")
Back to Journals » OncoTargets and Therapy » Volume 12
Diosgenin Suppresses Cholangiocarcinoma Cells Via Inducing Cell Cycle Arrest And Mitochondria-Mediated Apoptosis
Authors Mao XM, Zhou P, Li SY, Zhang XY, Shen JX, Chen QX, Zhuang JX, Shen DY
Received 6 August 2019
Accepted for publication 16 October 2019
Published 4 November 2019 Volume 2019:12 Pages 9093—9104
DOI https://doi.org/10.2147/OTT.S226261
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Xiao-Mei Mao,1,* Pan Zhou,1,* Si-Yang Li,2,* Xiao-Yun Zhang,2 Jin-Xing Shen,2 Qing-Xi Chen,1 Jiang-Xing Zhuang,3 Dong-Yan Shen2
1School of Life Sciences, Xiamen University, Xiamen 361102, People’s Republic of China; 2Biobank, The First Affiliated Hospital, School of Medicine, Xiamen University, Xiamen 361003, People’s Republic of China; 3Fujian Provincial Key Laboratory of Neurodegenerative Disease and Aging Research, Institute of Neuroscience, School of Medicine, Xiamen University, Xiamen, Fujian 361102, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dong-Yan Shen
Biobank, The First Affiliated Hospital, School of Medicine, Xiamen University, Xiamen 361003, People’s Republic of China
Tel +86 592 213 7507
Fax +86 592 213 7509
Email [email protected]
Jiang-Xing Zhuang
Fujian Provincial Key Laboratory of Neurodegenerative Disease and Aging Research, Institute of Neuroscience, School of Medicine, Xiamen University, Xiamen, Fujian 361102, People’s Republic of China
Email [email protected]
Purpose: Diosgenin (DSG) is the precursor of steroid hormones and plays a crucial part in the proliferation of various carcinomas including human colorectal cancer and gastric carcinoma. Nevertheless, its specific features and mechanisms in human cholangiocarcinoma (CCA) remain unknown.
Methods: MTS assay, colony-forming assay, and EdU assay were performed to determine the role of DSG on the progression of human CCA cells. The distributions of cell cycle, the ratio of apoptosis, and the mitochondrial membrane potential (ΔΨm) were studied by flow cytometry (FCM). AO/EB and Hoechst 33258 staining were performed to observe the morphological features of cell apoptosis. TEM was performed to observe the ultrastructures of QBC939 and HuCCT1 cells. The mRNA and protein expression of mitochondrial apoptotic pathway and GSK3β/β-catenin pathway were further confirmed by qPCR and Western blotting. The xenograft tumor model of HuCCT1 cells was built. Immunohistochemistry of tumor tissues was performed.
Results: Our results indicated that DSG inhibited the progression of six CCA cell lines. In vivo tumor studies also indicated that DSG significantly inhibited tumor growth in xenografts in nude mice. The expression of mitosis-promoting factor cyclinB1 was decreased along with the elevating level of cell cycle inhibitor p21, resulting in arresting CCA cell cycles at G2/M phase. Furthermore, DSG induced apoptosis with the increased expressions of cytosol cytochrome C, cleaved-caspase-3, cleaved-PARP1 and the Bax/Bcl-2 ratio. Mechanistically, our study showed that GSK3β/β-catenin pathway was involved in the apoptosis of CCA cells. Thus, DSG might provide a new clue for the drug therapy of CCA.
Conclusion: In our data, DSG was found to have efficient antitumor potential of human CCA cells in vitro and in vivo.
Keywords: diosgenin, cholangiocarcinoma, apoptosis, mitochondria, GSK3β/β-catenin pathway
Introduction
Cholangiocarcinoma (CCA) is one of the common malignant tumors. Its typical features include serious drug resistance and poor prognosis.1 Now surgery remains to be the primary treatment of CCA, along with radiotherapy and chemotherapy. Most patients are diagnosed in an advanced stage with metastasis.2,3 These observations emphasize the need to develop effective therapies against CCA.
Diosgenin (DSG) is a naturally occurring steroidal sapogenin, a major bioactive steroidal saponin found in natural plants, including tuber of wild yam (Dioscorea villosa Linn) and seed of fenugreek (Trigonella Foenum graecum Linn).4,5 As the main precursor of steroidal hormones, DSG is of great importance in pharmaceutical industry.6 Further, it is used in various human diseases, such as cancer, hypercholesterolemia, inflammation.7 Its functions include anti-inflammation, anti-infection, anticoagulation, anti-tumor, liver protection, and prevention of bone loss.8–11 In recent years, the anti-tumor role of DSG has been found in colon cancer, squamous cell carcinoma.12–19 However, the role of DSG on CCA is currently unknown.
Two common pathways of apoptosis are death receptor pathway and mitochondrial pathway. Mitochondrial dysfunction is an important marker. Stimulation caused the change of membrane permeability, then result in the loss of ΔΨm. Then, Bcl-2 family is regulated, followed by the cleavage of Caspase. With it, PARP proteins are cleaved, which induces the occurrence of apoptosis.
Substantial attention has been focused on the ectopic activation of the GSK3β⁄β-catenin signaling in various diseases, including CCA. In the existence of Wnt ligands, β-catenin accumulates, enters into the nucleus, and then initiates transcription. On the other hand, when without Wnt ligands, β-catenin binds to the destruction complex. Next, β-catenin is phosphorylated and degraded.
In this study, DSG was found to exert the remarkable antitumor potential of human CCA cells in vitro and in vivo.
Materials And Methods
Reagents
Diosgenin, RIPA buffer was obtained from Sigma Aldrich (St. Louis, MO, USA). RPMI-1640, DMEM medium, fetal bovine serum (FBS), trypsin, penicillin, and streptomycin were purchased from Gibco (Rockville, MD, USA). PI (propidium iodide), Annexin V-FITC/PI apoptosis detection kit, z-VAD-fmk, MTS, Hoechst 33258, and AO/EB (acridine orange/ethidium bromide) were purchased from Abcam (Cambridge, United Kingdom). 5-Ethynyl-2-deoxyuridine (EdU) was obtained from RiboBio (Guangzhou, China). The primary antibodies against GAPDH, Bax, Bcl-2, Caspase- 3, p21, PARP-1, cytochrome c (CYT C), COX IV, β-catenin and GSK3β were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Other antibodies were from Abcam. Hematoxylin–Eosin Staining Kit was from Solarbio (Beijing, China). The EliVision kit was from Maixin Biotech (Fuzhou, China).
Cell Culture And Treatment
The human CCA cell lines HuCCT1, QBC939, SK-ChA-1, HuH28, RBE, and Mz-ChA-1 were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cell lines were cultured in RPMI-1640 or DMEM medium with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were incubated under 37°C, 5% CO2. DSG was dissolved with anhydrous ethanol, and diluted to different concentrations with medium.
Cell Viability Analysis
Cell survival was analyzed by MTS assay. Briefly, cells were seeded at the density of 5 × 103 cells/well on 96-well microplates. Next day, cells were incubated with DSG of different concentrations (0, 10, 20, 40, 80 μM). After 24 h, 48 h or 72 h, 20 µl MTS was added to each well containing 100 µl medium. The cells were incubated at 37°C for 4 hrs. Finally, the absorbance values were measured at 490 nm by a microplate reader (Molecular Devices).
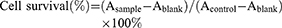
AO/EB And Hoechst 33258 Staining
QBC939 and HuCCT1 cells were seeded on the six-well plates overnight and then treated with DSG of different concentrations (0, 5, 10, 20, 40 μM). After 24 h, cells were washed with PBS and incubated with 10 μg/mL AO/EB. For another staining, cells were exposed to 10 μg/mL Hoechst 33258. Then, morphological changes of cells were photographed.
Cell Colony Formation Assay
Cells were seeded at a density of 500 cells/well on 6 cm plates. Cells of control group were incubated with fresh medium, and the other cells were treated with DSG (0, 5, 10, 20, 40 μM), respectively. Twelve days later, cells were fixed with methanol for 15 min and then stained with 0.1% crystal violet, and counted.
EdU Assay
A total of 5 × 103 cells were seeded on each well of a 96-well plate overnight. Next day, cells were incubated with 100 μL EdU solution (50 μM) for 2 h at 37°C. Then, cells were fixed in 4% paraformaldehyde at room temperature for 30 min, followed by permeabilization with 0.5% Triton X-100. Subsequently, the cells were stained with 1 × Apollo reaction mixture for 30 min. Finally, the nuclei were stained with Hoechst 33342. Images were taken using a fluorescence microscope.
Cell Cycle Analysis
CCA cells were treated with DSG for 24 h, then collected and washed. Then, 70% ethanol was added in cells for fixation. Next, cells were resuspended, and incubated with 5 μL of 10 mg/mL RNase at 37°C for 30 min. After that, DNA was treated with 5 μL of 10 mg/mL PI for 30 min at 4°C without light. The DNA content were monitored by a flow cytometer (FC500, Beckman Coulter), and the data were performed with ModFit LT 3.3 software.
Annexin V-FITC/PI Staining
Cell apoptosis was evaluated using Annexin V/PI apoptosis detection kit (Abcam) using the instruction book and examined by flow cytometry. Briefly, cells were seeded on the six-well plates and incubated with DSG for 24 h, then trypsinized, and resuspended using the binding buffer. Subsequently, 5 μL of Annexin V-FITC and 5 μL of PI were added and incubated for 15 min. Apoptotic cells were analyzed by an FC500 flow cytometer.
Determination Of Δψm
The treated cells were collected and incubated with JC-1 dye for 1 h. Then, the ΔΨ m was quantitatively measured and analyzed by an FC500 flow cytometer. In a general way, the proportion of JC-1 fluorescence by red/green reflects the change of ΔΨm.
Observation Of Cells With Transmission Electron Microscopy (TEM)
Cells were fixed and dehydrated, then cut into ultrathin sections. Then, the sections were stained with saturated uranyl acetate and lead citrate. The ultrastructure of cells and mitochondria was photographed under TEM (Tecnai G2 Spirit, FEI).
Cytochrome C (CYT C) Release Assay
Mitochondria were isolated from cells according to Cell Mitochondria Isolation Kit (Beyotime, China).
Western Blot
The CCA cells were collected and lysed. The proteins were separated by SDS-PAGE and transferred to PVDF membranes. Then, primary antibodies and secondary antibodies were added. The immunoreactive products were detected using enhanced chemiluminescence (Advansta).
Quantitative Real-Time PCR (qPCR)
Total RNA was extracted from the CCA cells and reversely transcribed into cDNA. The measurement was operated on an ABI Prism7500 system (Bio-Rad) using the Fast SYBR Green Master Mix kit (Tiangen, China). The primers used are in Table 1.
 |
Table 1 Primers Of Real-Time Fluorescence Quantitative PCR |
Animal Model
HuCCT1 cells were used to build animal model. Nude mice (BALB/c) were purchased from SLAC Laboratory Animal (Shanghai, China). HuCCT1 cells (3 × 106) were subcutaneously injected into the right oxter of mice, respectively. Tumor volumes were calculated: A×B2/2(A and B, respectively, represent the long and short diameter of the transplanted tumor). The control groups and treated groups received solvent and 50 mg/kg DSG, respectively (every 2 days, intragastric administration, n = 6). All of the animal experiments were approved by the Animal Ethics Committee of Xiamen University. All experimental procedures involving animals were performed in accordance with animal protocols approved by the Laboratory Animal Center of Xiamen University.
The tumor volume (day n) was calculated using relative tumor volume (RTV=Vt/V0; Vt is the volume of the transplanted tumor measured each time, and V0 is the volume of transplanted tumor measured when grouping drugs.). T/C (%), relative tumor proliferation rate used the formula T/C (%) = mean RTV of the treated group/mean RTV of the control group × 100%.
Statistical Analysis
All the experiments were repeated three times, and all the data were expressed as the mean ± standard deviation (SD) and studied using IBM SPSS 22.0 software.
Results
DSG Inhibited CCA Cells Proliferation And Colony Formation
The structure of DSG was shown in Figure 1A.12 We examined the efficacy of DSG on CCA cell lines (Figure 1B), and the data exhibited that DSG suppressed all six CCA cell lines in a dose- and time-dependent manner. QBC939 and HuCCT1 cell lines were employed for further study. The IC50 values were shown in Table 2. Furthermore, the colony-forming number of these two cell lines was significantly reduced by DSG compared with control (Figure 1C). After the initial cytotoxicity, DSG did not seem to affect the capability of the cells that still alive to grow and form the colony. Moreover, the results of EdU assays illustrated that the DNA synthesis of CCA cells was remarkably decreased in DSG-treated cells (Figure 1D). Taken together, those data indicated that DSG significantly suppressed the malignancy of CCA cells in vitro.
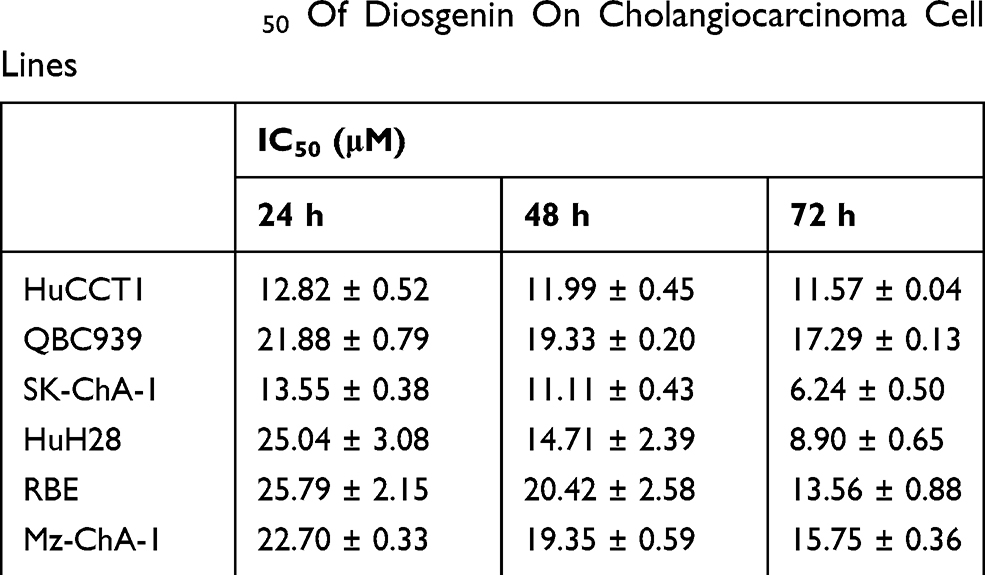 |
Table 2 The IC50 Of Diosgenin On Cholangiocarcinoma Cell Lines |
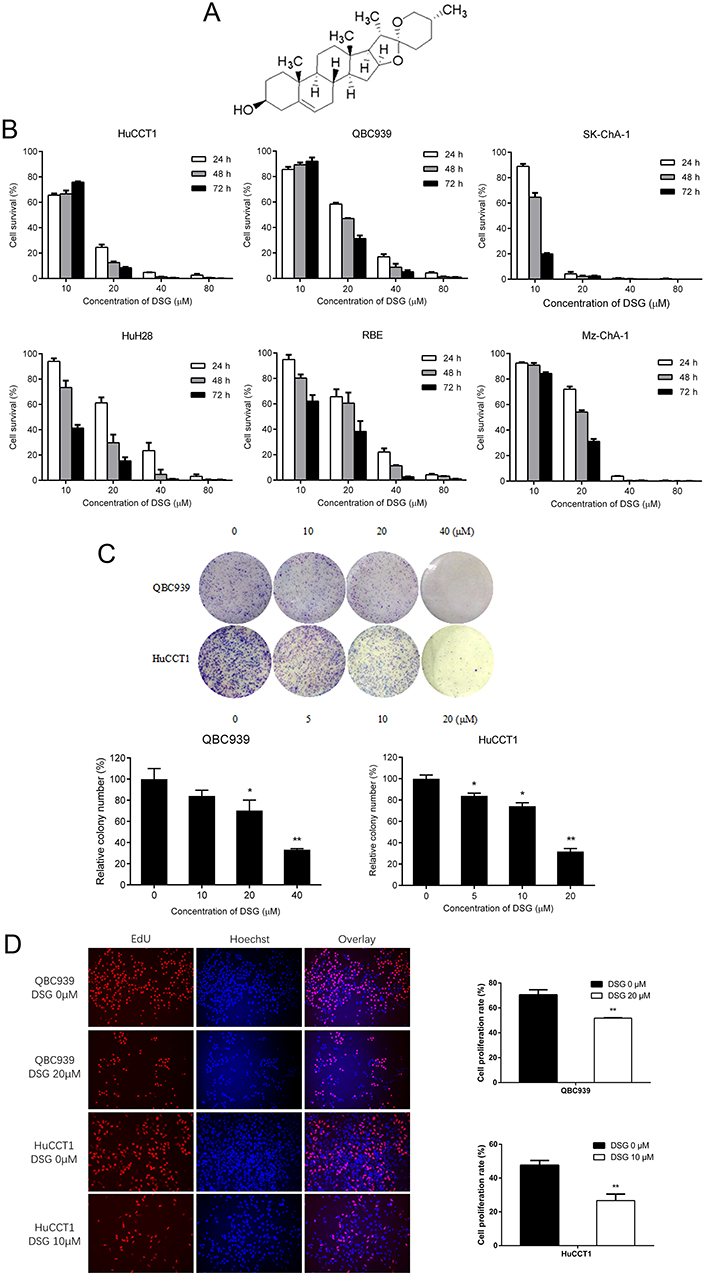 |
Figure 1 Diosgenin (DSG) inhibited the cell proliferation of cholangiocarcinoma (CCA). (A) The structure of DSG. (B) The role of DSG on the cell survival of CCA cells using MTS assay. (C) Colony-formation assays were performed in QBC939 and HuCCT1 cells treated with DSG. The relative number of colonies was quantified. (D) EdU assay was used to assess DNA synthesis of DSG-treated cells (left). Random 10 visions were quantified (right). *P < 0.05 and **P < 0.01. |
DSG Induced Cell Cycle Arrest In CCA Cells
The distributions of cell cycle were studied by flow cytometry (FCM). The ratio of cells in G2/M phase increased, implying that DSG arrested CCA cells at G2/M phase (Figure 2A). For QBC939 and HuCCT1 cell lines, the percentages of cells in G2/M phase increased from 8.06 ± 1.99% to 20.52 ± 2.17%, and 7.79 ± 0.56% to 16.70 ± 3.16%, respectively. Meanwhile, the protein and mRNA levels of cyclinB1 decreased after the treatment of DSG (Figure 2B and C), which were necessary for the transition of G2/M phase. Besides, the expression of cell cycle inhibitor P21 increased slightly in QBC939 cells, but had no significant differences in HuCCT1 cells.
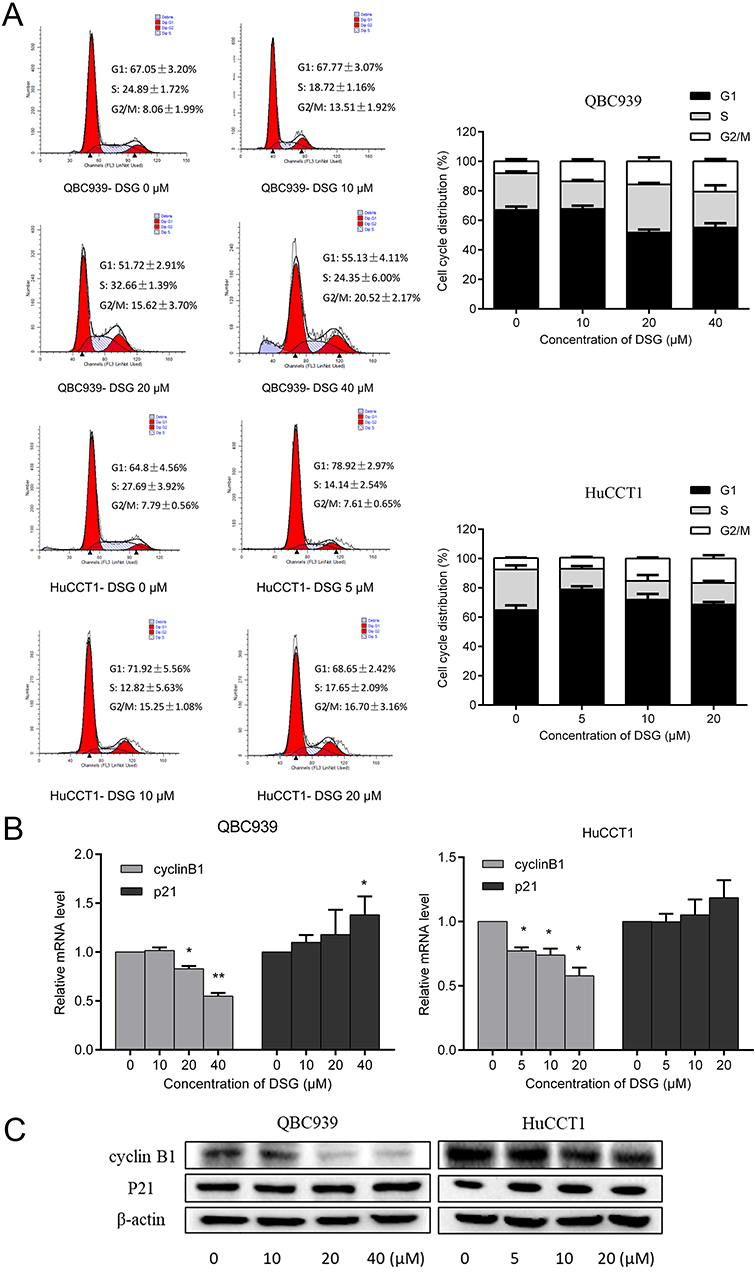 |
Figure 2 The change of cell cycle distribution after treatment with DSG. (A) Cells were treated with DSG at various concentrations for 24 h, and examined by FCM. Representative results were shown (left). Histogram showed the quantified data (right). (B, C) The qPCR and Western blot analysis for the expression of cyclinB1 and P21. *P < 0.05 and **P < 0.01. |
DSG Induced Cell Apoptosis In Vitro
AO/EB and Hoechst 33258 staining indicated the typical morphological features of cell apoptosis with the treatment DSG (Figure 3A and B). For Hoechst 33258 staining, DSG-treated cells exhibited brighter blue light than control, suggesting the chromatin condensation of nuclei. For AO/EB staining, the control cells showed green fluorescence and cell structures were intact, while treated cells emitted orange and red fluorescence.
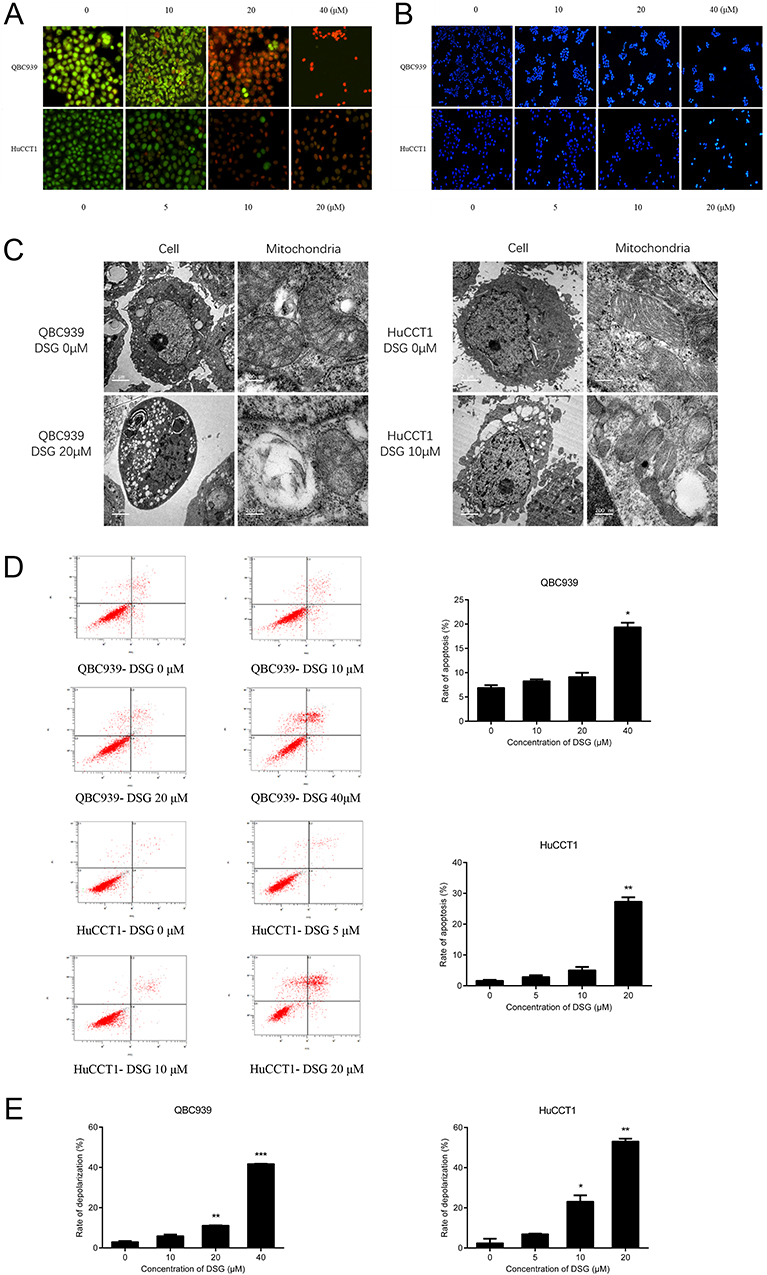 |
Figure 3 Cell apoptosis induced by DSG in CCA cells. (A, B) AO/EB and Hoechst 33258 staining of QBC939 and HuCCT1 cells. (C) The ultrastructures of cells and mitochondria in CCA cells were observed by TEM after DSG treatment. (D) FCM analysis of apoptosis using Annexin V-FITC/PI staining. Histogram showed the rates of apoptotic cells. (E) FCM analysis of ΔΨm. *P < 0.05, **P < 0.01, and ***P < 0.001. |
TEM was performed to observe the ultrastructures of QBC939 and HuCCT1 cells. For both cell lines, we could observe the normal cell morphology in the control sample: integrated cell nucleus and diffused chromatin. On the other hand, DSG-treated sample exhibited typical apoptotic morphology: cell body and nucleus shrinkage, the chromatin condensed, separated and moved to the inside edge of nuclear envelope (Figure 3C). What is more, mitochondria were swollen and their cristae were broken after DSG treatment.
The FCM data were used to determine the ratio of apoptosis with double staining. With the increasing concentrations of DSG, the rates of apoptosis of QBC939 cells were raised from 6.90 ± 0.48% to 19.38 ± 1.27%, and HuCCT1 cells were from 1.67 ± 0.33% to 27.33 ± 1.97% (Figure 3D). The influence of DSG on ΔΨm was also studied using FCM. With the DSG concentration increased, the rates of depolarization raised from 3.04 ± 0.71% to 41.79 ± 1.79%, and from 2.48 ± 0.47% to 53.13 ± 1.78%, respectively (Figure 3E). It suggested the collapse of ΔΨm in CCA cells.
The Mitochondria-Mediated Intrinsic Pathway And GSK3β/Β-Catenin Pathway Involved In The Antitumor Activity Of DSG In CCA Cells
Based on the changes in morphology and ΔΨm of mitochondria with DSG treatment, we further evaluated the expression of proteins. Western blot data indicated that DSG treatment elevated the ratio of Bax/Bcl-2 and activated PARP-1, Caspase-3. Besides, CYT C released from mitochondria to cytoplasm (Figure 4A and B). The Caspase inhibitor z-VAD-fmk (20 μM) significantly attenuated the DSG-induced cell apoptosis (Figure 4C). Our results suggested that DSG induced apoptosis of CCA cells via activation of Caspases.
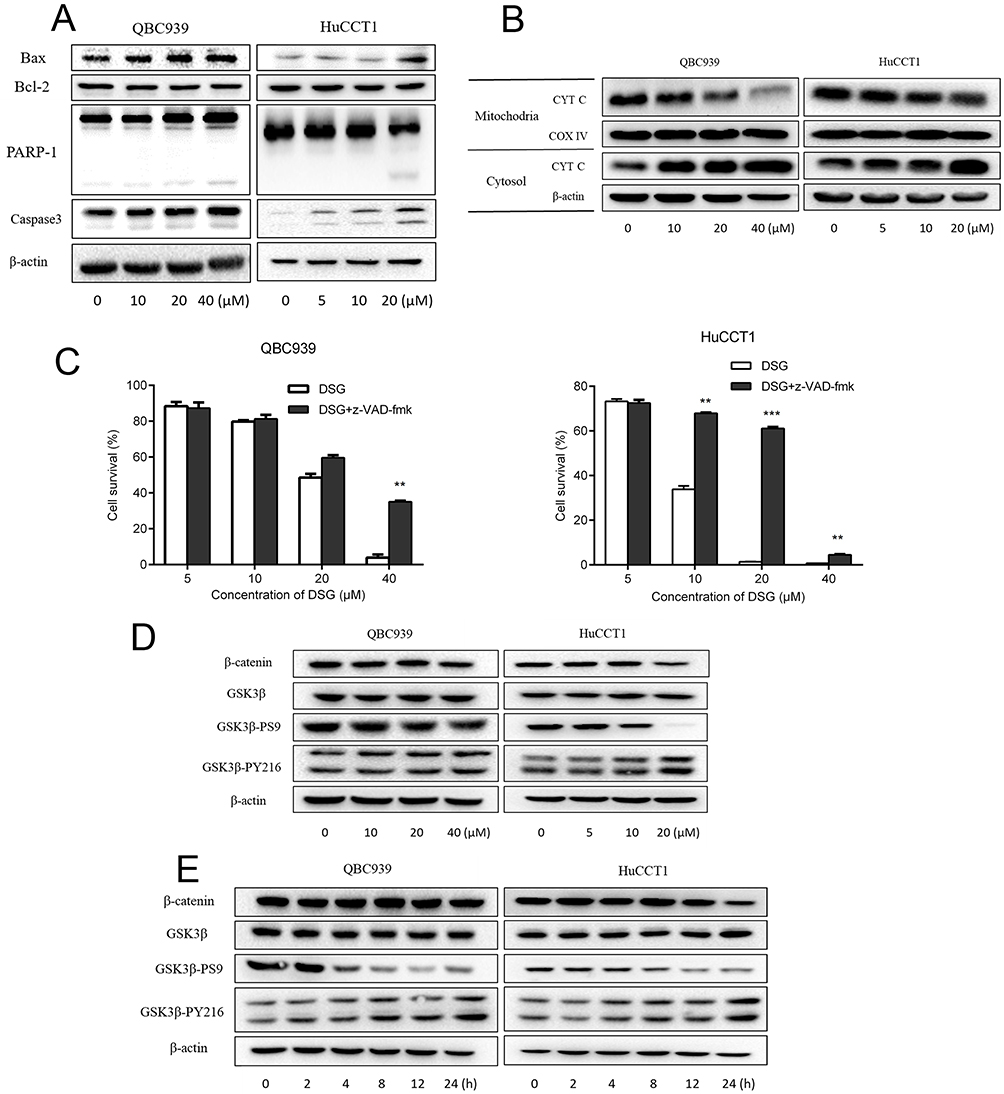 |
Figure 4 The involvement of intrinsic mitochondria pathway and GSK3β/β-catenin pathway in the DSG treatment. (A, B) Western blot for cell lysis collected to analyze the expression of mitochondria pathway–related proteins. (C) Cell survival rates were studied after treating with DSG in the presence or absence of 20 μM z-VAD-fmk. (D, E) GSK3β/β-catenin pathway was detected. **P < 0.01, and ***P < 0.001. |
A large amount of oncogenic factors induce ectopic activation of the GSK3β⁄β-catenin signaling in various tumors, including CCA. To characterize the effect of GSK3β/β-catenin signaling on the antitumor activity induced by DSG, we detected related proteins of this pathway (Figure 4D and E). The results showed that the phosphorylation level of GSK3β at Ser-9 decreased and the phosphorylation at Tyr-216 increased, indicating its activation, which could cause the degradation of β-catenin. Some results of QBC939 cells were not as obvious as HuCCT1 cells. We came to a conclusion that the suppression of GSK3β/β-catenin pathway might be responsible for the DSG-induced apoptosis.
DSG Suppressed The Proliferation Of CCA Cell In Xenograft
We built the xenograft tumor model of HuCCT1 cells. The body weight of mice showed no remarkable changes (Figure 5A), while tumor weights and volumes decreased apparently (Figure 5D and E). To verify the non-toxicity of DSG, the internal organs of animal were weighed and stained with HE staining (Figure 5B and C). The data indicated that viscus had no significant changes after DSG treatment. In the treatment group, for example, all the hepatocytes showed obvious nucleus structure, no punctate or patchy necrosis, and swelling. The typical alveolar reticular structure was seen in all lung tissues. The contrast between the nuclei and cytoplasm was obvious, and the nuclei were clearly visible. By contrast, HE staining of xenograft tumors in DSG-treated group exhibited classic apoptotic trait: condensed chromatin and pyknotic nuclei (Figure 5F). Besides, IHC analysis indicated that PCNA and Ki67 positive cells were significantly decreased in the xenograft tumor from DSG-treated group (Figure 5G). Furthermore, our data revealed that DSG greatly inhibited the progression of HuCCT1 xenograft tumors (T/C% = 36.6%). The results of Western blot (Figure 5H) are in line with our previous results in cell culture, showing the involvement of mitochondria-mediated apoptosis pathway and GSK3β/β-catenin pathway in DSG treatment.
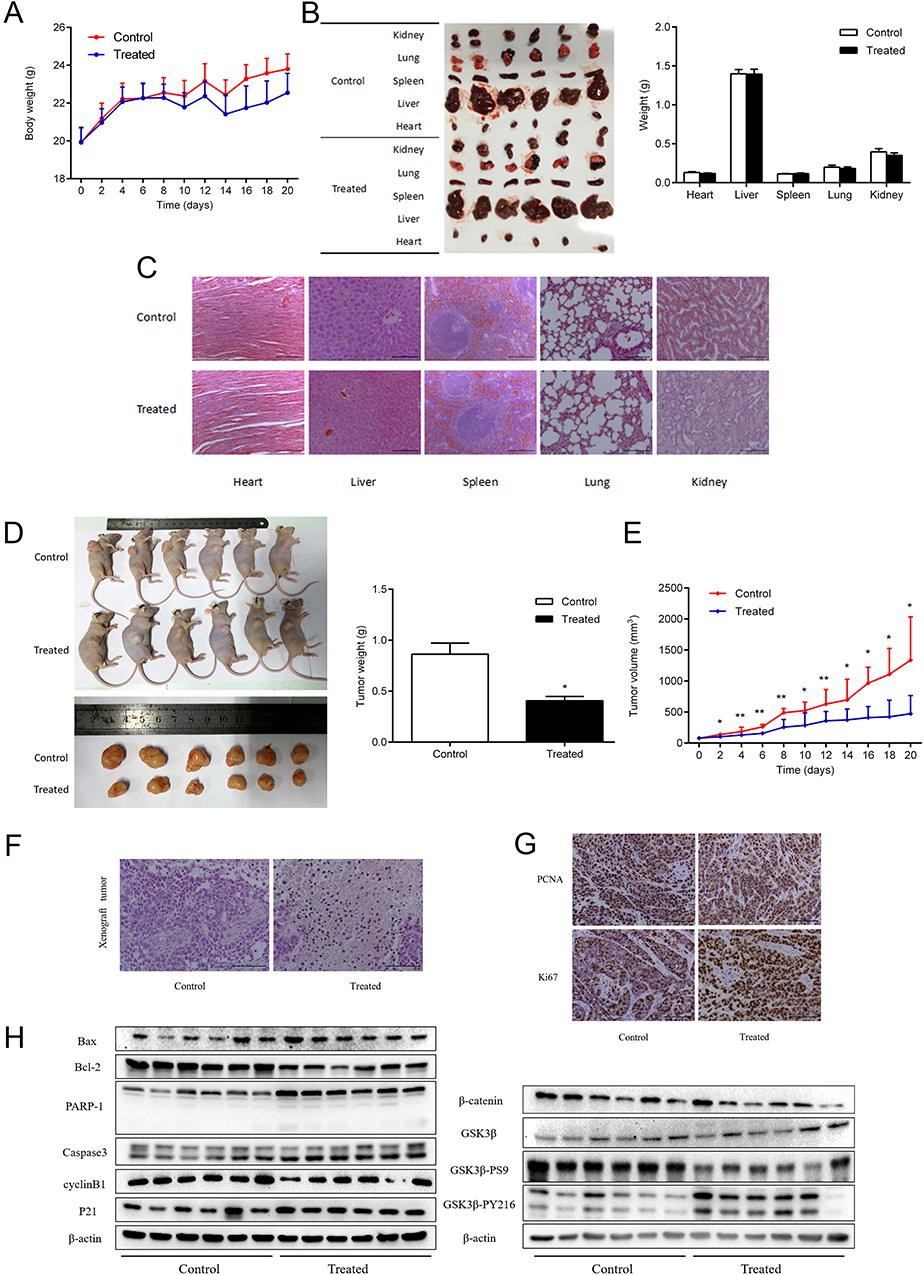 |
Figure 5 DSG suppressed the growth of CCA in vivo without toxic to viscera. (A) The variational curve of nude mice body weight. (B) The mean weights of viscera after administration. (C, F) The HE staining of viscera and tumors. (D) Mean tumor weight at the end of 20 days. (E) Mean tumor volume on the indicated days. (G) IHC analysis of PCNA and Ki67 in the xenograft tumor of nude mice. (H) Western blot assay to confirm the expression of proteins. *P < 0.05 and **P < 0.01. |
Discussion
Previously reports have demonstrated that DSG inhibits the malignancy of various carcinoma.20–22 However, its functions on CCA need to be studied further. DSG, a natural steroidal saponin, is an important precursor for the synthesis of steroidal hormone, which has high medicinal value.23,24 This experiment proved that DSG inhibited the malignancy of CCA cells through inducing G2/M phase arrest and mitochondria-mediated apoptosis.
CyclinB1 is a regulatory protein in G2 checkpoint and plays important roles in mitotic initiation regulation and cell cycle regulation.25 CyclinB1 overexpression can promote G2/M phase transformation, accelerate cell cycle progression, and contribute to malignant cell transformation. P21 is a cell cycle protein-dependent kinase inhibitor, which can inhibit the activity of most cyclin-dependent kinase (CDK).26 It can not only inhibit cell proliferation but also trigger cell cycle arrest. P21 can promote cyclinB1 nuclear transposition and block cells at G2/M stage.27 The expression changes of p21 and cyclinB1 in DSG-treated CCA cells were studied by Western blot and qPCR. The results illustrated that DSG remarkably decreased the expression of cyclinB1. So we speculated that DSG induced G2/M phase arrest, affected the smooth progress of the cell cycle, and thus suppressed the progression of CCA cells. However, the exact mechanism needs further exploration and verification. The cell proliferation is regulated through cell cycle and the occurrence of tumor may lie in deregulation of cell cycle, directly resulted from the abnormal of the complex of CDK. The catalytic activities of CDKs are modulated by interactions with cyclins and CDK inhibitors (CKIs).28 Whereas most cyclins promote CDK activity, CKIs suppress CDK activity. CKIs are divided into two classes based on their structure and specificity. The Ink4 family members, including p16, p15, p18, and p19, mainly target Cdk4 and Cdk6. On the other hand, p21, p27, and p57 are the members of Cip/Kip family. They are more promiscuous and broadly interfere with the activities of cyclin D-, E-, A- and B-dependent kinase complexes.29 In addition, previous studies have found that more than 10 microRNAs were involved in cell cycle regulation. Among them, mir-1-2 and mir-34 targeted CDK4, which induced cell cycle arrest in G1 phase.30 Therefore, the regulation of cell cycle is a complex process, and many studies need to be carried out.
Apoptosis is a process of multi-pathway regulation. For example, it includes Caspase activation, cleaved-PARP elevation, phosphatidylserine overturning on cell membrane, and chromatin condensation.31–36 According to the results of AO/EB, Hoechst 33258 fluorescent staining and Annexin V-FITC/PI staining, DSG inhibited the progression and induced apoptosis of CCA cells in vitro.
Endogenous mitochondrial pathway is crucial to cell apoptosis regulation. The endogenous approach in many events is closely related to mitochondrial, such as the destruction of mitochondria outer membrane, the release of Cyt C, the participation of apoptosis-related proteins.37 Both Caspase family proteins and Bcl-2 family proteins are crucial in this process. Detecting the apoptosis-related proteins by Western blot, the proportion of Bax/Bcl-2, Caspase-3 and Cleaved-PARP was increased in a dose-dependent manner. The data also showed that CYT C released from mitochondria to the cytoplasm. Under the existence of Caspase inhibitors z-VAD-fmk, MTS results revealed remarkably increase cell vitality of HuCCT1 cells. Above all, we speculated that DSG induced CCA cell apoptosis through the intrinsic mitochondrial pathway.
Interestingly, we found that GSK3β/β-catenin pathway involved in apoptosis inducing of CCA cells. The results showed that the phosphorylation level of GSK3β at Ser-9 reduced and the phosphorylation at Tyr-216 increased, indicating its activation. Therefore, this activation promoted the ubiquitin and degradation of β-catenin, and ultimately affected the CCA cell proliferation. To sum up, DSG might influence the progression and apoptosis of CCA cells through GSK3β/β-catenin signaling.
The xenograft assay in nude mice demonstrated DSG effectively inhibited the growth of CCA in vivo without obvious toxic and side effects. Down-regulated PCNA and Ki67 protein expression of DSG implied decreased proliferation ability. The expression of p21 and the Bax/Bcl-2 proportion was increased. And the expressions of cleaved Caspase-3 and PARP-1 were significantly enhanced by DSG too. Besides, the changes in GSK3β/β-catenin pathway were consistent with the results in vitro. Generally, our study proved that the DSG suppressed CCA cells and induced apoptosis in vitro and in vivo. From a natural source, DSG had remarkable potential to become one of the chemotherapy drugs in clinical.
Acknowledgment
This study was supported by the National Natural Science Foundation of China (Grant No. 81870435).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Khan SA, Thomas HC, Davidson BR, Taylorrobinson SD. Cholangiocarcinoma. Lancet. 2005;366(9493):1303–1314. doi:10.1016/S0140-6736(05)67530-7
2. Patel T. Cholangiocarcinoma. Nat Clin Pract Gastroenterol Hepatol. 2006;3(1):33–42. doi:10.1038/ncpgasthep0389
3. Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935):2168. doi:10.1016/S0140-6736(13)61903-0
4. Chen Y, Tang YM, Su-Lan YU, et al. Advances in the pharmacological activities and mechanisms of diosgenin. Chin J Nat Med. 2015;13(8):578–587. doi:10.1016/S1875-5364(15)30053-4
5. Selim S, Jaouni SA. Anticancer and apoptotic effects on cell proliferation of diosgenin isolated from Costus speciosus (Koen.) Sm. BMC Complement Altern Med. 2015;15(1):301. doi:10.1186/s12906-015-0836-8
6. Salvador JA, Carvalho JF, Neves MA, et al. Anticancer steroids: linking natural and semi-synthetic compounds. Nat Prod Rep. 2013;30(2):324–374.
7. Raju J, Rao CV. Diosgenin, a Steroid Saponin Constituent of Yams and Fenugreek: Emerging Evidence for Applications in Medicine. Bioactive Compounds in Phytomedicine. 2012:125–142.
8. Mayakrishnan T, Nakkala JR, Jeepipalli SPK, et al. Fenugreek seed extract and its phytocompounds- trigonelline and diosgenin arbitrate their hepatoprotective effects through attenuation of endoplasmic reticulum stress and oxidative stress in type 2 diabetic rats. Eur Food Res Technol. 2015;240(1):223–232. doi:10.1007/s00217-014-2322-9
9. Zhang Z, Song C, Fu X, et al. High-dose diosgenin reduces bone loss in ovariectomized rats via attenuation of the RANKL/OPG ratio. Int J Mol Sci. 2014;15(9):17130–17147. doi:10.3390/ijms150917130
10. Tikhonova MA, Ting CH, Kolosova NG, et al. Improving bone microarchitecture in aging with diosgenin treatment: a study in senescence-accelerated OXYS rats. Chin J Physiol. 2015;58(5):10.
11. Kalailingam P, Kannaian B, Tamilmani E, Kaliaperumal R. Efficacy of natural diosgenin on cardiovascular risk, insulin secretion, and beta cells in streptozotocin (STZ)-induced diabetic rats. Phytomed Int J Phytother Phytopharmacol. 2014;21(10):1154–1161. doi:10.1016/j.phymed.2014.04.005
12. Jiang SS, Fan JJ, Wang Q, et al. Diosgenin induces ROS-dependent autophagy and cytotoxicity via mTOR signaling pathway in chronic myeloid leukemia cells. Phytomedicine. 2016;23(3):243–252. doi:10.1016/j.phymed.2016.01.010
13. Légera DY, Liagrea B, Cardot PJP, Beneytout J-L, Battub S. Diosgenin dose-dependent apoptosis and differentiation induction in human erythroleukemia cell line and sedimentation field-flow fractionation monitoring. Anal Biochem. 2004;335(2):267–278. doi:10.1016/j.ab.2004.09.008
14. Das S, Dey KK, Dey G, et al. Antineoplastic and apoptotic potential of traditional medicines thymoquinone and diosgenin in squamous cell carcinoma. PLoS One. 2012;7(10):e46641. doi:10.1371/journal.pone.0046641
15. Yongjian L, Xiaorong W, Silu C, et al. Diosgenin induces G2/M cell cycle arrest and apoptosis in human hepatocellular carcinoma cells. Oncol Rep. 2015;33(2):693–698. doi:10.3892/or.2014.3629
16. Mao ZJ, Tang QJ, Zhang CA, et al. Anti-proliferation and anti-invasion effects of diosgenin on gastric cancer BGC-823 cells with HIF-1α shRNAs. Int J Mol Sci. 2012;13(5):6521–6533. doi:10.3390/ijms13056521
17. Rahmati Yamchi M, Ghareghomi S, Haddadchi G, Mobasseri M, Rasmi Y. Diosgenin inhibits hTERT gene expression in the A549 lung cancer cell line. Asian Pac J Cancer Prev. 2013;14(11):6945–6948. doi:10.7314/APJCP.2013.14.11.6945
18. Sung B, Prasad S, Yadav VR, Aggarwal BB. Cancer cell signaling pathways targeted by spice-derived nutraceuticals. Nutr Cancer. 2012;64(2):173–197. doi:10.1080/01635581.2012.630551
19. Patel K, Gadewar M, Tahilyani V, Patel DK. A review on pharmacological and analytical aspects of diosgenin: a concise report. Nat Prod Bioprospect. 2012;2:46–52. doi:10.1007/s13659-012-0014-3
20. Manivannan J, Arunagiri P, Sivasubramanian J, Balamurugan E. Diosgenin prevents hepatic oxidative stress, lipid peroxidation and molecular alterations in chronic renal failure rats. Int J Nut Pharmacol Neurol Dis. 2013;3(3):289. doi:10.4103/2231-0738.114870
21. Rahmati-Yamchi M, Ghareghomi S, Haddadchi G, Milani M, Aghazadeh M, Daroushnejad H. Fenugreek extract diosgenin and pure diosgenin inhibit the hTERT gene expression in A549 lung cancer cell line. Mol Biol Rep. 2014;41(9):6247–6252. doi:10.1007/s11033-014-3505-y
22. Li F, Fernandez PP, Rajendran P, Hui KM, Sethi G. Diosgenin, a steroidal saponin, inhibits STAT3 signaling pathway leading to suppression of proliferation and chemosensitization of human hepatocellular carcinoma cells. Cancer Lett. 2010;292(2):197–207. doi:10.1016/j.canlet.2009.12.003
23. Dong J, Lei C, Lu D, Wang Y. Direct biotransformation of dioscin into diosgenin in rhizome of dioscorea zingiberensis by penicillium dioscin. Indian J Microbiol. 2015;55(2):200–206. doi:10.1007/s12088-014-0507-3
24. Huang BZ, Xin G, Ma LM, et al. Synthesis, characterization and biological studies of diosgenyl analogues. J Asian Nat Prod Res. 2012;19(3):272–298. doi:10.1080/10286020.2016.1202240
25. Ying W, Yamaguchi Y, Watanabe H, Ohtsubo K, Wakabayashi T, Sawabu N. Usefulness of p53 gene mutations in the supernatant of bile for diagnosis of biliary tract carcinoma: comparison with K-ras mutation. J Gastroenterol. 2002;37(10):831–839. doi:10.1007/s005350200137
26. Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7(5):812–821. doi:10.1101/gad.7.5.812
27. Furubo S, Harada K, Shimonishi T, Katayanagi K, Tsui W, Nakanuma Y. Protein expression and genetic alterations of p53 and ras in intrahepatic cholangiocarcinoma. Histopathology. 2010;35(3):230–240. doi:10.1046/j.1365-2559.1999.00705.x
28. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140(15):3079–3093. doi:10.1242/dev.091744
29. Sherr CJ, Roberts J. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512. doi:10.1101/gad.13.12.1501
30. Freemantle SJ, Liu X, Feng Q, et al. Cyclin degradation for cancer therapy and chemoprevention. J Cell Biochem. 2007;102(4):869–877. doi:10.1002/(ISSN)1097-4644
31. Guo LL, Xiao S, Guo Y. Detection of bcl-2 and bax expression and bcl-2/JH fusion gene in intrahepatic cholangiocarcinoma. World J Gastroenterol. 2004;10(22):3251–3254. doi:10.3748/wjg.v10.i22.3251
32. Ito Y, Takeda T, Sasaki Y, et al. Bcl-2 expression in cholangiocellular carcinoma is inversely correlated with biologically aggressive phenotypes. Oncology. 2000;59(1):63–67. doi:10.1159/000012139
33. Li SM, Yao SK, Yamamura N, Nakamura T. Expression of Bcl-2 and Bax in extrahepatic biliary tract carcinoma and dysplasia. World J Gastroenterol. 2003;9(11):2579–2582. doi:10.3748/wjg.v9.i11.2579
34. Okaro AC, Deery AR, Hutchins RR, Davidson BR. The expression of antiapoptotic proteins Bcl-2, Bcl-X(L), and Mcl-1 in benign, dysplastic, and malignant biliary epithelium. J Clin Pathol. 2001;54(12):927–932. doi:10.1136/jcp.54.12.927
35. Yuen MF, Wu PC, Lai VC, Lau JY, Lai CL. Expression of c-Myc, c-Fos, and c-jun in hepatocellular carcinoma. Cancer. 2015;91(1):106–112. doi:10.1002/1097-0142(20010101)91:1<106::AID-CNCR14>3.0.CO;2-2
36. Voravud N, Foster CS, Gilbertson JA, Sikora K, Waxman J. Oncogene expression in cholangiocarcinoma and in normal hepatic development. Hum Pathol. 1989;20(12):1163–1168. doi:10.1016/S0046-8177(89)80006-1
37. Yadav N, Chandra D. Mitochondrial and postmitochondrial survival signaling in cancer. Mitochondrion. 2014;16(5):18–25. doi:10.1016/j.mito.2013.11.005
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.