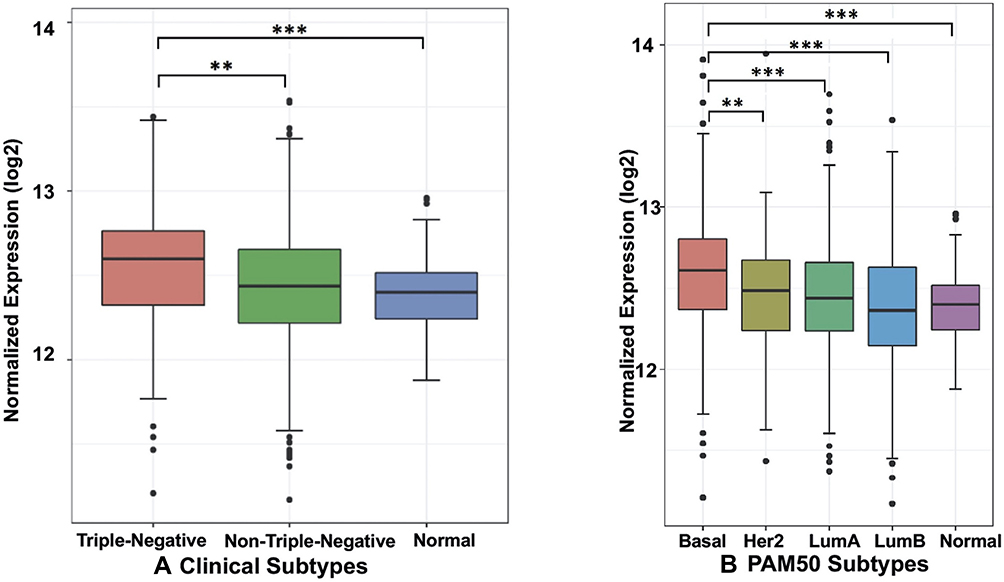 |
Figure 1 The boxplot of gene expression of RELA in breast cancer from TCGA: (A) clinical tumor subtype and (B) PAM50 subtype. The gene expression is presented as normalized counts using DESeq2 with variance stabilizing transformations (VST) on log2 scale. The comparison results between subtypes are presented as **P < 0.01, ***P < 0.001; 2-tailed unpaired t test with Bonferroni correction. |
Design and Development of RelA Inhibitors by a Structure-Based Approach
We used a structure-based approach to test whether a small molecule can be developed to block the nuclear translocation of RelA. In the inactive state of NF-κB, the RelA-p50-IκBα protein complex is located in the cytoplasm. In the active state, phosphorylation of IκBα leads to its degradation, resulting in RelA-p50 translocation to the nucleus. The crystal structure of RelA complexed with p50 and IκBα (Figure 2A) was determined by Jones et al.25 and showed that the inactive complex is held together through a network of hydrogen bonds that keep the NLS masked by p50 (Figure S2). During this process, the nuclear localization signal (NLS), located after the DNA binding domain in C-terminus of RelA, drives nuclear translocation of RelA. Upon NF-κB activation (IκBα degrade), a large conformational change (relaxed by the network of hydrogen bonds) exposes the nuclear localization signal (NLS) motif (Figure 2B).
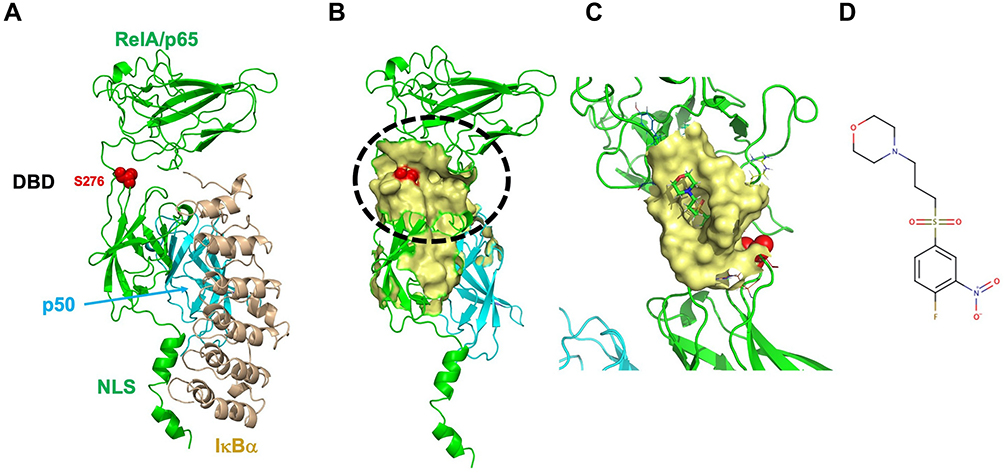 |
Figure 2 Three-dimensional structure of RelA with putative binding of CRL1101: (A) Resting state of RelA (green) is found in complex with p105 (p50 is shown (cyan)) and the Ankyrin repeat of IκBα (light brown color) which sequesters the NLS of RelA preventing nuclear translocation of RelA-p50 protein complex. The DNA binding domain (DBD) is located at the N-terminus, while the nuclear localization signal (NLS) motif is located at the C-terminus. Key phosphorylation site, S276 is shown as red sphere model (B) Inhibitor-binding pocket is show in surface representation (Yellow). Location of S276 is shown in red (C) Closeup look at the binding of CRL1101 (shown in stick representation; carbon, nitrogen and oxygen are shown in green, blue and red colors, respectively) binding to RelA (yellow surface). Partial structure of p50 is shown in cyan color (D) Chemical structure of CRL1101 is shown. Pictures were created using Pymol.27,46 |
Previously, we used CIAM to identify allosteric sites in protein.22,23 Using the modified approach we identified a distal inhibitor-binding site from the NLS motif (Figure 2A and B) proximal to S276, a known phosphorylation site. To block nuclear translocation of RelA, structural changes in RelA must be restrained so that NLS motif remains unchanged. For this purpose, the inhibitor-induced conformational changes (ie, network of hydrogen bonds shown in Figure S2) need to be restrained. Hence, rigidity of the cavity was assessed by monitoring cavity size calculated using sitemap (Schrodinger, Inc) from molecular simulation results. Binding site proximal to S276 (Figure 2C) (cavity volume 442Å3 and Dscore=0.955) was used from an average structure from the molecular simulation for subsequent virtual screening. We selected 15 compounds based on change in binding energy (∆G bind ranging from 3–8 Kcal/mol) from MMGBSA analysis. One of the compounds, CRL1101 (Figure 2D) was identified (∆G bind =7.8 Kcal/mol) as the most potent compound based on the combination of breast cancer cell proliferation assays and differential scanning fluorimetry (DSF).32
CRL1101 Inhibits Nuclear Translocalization of RelA
Next, we tested if CRL1101 is specific for RelA. For this purpose, we used RelA-null cells. Cells treated with CRL1101 were unaffected based on cell survival assays compared to the wild-type suggesting that CRL1101 is specific to RelA. (Figure 3A). Furthermore, to check whether CRL1101 can sequester RelA in the cytoplasm, two TNBC cell lines, MDA-MB-231 and MDA-MB-468 were treated with 15 μM of CRL1101. Immunofluorescence showed that CRL1101 diminished nuclear localization of RelA (Figure 3B).
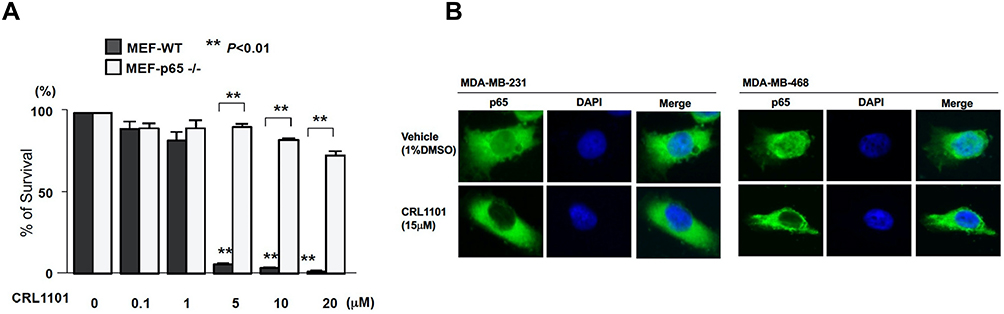 |
Figure 3 CRL1101 is specific to RelA: (A) CRL1101 affected RelA-null cells viability. Cell survival of RelA-null MEF cells were measured by MTT assay after treatment of CRL1101 at different concentrations. **P < 0.01 (B) CRL1101 sequestered RelA in the cytoplasm. Localization of RelA in TNBC breast cancer cells, MDA-MB-231 and MDA-MB-468, measured by fluorescence microscopy. TNBC cells treated with CRL1101 retained RelA (p65, green) in the cytoplasm compared to cells treated with vehicle. Cellular nuclei are stained with DAPI (blue). |
Biophysical Characterization of CRL1101 Directly Binding to RelA
We wanted to verify whether CRL1101 could directly bind to RelA protein. First, we tried to measure the binding of CRL1101 by Surface Plasmon Resonance (SPR). However, the resonance signal was insufficient to measure kinetic parameters. We reasoned that immobilization of RelA might be interfering with ligand binding. Hence, we tried two techniques that do not require immobilization: (1) In microscale thermophoresis (MST) assay there is no immobilization involved and both ligand and target can freely move in the favored buffer. In this technique either target or ligand can be labeled. Here, we labeled the target protein RelA with fluorescent dye. CRL1101 bound to RelA with a binding constant (KD) of 2.3 μM (Figure 4A). (2) Similar to MST, in isothermal titration calorimetry (ITC), protein and ligand are free and provide information related to thermodynamics of the interaction. In this assay, we observed CRL1101 binding to recombinant RelA with an affinity of 2.2μM (∆H= −1734 Kcal/mol and ∆S= 14.1cal/mol/deg; N=2) (Figure 4B) confirming direct binding of CRL1101 to RelA.
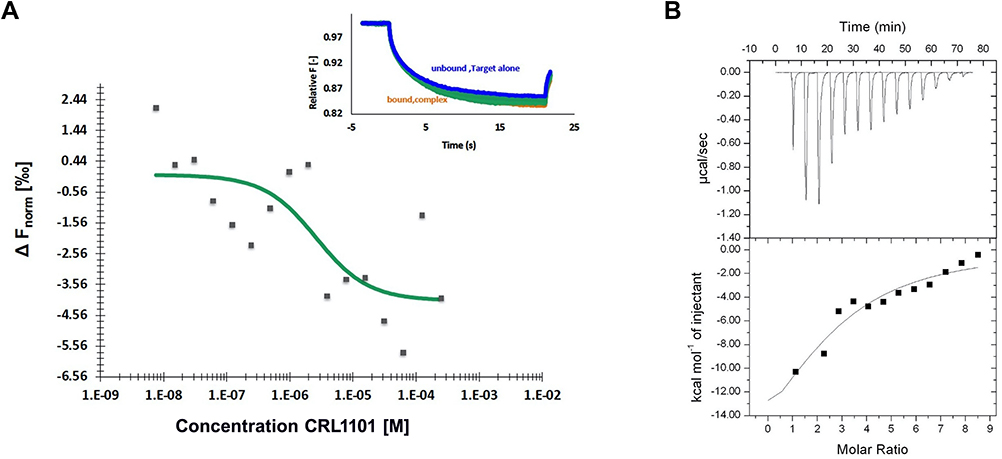 |
Figure 4 Analysis of CRL1101 binding to recombinant RelA: (A) Microscale Thermophoresis technique (MST) Dose response curve upon titrating CRL1011 from 0.0076 to 250 µM against 0.55 µM RelA. The KD obtained was 2.3 µM. Insert represents MST traces depicting fluorescence change over time at different concentrations of CRL1011. Traces corresponding to unbound and bound protein are shown in blue and green respectively. (B) Isothermal titration calorimetry: Serial dilution of 10μM CRL1101 was titrated against recombinant RelA and heat generated from the interaction is shown. Affinity of CRL1101 to RelA is 2.2μM. |
Biological Activity of CRL1101
Having demonstrated that CRL1101 specifically binds to RelA, we tested its biological activity in a series of in vitro assays. First, we measured the effect of CRL1101 on cell proliferation in TNBC breast cancer cell-lines, MDA-MB-231 and MDA-MB-468. IC50 of CRL1101 in MDA-MB-231 and MDA-MB-468 was 12.76 mM and 12.33 mM respectively. As expected, CRL1101 significantly inhibited cell proliferation with an inhibition constant of 15 μM in both cell lines (Figure 5A). Next, we tested whether CRL1101 can block constitutive activation of NF-κB. Indeed, CRL1101 inhibited NF-κB activation as measured by decrease in phosphorylation of IκBα in both tumor cell lines (Figure 5B). However, there was a small reduction in expression of RelA in MDA-MB-231 at the 60 min time point as opposed to MDA-MB-468. The significance of this reduction of RelA expression in MDA-MB-231 is yet to be determined. Next, we examined the CRL1101 effect on colony formation. As shown in Figure 5C, CRL1101 inhibited colony formation. These results show that CRL1101 limits tumor cell growth.
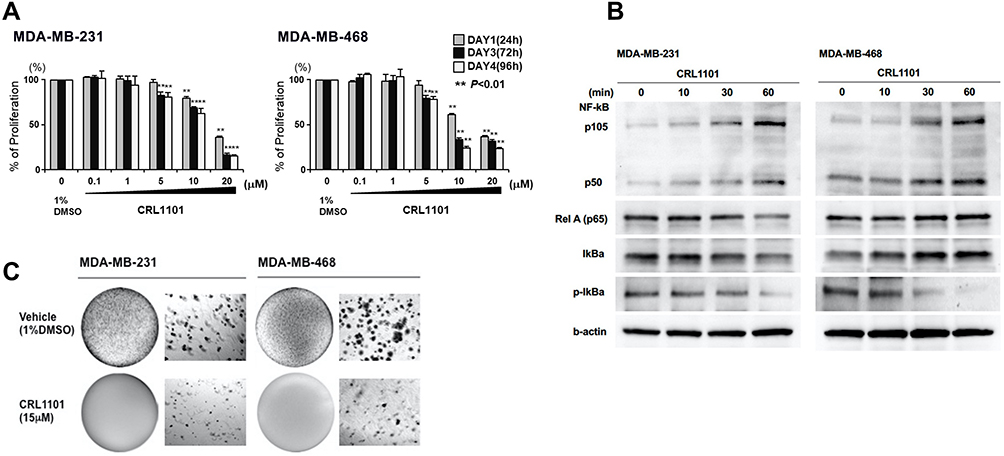 |
Figure 5 Anti-tumor effects of CRL1101 in TNBC: (A) Anti-proliferation effects of CRL1101. Cells were treated with different concentations of CRL1101, as shown in figure. Proliferation of the cells were investigated by MTT assay at 24, 72 and 96 h after CRL1101 treatment. Proliferation was inhibited in a dose-dependent manner (**P<0.01). (B) Time series of the effects of CRL1101 on expression and activation of NF-κB and related molecules by Western blot analysis. The effects of 15µM of CRL1101 on NF-κB signal related molecules, p105, p50, RelA, IκBα and p- IκBα, is shown after 0, 10, 30 and 60 minutes in MDA-MB-231 and MDA-MB-468 cells. β-actin was used as an internal expression standard. (C) Inhibition of tumor growth by anchorage independent growth assay. The effects of CRL1101 on clonogenic survival is shown after 24 h. CRL1101 inhibited colony formation in MDA-MB-231 and MDA-MB-468 cells after cells were treated with 15µM of CRL1101. |
Using a NF-κB-luciferase vector which contains tandem RelA-binding sites in the promoter governing the luciferase gene, we also found that CRL1101 inhibited the activity of luciferase reporter induced by RelA binding in MDA-MB-231 cells (Figure 6A). Next, we checked whether CRL1101 affected RelA-regulated gene expression. In breast cancer, the expression of interleukin-8 (IL-8)33,34 and EZH2 has been shown to depend on RelA.35 Thus, we examined the expression of IL8 and EZH2 in MDA-MB-231 cells treated with CRL1101. RT-PCR showed that IL-8 and EZH2 mRNA levels were significantly reduced while p65 and p50 levels remained unaffected (Figure 6B). We also tested if CRL1101 at 10 μM can affect cell migration which is known to be regulated by NF-κB. In a wound-healing assay, CRL1101 showed reduced cell migration in TNBC cells (Figure 6C and D). These results suggest that targeting RelA nuclear translocation impairs NF-κB-mediated cell function.
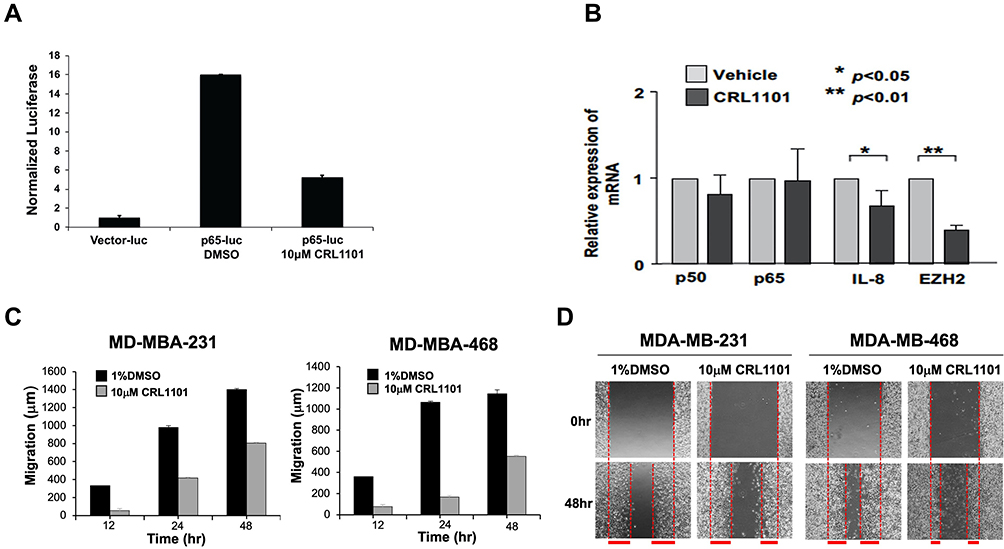 |
Figure 6 (A) Regulation of NF-κB-responsive reporter activity by CRL1101: MDA-MB-231 cells transfected with the pGL3-luciferase (empty vector control) or pGL3-NF-κB binding luciferase vector were treated with 10 µM CRL1101 or DMSO vehicle for 24 h the day after transfection. Reporter activity was measured by luciferase assays and was normalized to β-galactosidase activity. (B) RelA inhibitor, CRL1101 reduced pro-inflammatory cytokines in MDA-MB-231 cells. The inhibitory effects of CRL1101 (5 μM) on RelA-dependent gene regulation in MDA-MB-231 cells was assayed by using quantitative real time PCR. Two key players, IL8 and EZH2, regulated by nuclear RelA decreased after 48 h incubation with CRL1101. *P<0.05 and **P<0.01 (C) CRL1101 diminish tumor cells’ migration. Cells treated with CRL1101 have reduced cell migration as compared to the vehicle control (1% DMSO). Migration results are expressed as the average migration distance (μm±SD). The cells were monitored by phase contrast microscopy on an inverted microscope at 12, 24 and 48 h. All the data presented are from at least 3 independent experiments performed in duplicate. (D) Representative images for the wound healing assay. Control (1% DMSO) and CRL1101 (10 μM) treated MDA-MB-231 and MDA-MB-468 cells are shown at 0 and 48 h. Verticle red lines are drawn along wound edges and migration distance is depicted as thick horizontal red lines. |
Therapeutic Efficacy of CRL1101 in a Pre-Clinical TNBC Mouse Model
We examined the therapeutic efficacy of CRL1101 in a common TNBC xenograft model.36 Briefly, MDA-MB-231 cells were used to grow tumors in athymic mice. Mice were treated with 25 mg/kg/day (IP) CRL1101 for 4 weeks. The control group was treated with vehicle. Mice treated with CRL1101 showed significant reduction in tumor growth (Figure 7A and B). At day 31, the CRL1101-treated group had reduced tumor volume by 50% compared to the PBS group (P<0.001). During the treatment there was no significant change in body weight and the change was within the margin of error due to differences in tumor size. These results suggest that CRL1101 has a potent anti-tumor effect in vivo and does not show severe toxicity in mice (Figure 7C).
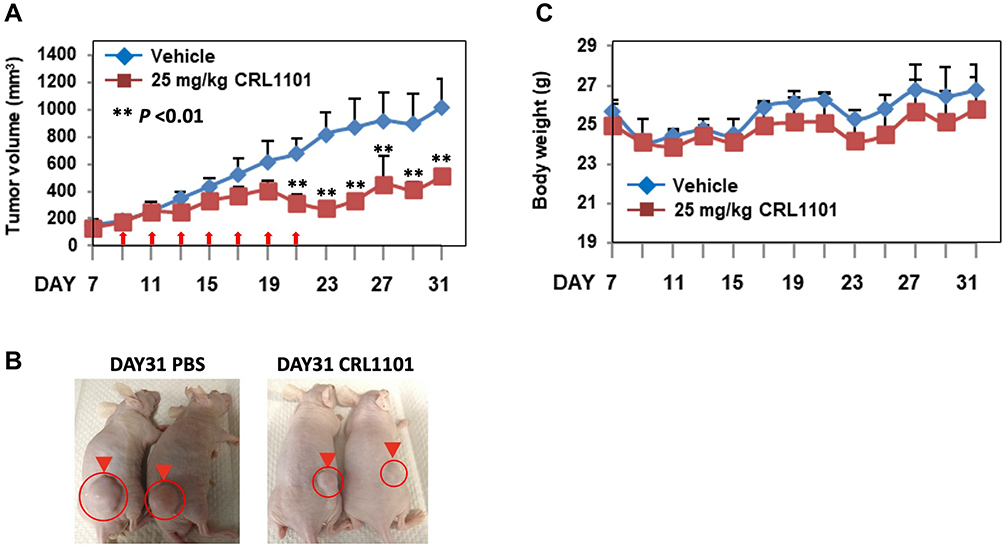 |
Figure 7 CRL1101 limits tumor growth breast in a TNBC mouse model: Tumor growth mediated by MDA-MB-231 cells was assessed in athymic mice. (A) Mice were administered with 25mg/kg/day of CRL1101 or vehicle (PBS) by IP injection, as indicated by arrows. Results are given as mean tumor volume (mm3) ±S.E. Treatment days are shown by arrows. Tumor growth in animals treated with CRL1101 was significant (**P<0.01) compared to vehicle. N = 6. (B) Visual of tumor size at the end of experiment is shown. (C) Body weight during the treatment is shown. Body weight during the treatment was measured and no significant loss of weight was observed. |
Discussion
RelA/p65 of NF-κB has been shown to promote cancer cell growth and resistance to therapy in many cancers including breast cancer.10,12,29 There have been many inhibitors developed against NF-κB that mainly target IKK.37 To date, clinical trials with NF-κB inhibitors have not been successful largely due to the essential role of the NF-κB family transcription factors in pleiotropic physiological functions and thus ubiquitous supression of total NF-κB activity leads to undesirable toxicity.38 In contrast, we demonstrated a novel way to disable constitutively activated NF-κB mediated tumor growth by targeting RelA, the dominant TNBC-associated NF-κB transcription factor subunit, using a small-molecule inhibitor to prevent its nuclear translocation.
RelA-specific inhibitors have not been developed. As mentioned before, the irreversible inhibitor, DHMEQ binds to Rel-family proteins and is not specific to RelA.39 To develop a RelA-specific inhibitor we used a non-canonical approach. Previously, we developed a novel computer algorithm termed “Cavity-induced allosteric modification (CIAM)”.40 This approach was used to disable protein–protein interactions such as TNFR-TNFα receptor complex,22 and survivin.23 The algorithm is based on the identification of molecular/atomic determinants responsible for flexibility in the protein molecule. The crystal structure of RelA/p50 protein complex revealed that large conformational and structural changes are needed for RelA nuclear localization. We exploited this structural aspect of RelA and identified CRL1101. In a series of in vitro and in vivo studies, we demonstrated that CRL1101 is a novel agent that binds to RelA with an affinity of 2.2 μM. CRL1101 is also specific to RelA based on the observation from RelA-null MEF cells. Of note, it has been shown that tumor cells lacking of either cRel or RelB show increased proliferation in tumor cells.41,42 Since we observed wild-type cells treated by the inhibitor undergo apoptosis, we believe CRL1101 is specific to RelA. We plan to explore the specificity of CRL1101 in next set of studies by expressing cRel and RelB recombinant proteins.
CRL1101 as an allosteric inhibitor shows anti-tumor effect in vitro and in vivo. Treatment of TNBC cells, MDA-MB-231 and MDA-MB-468, with CRL1101 also diminished nuclear localization of RelA (Figure 3B). By sequestering RelA in the cytoplasm, CRL1101 inhibited NF-κB activation and concomitantly reduced the expression of pro-metastatic genes such as IL-8 and EZH2, consistent with ability of CRL1101 to limit cell migration, suggesting that our molecule might also aid in delaying/preventing cancer metastasis. This allosteric inhibitor reduced tumor growth in a breast cancer mouse model (Figure 7A) suggesting the biological effect observed is due to blocking RelA nuclear transport.
Activation of NF-κB is critical for adaptive/innate immunity. In this context, p50, RelB and cRel play a dominant role. Lack of expression of RelA in immune cells such as lymphocytes and macrophages has been shown to promote suppressive phenotype. It can be argued that CRL1101 can promote immunosuppresive environment in cancer. However, when we used CRL1101 in pancreatitis mouse model as an anti-inflammatory study using wild-type B6 mice, it was found that CRL1101 did not have any effect on immune cell function (ie, promote immunosuppression) based on the cytokine profiles (unpublished data, personal communication with Drs. Pandol and Edderkaoui). Thus, we believe that CRL1101 will have minimal or no effect on tumor microenvironment of cancer. However, effect of CRL1101 on T-cells or macrophage cannot be ruled out. This aspect requires further investigations that may involve targeted delivery of CRL1101 by nanoparticles or as drug-conjugates used by us before.43–45
In summary, we have developed a novel way to disable NF-κB activation by selectively targeting nuclear localization of RelA in breast cancer. This new approach will be a new tool to target cancer that depends on NF-кB function for growth and metastasis. The approach can be considered as an alternate strategy to disable certain transcription factor-mediated tumor growth.
Acknowledgments
This study was partially supported by the Translational Oncology Program Discovery Fund Award, Samuel Oschin Comprehensive Cancer Institute (SOCCI), Cedars-Sinai Medical Center (RM and XC),NIH R01CA151610 (XC) and BCRF (MIG). We would like to thank Dr. Akashi Otaki for the construct and preparations of recombinant RelA protein.
Disclosure
HK and RM are inventors in patent application related to the work and RM reports a US patent issued. The work has been licensed to Kairos Pharma, Ltd. The authors reported no other potential conflicts of interest for this work.
References
1. Keenan TE, Tolaney SM. Role of immunotherapy in triple-negative breast cancer. J Natl Compr Canc Netw. 2020;18(4):479–489. doi:10.6004/jnccn.2020.7554
2. Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321(3):288–300. doi:10.1001/jama.2018.19323
3. Cao Y, Karin M. NF-kappaB in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia. 2003;8(2):215–223. doi:10.1023/A:1025905008934
4. Heiser LM, Sadanandam A, Kuo WL, et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci U S A. 2012;109(8):2724–2729. doi:10.1073/pnas.1018854108
5. Subbaramaiah K, Howe LR, Bhardwaj P, et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev Res. 2011;4(3):329–346. doi:10.1158/1940-6207.CAPR-10-0381
6. Egusquiaguirre SP, Yeh JE, Walker SR, Liu S, Frank DA. The STAT3 target gene TNFRSF1A modulates the NF-kappaB pathway in breast cancer cells. Neoplasia (New York, NY). 2018;20(5):489–498. doi:10.1016/j.neo.2018.03.004
7. Li HH, Zhu H, Liu LS, et al. Tumour necrosis factor-alpha gene polymorphism is associated with metastasis in patients with triple negative breast cancer. Sci Rep. 2015;5:10244. doi:10.1038/srep10244
8. Wee ZN, Yatim SM, Kohlbauer VK, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun. 2015;6:8746. doi:10.1038/ncomms9746
9. Wang W, Nag SA, Zhang R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr Med Chem. 2015;22(2):264–289. doi:10.2174/0929867321666141106124315
10. Kanzaki H, Mukhopadhya NK, Cui X, Ramanujan VK, Murali R. Trastuzumab-resistant luminal b breast cancer cells show basal-like cell growth features through NF-kappaB-activation. Monoclon Antib Immunodiagn Immunother. 2016;35(1):1–11. doi:10.1089/mab.2015.0056
11. Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castillo B, et al. Pathway-focused proteomic signatures in HER2-overexpressing breast cancer with a basal-like phenotype: new insights into de novo resistance to trastuzumab (Herceptin). Int J Oncol. 2010;37(3):669–678. doi:10.3892/ijo_00000716
12. Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ
13. Yamaguchi N, Ito T, Azuma S, et al. Constitutive activation of nuclear factor-κB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 2009;100(9):1668–1674. doi:10.1111/j.1349-7006.2009.01228.x
14. Baldwin AS
15. Karin M, Delhase M. The IκB kinase (IKK) and NF-κB: key elements of proinflammatory signalling. Semin Immunol. 2000;12(1):85–98. doi:10.1006/smim.2000.0210
16. Kim JY, Jung HH, Ahn S, et al. The relationship between nuclear factor (NF)-kappaB family gene expression and prognosis in triple-negative breast cancer (TNBC) patients receiving adjuvant doxorubicin treatment. Sci Rep. 2016;6:31804. doi:10.1038/srep31804
17. Umezawa K. Inhibition of tumor growth by NF-kappaB inhibitors. Cancer Sci. 2006;97(10):990–995. doi:10.1111/j.1349-7006.2006.00285.x
18. Lin Y, Ukaji T, Koide N, Umezawa K. Inhibition of late and early phases of cancer metastasis by the NF-κB inhibitor DHMEQ derived from microbial bioactive metabolite epoxyquinomicin: a review. Int J Mol Sci. 2018;19(3):729. doi:10.3390/ijms19030729
19. Netanely D, Avraham A, Ben-Baruch A, Evron E, Shamir R. Expression and methylation patterns partition luminal-A breast tumors into distinct prognostic subgroups. Breast Cancer Res. 2016;18(1):74. doi:10.1186/s13058-016-0724-2
20. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
21. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300.
22. Murali R, Cheng X, Berezov A, et al. Disabling TNF receptor signaling by induced conformational perturbation of tryptophan-107. Proc Natl Acad Sci U S A. 2005;102(31):10970–10975. doi:10.1073/pnas.0504301102
23. Berezov A, Cai Z, Freudenberg JA, et al. Disabling the mitotic spindle and tumor growth by targeting a cavity-induced allosteric site of survivin. Oncogene. 2012;31(15):1938–1948. doi:10.1038/onc.2011.377
24. Chen Y-Q, Ghosh S, Ghosh G. A novel DNA recognition mode by the NF-κB p65 homodimer. Nat Struct Biol. 1998;5(1):67–73. doi:10.1038/nsb0198-67
25. Jacobs MD, Harrison SC. Structure of an IκBα/NF-κB complex. Cell. 1998;95(6):749–758. doi:10.1016/S0092-8674(00)81698-0
26. Chen FE, Huang D-B, Chen Y-Q, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature. 1998;391(6665):410–413. doi:10.1038/34956
27. Morin A, Eisenbraun B, Key J, et al. Collaboration gets the most out of software. eLife. 2013;2:e01456. doi:10.7554/eLife.01456
28. Pratt MA, Tibbo E, Robertson SJ, et al. The canonical NF-kappaB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor population. Oncogene. 2009;28(30):2710–2722. doi:10.1038/onc.2009.131
29. Shostak K, Chariot A. NF-kappaB, stem cells and breast cancer: the links get stronger. Breast Cancer Res. 2011;13(4):214. doi:10.1186/bcr2886
30. Jones RL, Rojo F, A’Hern R, et al. Nuclear NF-kappaB/p65 expression and response to neoadjuvant chemotherapy in breast cancer. J Clin Pathol. 2011;64(2):130–135. doi:10.1136/jcp.2010.082966
31. Wang L, Kang F, Li J, Zhang J, Shan B. Overexpression of p65 attenuates celecoxib-induced cell death in MDA-MB-231 human breast cancer cell line. Cancer Cell Int. 2013;13(1):14. doi:10.1186/1475-2867-13-14
32. Jubb H, Higueruelo AP, Winter A, Blundell TL. Structural biology and drug discovery for protein-protein interactions. Trends Pharmacol Sci. 2012;33(5):241–248. doi:10.1016/j.tips.2012.03.006
33. Hartman ZC, Poage GM, den Hollander P, et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013;73(11):3470–3480. doi:10.1158/0008-5472.CAN-12-4524-T
34. Lee ST, Li Z, Wu Z, et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43(5):798–810. doi:10.1016/j.molcel.2011.08.011
35. Li S, Kendall SE, Raices R, et al. TWIST1 associates with NF-kappaB subunit RELA via carboxyl-terminal WR domain to promote cell autonomous invasion through IL8 production. BMC Biol. 2012;10:73. doi:10.1186/1741-7007-10-73
36. Park B-W, Zhang H-T, Wu C, et al. Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo. Nat Biotechnol. 2000;18(2):194–198. doi:10.1038/72651
37. Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799(10–12):775–787. doi:10.1016/j.bbagrm.2010.05.004
38. Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8(1):33–40. doi:10.1038/nrd2781
39. Umezawa K. Possible role of peritoneal NF-κB in peripheral inflammation and cancer: lessons from the inhibitor DHMEQ. Biomed Pharmacother. 2011;65(4):252–259. doi:10.1016/j.biopha.2011.02.003
40. Murali R, Greene MI, Inventors; (The Trustees of the University of Pennsylvania, USA). assignee. Cavity-induced allosteric modification of intermolecular interactions and methods of identifying compounds that effect them. US patent 99-US15062 2000001349. 19990701. 2000.
41. Chen X, Kandasamy K, Srivastava RK. Differential roles of RelA (p65) and c-Rel subunits of nuclear factor kappa B in tumor necrosis factor-related apoptosis-inducing ligand signaling. Cancer Res. 2003;63(5):1059–1066.
42. Jacque E, Billot K, Authier H, Bordereaux D, Baud V. RelB inhibits cell proliferation and tumor growth through p53 transcriptional activation. Oncogene. 2013;32(21):2661–2669. doi:10.1038/onc.2012.282
43. Ding H, Gangalum PR, Galstyan A, et al. HER2-positive breast cancer targeting and treatment by a peptide-conjugated mini nanodrug. Nanomedicine. 2017;13(2):631–639. doi:10.1016/j.nano.2016.07.013
44. Levine DH, Ghoroghchian PP, Freudenberg J, et al. Polymersomes: a new multi-functional tool for cancer diagnosis and therapy. Methods. 2008;46(1):25–32. doi:10.1016/j.ymeth.2008.05.006
45. Guillemard V, Nedev HN, Berezov A, Murali R, Saragovi HU. HER2-mediated internalization of a targeted prodrug cytotoxic conjugate is dependent on the valency of the targeting ligand. DNA Cell Biol. 2005;24(6):350–358. doi:10.1089/dna.2005.24.351
46. DeLano WL The PyMOL Molecular Graphics System. Palo Alto, CA, USA: DeLando Scientific LLC; 2007. Available from: http://www.pymol.org.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.