")
Back to Journals » Psoriasis: Targets and Therapy » Volume 10
Dimethyl Fumarate Targets MSK1, RSK1, 2 and IKKα/β Kinases and Regulates NF-κB /p65 Activation in Psoriasis: A Demonstration of the Effect on Peripheral Blood Mononuclear Cells, Drawn from Two Patients with Severe Psoriasis Before and After Treatment with Dimethyl Fumarate
Authors Gesser B , Rasmussen MK , Iversen L
Received 8 January 2020
Accepted for publication 3 March 2020
Published 31 March 2020 Volume 2020:10 Pages 1—11
DOI https://doi.org/10.2147/PTT.S234151
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Uwe Wollina
Borbala Gesser,1 Mads K Rasmussen,1,2 Lars Iversen1,2
1Department of Dermatology, Aarhus University Hospital, Aarhus, Denmark; 2Department of Clinical Medicine, Aarhus University, Aarhus, Denmark
Correspondence: Borbala Gesser
Email [email protected]
Background: Dimethyl fumarate (DMF) has an inhibitory effect on the production of pro-inflammatory proteins from different cells which participate in the immune reaction in psoriatic skin. Most recently it was shown that DMF is an allosteric covalent inhibitor of the p90 ribosomal S6 kinases (RSK1, 2), determined by X-ray crystallography. DMF binds to a specific cysteine residue in RSK2 and in the closely related mitogen and stress-activated kinases 1 (MSK1) which inhibits further downstream activation.
Objectives: The aim of this study was to review the literature on the effects of DMF on activation of MSK1, RSK1, 2 kinases, and downstream transcription factors NF-κB/p65 and IκBα in cells contributing to the pathogenesis of psoriasis. We also hypothesized and studied if treatment with DMF would inhibit the activation of MSK1, RSK1, 2 kinases in peripheral blood mononuclear cells (PBMCs) in psoriatic patients.
Methods: PBMCs were purified from patients with severe psoriasis before and after 90 days of treatment with DMF. Cells were stimulated with anisomycin, IL-1β or EGF for 10 and 20 minutes. The levels of phosphorylation of MSK1, RSK1, 2 or NF-κB/p65, IκBα were analyzed by Western blotting.
Results: Our case study showed that treatment with DMF inhibited the activation of MSK1 and RSK1, 2 kinases in PBMCs in patients. This supports that DMF is the active metabolite in vivo in psoriatic patients during DMF treatment.
Conclusion: Pro-inflammatory proteins are induced through activation of MSK1 and NF-κB/p65 at (S276). The extracellular signal-regulated kinases (ERK1/2) control cell survival by activating both MSK1 and RSK1, 2 kinases. P-RSK1, 2 activates P-κBα and NF-κB/p65 at (S536). The phosphorylation of NF-κB/p65 at (S276) and (S536) controls different T cell and dendritic cell functions. DMF´s inhibitory effect on MSK1 and RSK1, 2 kinase activations reduces multiple immune reactions in psoriatic patients.
Keywords: psoriasis, DMF, MSK1, RSK1, 2, IKKα, IKKβ, NF-κB/p65, IκBα
Introduction
Fumaderm® is registered in Germany for systemic treatment of severe psoriasis.1 The main active ingredient is Dimethyl fumarate (DMF) but the mixture contains also monomethyl fumarate (MMF) and fumaric acid (FA). The European Medicines Agency (EMA) has currently approved two formulation containing DMF; Tecfidera® is registered for systemic treatment of multiple sclerosis.2 Skilarence® is registered for the treatment of moderate and severe psoriasis.3
Psoriasis vulgaris is an auto-inflammatory skin disease with an immune reaction mediated by T cells and dendritic cells. DMF changes the phenotype of the dendritic cells. Peripheral blood lymphocyte subsets composed of CD4+T helper cells, CD8+ cytotoxic T cells, activated Th1, Th17 and Th22 cells are all participating in the inflammatory immune reaction in psoriasis. The mature dendritic cells (DCs) have upregulated MHC class II and costimulatory molecules and release IL-23 and IL-12. Mature DCs make complex formation with the activated skin resident T cells. This stimulates Th1 cells to produce TNF-α, IFN-γ, Th17 cells to produce IL-17, and Th22 cells to produce IL-22.4 Lipopolysaccharides (LPS) stimulates DC maturation. DMF inhibited the LPS induced maturation of bone marrow derived dendritic cell (BMDCs) by inhibiting the expression of P-NF-κB/p65 (S276) which reduced the production of IL-23 and IL-12. DMF also inhibited the DC mediated T cell response by reducing the expression of MHC class II, CD80 and CD86 on DCs which also reduced the complex formation with CD4+ T cells. The immature DC phenotype generates fewer activated T cells leading to a decreased amount of IFN-γ and IL-17. The maturation of DCs was inhibited by DMF through suppressing the activation of P-ERK1/2, P-MSK1 and both P-NF-κB/p65 (S276) and (S536).5 The phosphorylation of the two serine sites in NF-κB/p65 at (S276) and (S536) control different T cell and DC functions. LPS activates the ERK1, 2 kinase pathways in DCs which supports DC survival. LPS also activates the p38α MAPK/NF-κB pathway which regulates DC maturation.6 Furthermore DMF inhibited in LPS stimulated bone marrow derived macrophages (BMDMs) the activation of P-ERK1/2 and P-NF-κB which reduced the production of pro-inflammatory cytokines.7 Stimulation of p38α MAPK pathway activates P-MSK1 while stimulation of ERK1/2 activates both P-MSK1 and P-RSK1, 2 kinases.8
DMF Inhibits the Production of Pro-Inflammatory Cytokines from Psoriatic T-Cells, Keratinocytes and Endothelial Cells, Which Inhibits Amplification of the Pro-Inflammatory Immune Reaction
DMF modulates T-cell cytokine secretion. In co-cultures of skin biopsies from lesional psoriatic skin and HUT78 T-cells, DMF diminished IFN-y but stimulated IL-10 secretion.9 In PBMCs isolated from psoriasis patients or healthy controls, stimulation with phytohemagglutinin (PHA), induced IL-17 and IL-22 mRNA expression which was higher in the patient group compared with controls. Treatment with DMF significantly reduced IL-17 and IL- 22 mRNA expression in the patient group, compared with healthy controls.10 IL-20 is produced by supra-papillary keratinocytes in lesional psoriatic skin.11 DMF inhibited in human keratinocytes the IL-1β induced activation of P-MSK1 (S376), P-NF-κB/p65 (S276) and the NF-κB p65/p50 DNA binding in human keratinocytes. In accordance with this DMF also inhibited the IL-1β induced expression of IL-8 and IL-20 mRNA.12 In human endothelial cells, DMF suppressed the TNF-α induced nuclear translocation of P-NF-κβ/p65 (S536) and the secretion of inflammatory cytokines.13 Exposure of endothelial cells to DMF reduced TNF-α induced expression of E-selectin, ICAM-1, VCAM-1 and the interaction with lymphocytes which reduced the lymphocyte rolling by 85.9%.14 In human PBMCs DMF also inhibited the expression of adhesion molecules in vivo and thereby diminished leukocyte rolling.15
DMF Inhibits the Activation of MSK1 and RSK1, 2 Kinases and the Phosphorylation of NF-κB P65 at Two Different Sites, Separately Controlled by IKKα/β
MSK1 is a serine/threonine protein kinase that is activated by the p38α MAPK and the ERK1/2 signaling pathways. The levels of phosphorylated forms of p38 MAPK, ERK1/2 and MSK1 are increased in lesional compared with non lesional psoriatic skin.16,17 MSK1 regulates the transcription of pro-inflammatory cytokines by phosphorylating NF-κB/p65 (S276) in the Rel domain of p65, which binds to IκBα.18–20 In the inactive state, IκBα is masking the nuclear localization site (NLS) of p65 and inhibits the nuclear translocation of p65.19 DMF inhibited the IL-1β induced activation of P-MSK1 (S376) and the transactivation of P-NF-κB/p65 (S276) in keratinocytes.12 Box 1 shows the cytokines and growth factors induced phosphorylation of RelA p65 which is mediated by MSK1 and RSK1, 2 respectively.
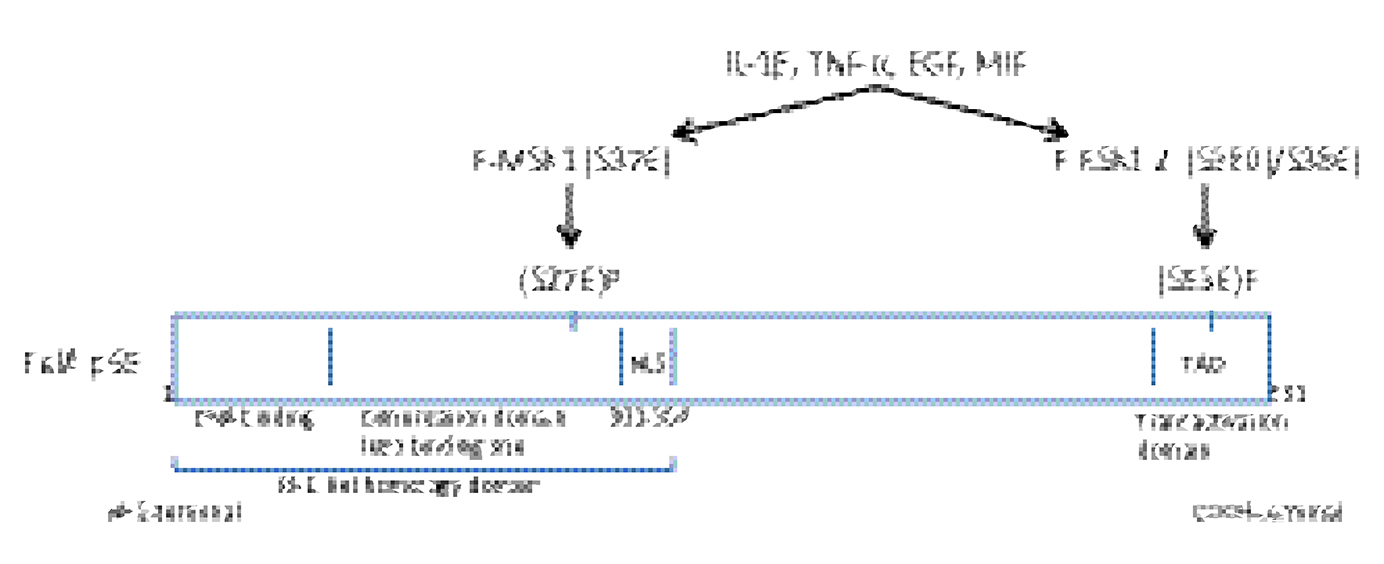 |
Box 1 Cytokine and growth factor activated Rel A p65. |
RSK1 and 2 are serine/threonine protein kinases that are activated by the ERK1/2 signaling pathways and play a critical role in cell proliferation. Elevated levels of growth factors in the serum of patients with psoriasis were suggested to activate RSK1, 2 which was demonstrated in biopsies from psoriatic skin.20 RSK1, 2 are directly involved in the regulation of cell-cycle progression by phosphorylating the transcription factor c-Fos which regulates the expression of cyclin D1 during G1/S transition.21 P-RSK2 phosphorylates histone H3 (S10) and several transcription factors that regulate gene expression through the RSK2/c-Fos/AP-1 signaling pathway.21 In COS1 cells TPA stimulation activated the expression of P-RSK1, 2 and P-IκBα (S32) which resulted in the rapid degradation of IκBα. In cells that express low level of RSK1, NF-κB could not be activated by TPA.22 RSK2 also inactivates proteins involved in apoptosis. EGF induced P-RSK2 in HEK293 cells which phosphorylated and stabilized P-Caspase-8 (Thr263) and resulted in inactivation of apoptosis.23,24 When RSK2 activation was blocked by kaempferol (a RSK2 inhibitor23 Suppl. Material 09–151290), the activity of Caspase-8 and −3 increased and led to apoptosis.23,24
Macrophage migration inhibitory factor (MIF) is a growth factor elevated in homogenates from lesional psoriatic skin compared with normal skin.25 There is a spontaneous higher MIF production by PBMCs from patients with extended psoriasis, than in normal controls.26 Stimulation with MIF induced proliferation in keratinocytes in vitro. Co-stimulation of keratinocytes with DMF and MIF inhibited the MIF induced activations of P-MSK1 (S376) and P-RSK1 (S380). This resulted in the inhibition of proliferation in keratinocytes mediated through the induction of the tumor suppressor protein P-p53 (S15)/p53.27 The induction of total p53 was significant after 96 hours when vehicle was compared with MIF stimulated cells. Epidermal growth factor (EGF) is also overexpressed in the serum of psoriasis patients.28 In keratinocytes stimulation with EGF induced activation of P-MSK1 (S376) and P-RSK1 (S380) which was also inhibited when keratinocytes were co-stimulated with DMF and EGF.27
ERK1/2 activate both IκB kinase α and IκB kinase β and both phosphorylate NF-κB/p65 at the C-terminal part (S536) of p65 which allows direct nuclear translocation of NF-κB/p65 (Box 1). IKKα and IKKβ induce separately the phosphorylation of P-IκBα (S32) which led to the degradation of IκBα.29–31 In HeLa cells stimulation with IL-1β or TNF-α activated P-IKKβ (S177/S181) which further phosphorylated P-NF-κB/p65 (S536).32 In the normal mouse IKKα suppressed NF-κB activity by accelerating the turnover of the NF-κB subunit RelA p65 and by removing it from the pro-inflammatory gene promoter.33 In human T-cells, stimulation with PMA induced nuclear translocation of P-NF-κB p65 (S536) without IκBα degradation which seems to be specific for T-cell activation.34
Transcription of the IκBα gene is also dependent on NF-κB p65 activation. In TNF-α or PMA stimulated T cells, degradation of IκBα started to occur after 40 min and re-synthesis of IκBα emerged at 2 hours. Following degradation, IκBα is resynthesized by an auto-regulatory pathway.35 In LPS stimulated DC’s DMF inhibited the activation of P-ERK1/2 and both P-NF-κB/p65 (S276) and (S536).5 This was probably achieved by the inhibition of P-IKKβ, similarly to BMDM cells where DMF inhibited the LPS induced activations of P-IKKβ (S177) (S181) and of P-ERK1/2 (Thr202/Tyr204) which resulted in the stabilization of IκBα.7
DMF Inhibited NF-κB Activation in CTCL Cell and Restored Apoptosis
DMF therapy has also been tested in cutaneous T-cell lymphoma (CTCL).36 This showed that DMF but not MMF was able to inhibit the number of malignant T-cells and metastases in cutaneous T cell lymphoma (CTCL) by inhibiting constitutive NF-κB activation and sensitized cells towards apoptosis. DMF induced cell death in primary patient-derived CD4+ T-cells and CTCL cell lines but did not affect T-cells from healthy donors. These effects were linked specifically to inhibition of NF-κB/p65 DNA-binding activity and caspase 3 cleavages.36
Cytoskeleton Proteins, β-Actin, γ-Actin Regulate Lymphocyte (T-Cell) Cell Motility
Cell growth, migration and the G-Actin pool are specifically controlled by β-Actin. Deletion of β-Actin in highly motile CD4+ T-cells caused migration defects and altered cell motility and changed the ratio of G-Actin.37 Protein tyrosine phosphatase (PTP) is expressed broadly in hematopoietic cells and was needed for full induction of the DC induced T cell dependent immune response.38 PTPs and protein tyrosine kinases (PTKs) function in a coordinated manner. The anti-inflammatory compound Bay 11–7082 has an α, β unsaturated electrophilic center and is a potent inhibitor of PTP, and inhibited IκB kinase activity.39 PTP has an active site at a cysteine which forms covalent binding with BAY 11–7082. Based on this fact, PTP may also be a target for inhibition by electrophilic drugs such as DMF.7
Dimethyl Fumarate Is an Allosteric Inhibitor of MSK1 and RSK1, 2 Kinases
Based on the observation that DMF inhibited IL-1β, MIF and EGF induced phosphorylation of MSK1/2 and RSK1 kinases in keratinocytes12,27 the theoretical binding sites of DMF at the C-terminal part of RSK2 kinase was investigated by X-ray crystallography. DMF indeed targeted covalently a cysteine residue in the C-terminal kinase domain (CTKD) of p90 RSK2 which is conserved in both RSK2 and MSK1 kinases.40 The binding of DMF to cysteine C599 in RSK2, was confirmed in vivo by mass spectroscopy analysis by isolating full length RSK2 from DMF treated HEK293 epithelial cells transfected with WT GST-RSK2 plasmids. Mutation analysis showed that binding of DMF to C599 prevented the auto-phosphorylation of P-RSK2 (S386). A homologous cysteine C603 at the CTKD of MSK1 was identified, binding DMF which inhibited the auto-phosphorylation of P-MSK1 (S376). To compare the efficacy of DMF and MMF as inhibitors against purified RSK CTKD, an evaluation with a FRET kinase activity assay was carried out.40 This showed that binding of DMF leads to full inhibition of RSK2 CTKD activity whereas MMF displayed no inhibition of RSK2 CTKD activity. This explains earlier results12,36 that only DMF but not MMF reacts covalently as a Michael acceptor to a cysteine residue, which is conserved in the αF-helix of RSKs/MSKs.40
Case Reports
It has been questioned for a long time whether DMF can stay stable and be transported in the blood and whether DMF is the active metabolite in vivo in the treatment of psoriasis and multiple sclerosis.41 We therefore studied the effects of DMF on MSK1 and RSK1, 2 activations in PBMCs isolated from two patients at day 0 and day 90 after treatment with DMF (Fumaderm®). Psoriatic patients with a Psoriasis Area and Severity Index (PASI) of 14 and 19.8 respectively were treated with DMF (Fumaderm®) according to the established treatment regimen42 with a gradual dose increase. The Regional Ethical Committee of Region Midtjylland, Denmark approved the experiment with psoriatic patients (M-20090102). A signed consent was obtained from each patient participating, according to the Declaration of Helsinki principles.
Materials and Methods
PBMCs were purified by Lymphoprep density gradient media (Axis-Shield) from EDTA blood collected before and after 90 days of treatment with DMF. Cells were washed with cold sterile Dulbecco´s PBS (Gibco) and seeded at 6 x 106/petri dish in 10 ml RPMI 1640 (Gibco) supplemented with penicillin (10,000 units/ml) streptomycin (10 mg/ml) and gentamycin (2.5 mg/ml) in 10 cm (diameter) petri dishes. Cells were stimulated either with anisomycin (0.2 µg/ml) or IL-1β (10 ng/ml) (R&D Systems) for 10 and 20 minutes. After stimulation, petri dishes were placed on ice and cells were collected by ice cold Dulbecco´s PBS and centrifuged 1400 rpm for 10 minutes. Cells were added 100 µl of 1 x cell Lysis sample buffer (Cell Signaling Technology)/sample. An amount of 20 µg protein/lane was separated on 8%-16 % SDS-Page Tris-glycin gels (Invitrogen) or on NuPAGE 4%-12% Bis-Tris gels (Novex; Life Technology; Thermo Fisher Scientific, Waltham, MA, USA). After Western blotting proteins were probed with antibodies for anti-p-MSK1 (S376) (#9591, 1:750), anti-p-MSK1 (S212) (AF1086 R&D Systems, 1:750), anti-p-RSK2 (S386) (#9341, 1:750), (S227) (#3536, 1:750), (Cell Signalling Technology, Beverly, MA, USA), anti-MSK1 (H19, sc:2033, 1:1000), anti-RSK2 (sc:9986, 1:1000) (Santa Cruz Biotechnology) and anti-β-Actin (Sigma-Aldrich) as described earlier Gesser et al (2007, 2011)12,27 and Rasmussen et al (2017).20 Additionally 20 µg protein/lane were also probed with anti-p-NF-κB/p65 (S536) (#3033, 1:750), anti-NF-κB p65 (#3034, 1:750) and anti-Pan-Actin (#4968, 1:1000). Anti-p-IκBα (S32) (#9241, 1:750) and anti-IκBα (#4814, 1:1000), HRP anti-rabbit (#7074, 1:2000) and HRP anti-mouse (#7076, 1:2000) antibodies were from Cell Signalling Technology (Beverly, MA, USA).
Results
The Psoriatic patient with a baseline PASI of 14 responded clinically well with a PASI reduction of more than 90% after 90 days of treatment with DMF. Protein samples isolated from PBMCs Day 0 and analyzed by Western blotting showed a clear activation induced by anisomycin and IL-1β of both sites of P-MSK1 (S376), (S212) and of P-RSK2 (S386), (S227) after 10 minutes (Figure 1A and B), which were inhibited after 90 days treatment with DMF. Proteins samples tested with anti-p-NF-κB/p65 (S536) antibodies showed similarly day 0 an anisomycin and IL-1β induced activation after 10 minutes before but not after 90 days of treatment with DMF (Figure 1A). The level of induction was normalized to β-Actin. We tested the level of activation of P-MSK1 (S376) and P-RSK1, 2 (S380)/(S386) in PBMCs from healthy individual, normalized to β-Actin as before (Figure 1C).
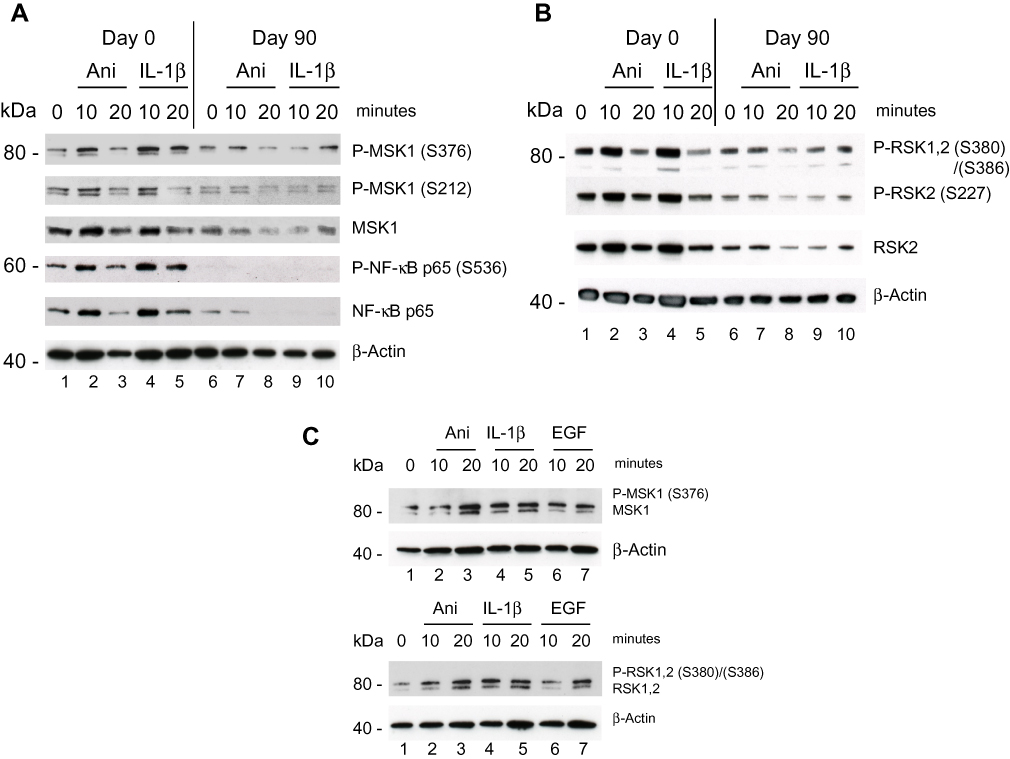 |
Figure 1 Effect of DMF on PBMCs isolated from psoriatic patient 1 responding to treatment with DMF. PBMCs were collected prior, Day 0 and 90 days after treatments start. (A) PBMCs were stimulated with anisomycin (0.2 µg/ml) or IL-1β (10 ng/ml) for 10 and 20 minutes. In a sample of 20 µg protein/lane the level of immune-staining with antibodies for P-MSK1 (S376), (S212) and total MSK1 or for P-NF-κB p-p65 (S536) and total NF-κB p65 was determined by Western Blotting. The blot was re-incubated with antibodies for β-Actin (B) The level of immune-staining with antibodies for P-RSK1, 2 (S380)/(S386) both, P-RSK2 (S227) and total RSK2 was determined by Western blotting. The blot was re-incubated with antibodies for β-Actin. (C) PBMCs were isolated from healthy individual and stimulated with anisomycin (0.2 µg/ml) or EGF (1 µg/ml) for 0 and 20 minutes. In a sample of 20 µg protein/lane the level of immune-staining with antibodies for P-MSK1 (S376), MSK1 and P-RSK1, 2 (S380)/(S386), RSK1, 2 was determined by Western blotting. The blot was re-incubated with antibodies for β-Actin. The phosphorylated band appears above the total protein. |
Because p-RSK1, 2 activation was shown earlier to induce the expression of P-IκBα (Ser32),22 proteins were tested by rabbit antibodies for P-IκBα (Ser32). This showed an induction of P-IκBα (S32) day 0 after stimulation with anisomycin and IL-1β for 10 minutes which was inhibited day 90 after treatment with DMF (Figure 2A). In a second step the blot was striped and re-incubated with a monoclonal mouse antibody for IκBα. This showed a highly elevated basal level of IκBα in PBMCs on day 0 before treatment which was substantially reduced day 90 after treatment with DMF. This is in agreement with the fact that new synthesis of IκBα is dependent on P-NF-κB activation which was elevated on day 0 (Figure 2A) while inhibited day 90 after treatment with DMF. The fold induction of P-IκBα/IκBα was normalized to day 0 and time 0.
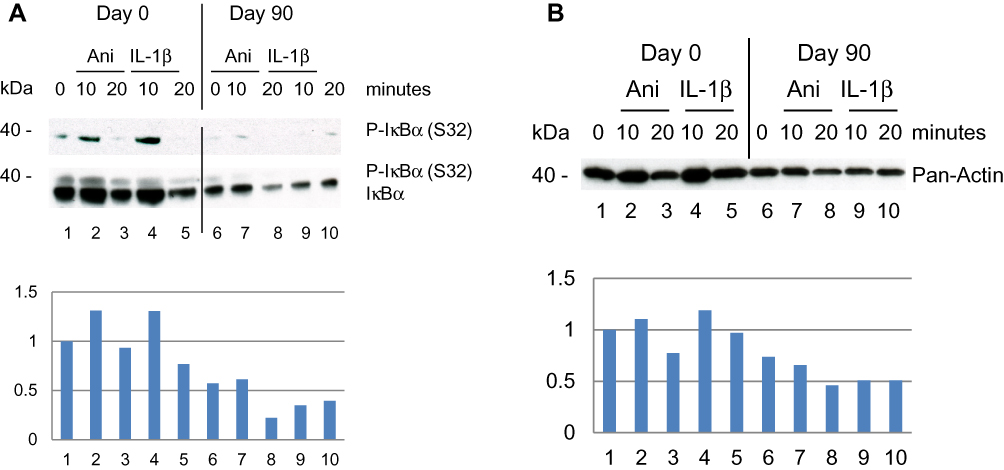 |
Figure 2 Effect of DMF on PBMCs isolated from psoriatic patient 1 responding to treatment with DMF. (A) PBMCs were collected prior, Day 0 and Day 90 after treatments start. PBMCs were stimulated as before and in a sample of 20 µg protein/lane the level of immune-staining was tested with rabbit anti-P-IκBα (S32) antibodies. Thereafter the blot was striped and re-incubated with mouse anti-IκBα antibodies to determine the tot amount of IκBα by Western blotting. (Below) The fold induction of IκBα was normalized to day 0 and the non-stimulated PBMCs. (B) The expression level of total Actin was determined by antibodies for Pan-Actin and Western blotting. (Below) The fold induction was normalized to the expression of Pan-Actin at Day 0 and the non-stimulated PBMCs. |
Antibodies for Pan-Actin identify all subtypes of Actin forms. The expression of Pan-Actin was activated by anisomycin and IL-1β day 0 after 10 minutes of stimulation while this was reduced at day 90 after treatment with DMF (Figure 2B). The activation of Pan-Actin was normalized to day 0 and time 0. Inhibition of P-Tyr by DMF was demonstrated earlier in P-ERK1/2 (Thr202/Tyr204).5,7 We observed that DMF reduced the activation of Pan-Actin via PTP38,39 after treatment with DMF (Figure 2B). This may indicate that DMF has an inhibitory effect on T cell motility in the serum of patients with psoriasis although this has to be further investigated.
The second Psoriatic patient with a baseline PASI of 19.8 did not respond clinically to the treatment. After 90 days of treatment the PASI had increased 27. PBMCs were purified day 0 and were stimulated with IL-1β (10 ng/ml) and EGF (1 ng/ml) for 10 and 20 minutes. (EGF was shown earlier to activate both P-MSK1 and P-RSK1.)27 Western Blotting showed a small activation of P-MSK1 (S376), MSK1, P-NF-κB/p65 (S536), NF-κB p65 (Figure 4A) and of P-RSK1, 2 (S380)/(S386), RSK2 (Figure 4B) induced by IL-1β or EGF at 10 minutes before treatment with DMF. Opposite to the first psoriasis patient, treatment for 90 days with DMF further induced the activation of P-MSK1 (S376), P-NF-κB/p65 (S536) and P-RSK1, 2(S380)/(S386) by IL-1β and EGF. These results are in accordance with the negative clinical response to DMF treatment. The level of expression of β-Actin was not changed by stimulation with IL-1β or EGF and was not influenced either by the treatment with DMF in the non-responding patient.
Discussion
DMF inhibited the activation of P-MSK1, P-RSK1, 2 and P-IκBα (S32) in PBMCs which led to inhibitions of P-NF-κB p65 at two phosphorylation sites. Induction of P-MSK1 (S376) and P-RSK1, 2 (S380)/(S386) by IL-1β, anisomycin and EGF is schematically presented in (Figure 3A) and the effects of DMF treatment in (Figure 3B). IKKα and IKKβ integrate the activations by MSK1 and RSK1, 2.31 The induction of P-NF-κB/p65 (S536) was mediated by P-IKKβ or P-RSK1, 2 which was also inhibited after treatment with DMF (Figure 3A and B). According to our hypothesis and to our most recently presented results,40 we saw that treatment with DMF inhibited the anisomycin and IL-1β induced P-MSK1 and P-RSK1, 2 kinases at several activation sites in PBMCs isolated from psoriatic patients (Figure 1A and B). We therefore believe that DMF is not completely hydrolyzed to MMF in vivo and DMF is the active metabolite in the blood of patients with psoriasis during treatment with DMF.
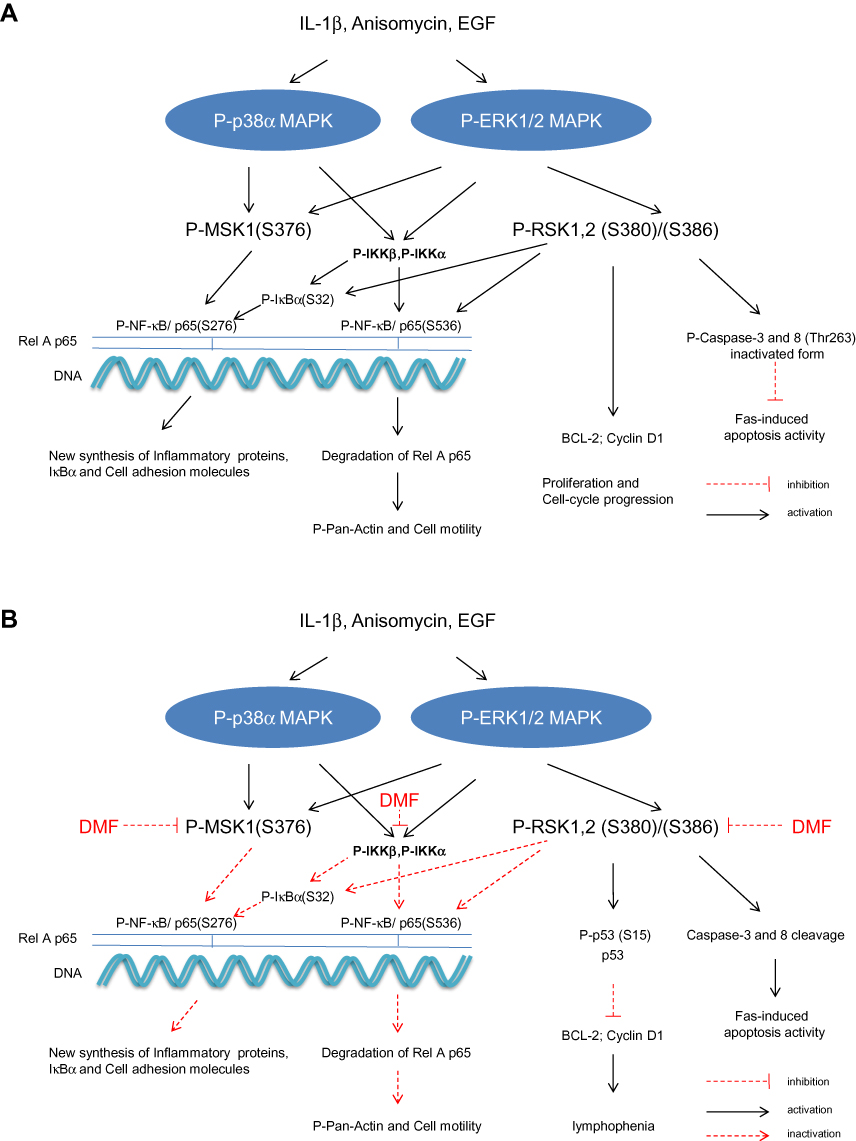 |
Figure 3 Schematic representation in PBMCs of the IL-1β, anisomycin induced activations of MSK1 and RSK1, 2 and IKKα/β kinases via the p38α MAPK and ERK 1/2 signalling pathways 9(A). Effect of DMF on these activations in PBMCs isolated from a patient with severe psoriasis and treated with DMF. DMF inhibited specifically the phosphorylation of MSK1 and RSK1, 2 kinases (B) P-MSK1 mediates the transactivation of P-NF-κB/(S276) while P-RSK1, 2 and P-IKKα mediate the transactivation of P-NF-κB (S536). P-RSK1, 2 and P-IKKβ can both induce the activation of P-IκB (S32). These were all inhibited by treatment with DMF. Inhibition of P-NF-κB/p65 (S276) and (S536) resulted in inhibition of the synthesis of inflammatory proteins and these resulted in the new synthesis of p53/p-p53 (S15).27 P53 DNA binding repressed BCL-2 and Cyclin D1.43,44 Inactivation of P-RSK2 leads also to apoptosis via the cleavage of Caspase-3 and 8 in the cytoplasm.23,36 |
RSK1, 2 play a critical role in cell proliferation21 and cell-cycle progression.44 Therefore inhibitions by DMF of P-RSK1, 2 and P-MSK1 activations in PBMCs (Figure 1B) will similarly result in the induction of P-p53 (S15)/p5327 which also lead to cell cycle arrest by inhibiting Cyclin D131,44 (Figure 3A and B). The newly induced P53 is an activator of transcription by activating P-RSK1 and P-NF-κB/(S536) which induces the pro-apoptotic gene Bax.45 In purified human T cells, treatment with DMF and IL-2 stimulation for 48 hours increased the number of apoptotic cells by repressing the anti-apoptotic protein Bcl-2.43 Activated p53 induces p21 protein (gene CDKN1A) which suppresses Cyclin D144, (Table 1). Growth factors like EGF activate P-RSK1, 2 which stimulate Cyclin D1 and the progression from G1 to S phase.44,48
 |
Table 1 Effects of the DMF mediated inhibition of P-NF-κB /p65 in T cells, dendritic cells, keratinocytes and endothelial cells. |
We observed a high basal level of IκBα in the psoriatic PBMCs on day 0 which is somewhat surprising (Figure 2A). Earlier it was pointed out that mutation in genes important for maintaining homeostasis might be associated with disease susceptibility. NFKBIA which encodes the protein IκBα is known as one of the genes suggested for predisposition to psoriasis.46 Mutation in NFKBIA which is a negative regulator of immune responses decreases the threshold for immune activation and lowers the level for onset of psoriasis. Loss of function caused by polymorphism in NFKBIA may result in the high basal level of IκBα in psoriatic PBMCs (Figure 2A).
It was shown earlier that wild-type p53-induced phosphatase 1 (Wip1) is a negative regulator of the NF-κB signaling pathway.47 Wip1 dephosphorylate P-NF-κB/p65 (S536) in normal mice and mice lacking Wip1 showed enhanced inflammation. WiP1 is regarded as a homeostatic factor in normal cells. Wip1 is expressed in psoriatic skin, forms complexes with P-RSK2 in lesional psoriatic skin and reduces P-RSK2 (S386) activation.20 DMF inhibited the MIF induced complex formation of P-Wip1 with P-RSK1, 2 by inhibiting activation of P-Wip1 and P-RSK1, 2 in keratinocytes in vitro. In the initial phase, MIF stimulation activated new synthesis of P-Wip1/Wip1 which de-phosphorylated P-p53 (S15) while co-stimulation with DMF and MIF inhibited the level of P-Wip1/Wip1 and re-activated P-p53 (S15).20 Dysregulation of WiP1 activity in PBMCs in some patients with psoriasis, similarly to our results in the second patient with the lack of regulation of P-NF-κB/p65 (S536) (Figure 4) will result in enhanced inflammation. Re-activation of p53 is necessary for controlling apoptosis through increased binding of p53 to the promoter of BAX.48
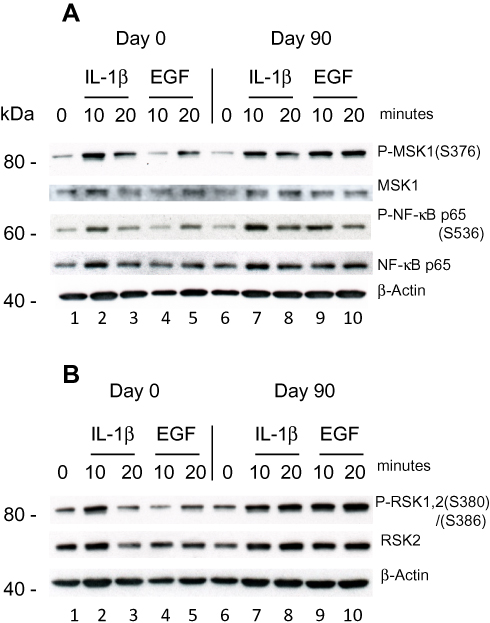 |
Figure 4 Effect of DMF on PBMCs isolated from psoriatic patient 2 not responding to treatment with DMF. PBMCs were isolated Day 0 and Day 90 as before and stimulated with IL-1β (10 ng/ml) or EGF (1 ng/ml) for 10 and 20 minutes. (A) In a sample of 20 µg protein/lane the level of immune-staining was tested with antibodies for P-MSK1 (S376), total MSK1, P-NF-κB p65 (S536) and tot NF-κB p65 were determined by Western blotting. (B) The level of immune-staining with antibodies for P-RSK1, 2 (S380)/(S386) and total RSK2 was determined by Western blotting. The blots were re-probed with antibody for β-Actin. |
Summary
DMF inhibits the activation of MSK1 and RSK1, 2 kinases and the transactivation of P-NF-κB/RelA/p65 at two different serine phospho-acceptor sites which lead to the induction of P-p53/p53 and the inhibition of cell growth. The inhibition of P-RSK1, 2 (S380)/(S386) lead also to the induction of apoptosis in PBMCs31,43 (Table 1). The activation of P-NF-κB/p65 (S276) and (S536) control different T cell functions and DMF inhibits the immune reaction in psoriatic skin. DMF also inhibits the activation of IKKα/β kinase but this is in an indirect manner through the PTP/PTK pathway which is connected to P-NF-κB/p65 (S536).7 IKKα activation accelerates the turnover of P-NF-κB/p65 (S536).33 New synthesis of IκBα is also regulated by P-NF-κB/p65 which restore the binding of IκBα to NF-κB/p65.35
Acknowledgments
The work was supported by research grants from the Danish Psoriasis Foundation and the Novo Nordisk Foundation.
Disclosure
Lars Iversen has served as a consultant and/or paid speaker for and/or participated in clinical trials sponsored by: AbbVie, Almirall, Amgen, Astra Zeneca, BMS, Boehringer Ingelheim, Celgene, Centocor, Eli Lilly, Janssen Cilag, Kyowa, Leo Pharma, MSD, Novartis, Pfizer, Samsung, UCB. MK Rasmussen has served as a consultant and/or paid speaker for and/or participated in clinical trials sponsored by: AbbVie, Almirall, Eli Lilly, Janssen Cilag, Leo Pharma, Novartis, UCB. The authors report no other conflicts of interest in this work.
References
1. Mrowietz U, Asadullah K. Dimethyl fumarate for psoriasis: more than a dietary curiosity. Trend Mol Med. 2005;11(1):43–48. doi:10.1016/j.molmed.2004.11.003
2. Smith TE. Dimethyl fumarate (tecfidera) is the first line treatment choice in patients with remitting multiple sclerosis. Zh Neurol Psikhiatr Im S S Korsakova. 2017;117(11):140–145. doi:10.17116/jnevro2017117111140-145
3. Blair HA. Dimethyl fumarate: a review in moderate to severe plaque psoriasis. Drugs. 2018;78(1):123–130. doi:10.1007/s40265-017-0854-6
4. Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–255.
5. Peng H, Guerau-de-Arello M, Metha VB, et al. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor κB (NF-κB) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and Mitogen Stress-activated Kinase 1 (MSK1) signaling. J Biol Chem. 2012;287(33):2817–2826.
6. Rescigno M, Martino M, Satherland C, et al. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med. 1998;188(11):2175–2180.
7. McGuire VA, Ruiz-Zorrilla Diez T, Emmerich CH, et al. Dimethyl fumarate blocks pro-inflammatory cytokine production via inhibition of TLR induced M1 and K63 ubiquitin chain formation. Sci Rep. 2016;6:31159.
8. Arthur JC, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679–692.
9. Ockenfels HM, Schulterwolter T, Ockenfels G, et al. The anti-psoriatic agent dimethyl fumarate immune-modulates T-cell cytokine secretion and inhibits cytokines of the psoriatic cytokine network. Br J Dermatol. 1998;139(3):390–395.
10. Tahvili S, Zandieh B, Amirghofran Z. The effect of dimethyl fumarate on gene expression and the level of cytokines related to different T helper cell subsets in peripheral blood mononuclear cells of patients with psoriasis. Int J Dermatol. 2015;54(7):e254–60. doi:10.1111/ijd.2015.54.issue-7
11. Bech R, Otkjær K, Birkelund S, et al. Interleukin 20 protein locates to distinct mononuclear cells in psoriatic skin. Exp Dermatol. 2014;23(5):349–352. doi:10.1111/exd.2014.23.issue-5
12. Gesser B, Johansen C, Rasmussen MK, et al. Dimethyl fumarate specifically inhibits the mitogen and stress-activated kinases 1 and 2 (MSK1/2): possible role for its anti-psoriatic effect. J Invest Dermatol. 2007;127(9):2129–2137. doi:10.1038/sj.jid.5700859
13. Gerhardt S, König VM, Doll M, et al. Dimethyl fumarate protects against TNF-α induced secretion of inflammatory cytokines in human endothelial cells. J Inflamm. 2015;12:49.
14. Walbrecht K, Drick N, Hund AC, Schön MP. Downregulation of endothelial adhesion molecules by dimethylfumarate, but not monomethylfumarate, and impairment of dynamic lymphocyte-endothelial cell interactions. Exp Dermatol. 2011;20(12):980–985. doi:10.1111/j.1600-0625.2011.01376.x
15. Rubant SA, Ludwig RJ, Diehl S, et al. Dimethylfumarate reduces leukocytes rolling in vivo through modulation of adhesion molecule expression. J Invest Dermatol. 2008;128(2):326–331. doi:10.1038/sj.jid.5700996
16. Johansen C, Kragballe K, Westergaard M, et al. The mitogen-activated protein kinases p38 and ERK1/2 are increased in lesional psoriatic skin. Br J Dermatol. 2005;152(1):37–42. doi:10.1111/bjd.2005.152.issue-1
17. Funding AT, Johansen C, Kragballe K, et al. Mitogen- and stress-activated protein kinase 1 is activated in lesional psoriatic epidermis and regulates the expression of pro-inflammatory cytokines. J Invest Dermatol. 2006;126(8):1784–1791. doi:10.1038/sj.jid.5700252
18. Vermeulen L, De Wilde G, Van Damme P, et al. Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 2003;22(6):1313–1324. doi:10.1093/emboj/cdg139
19. Chen LF, Greene WC. Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol. 2004;5(5):393–401. doi:10.1038/nrm1368
20. Rasmussen MK, Nielsen J, Kjellerup RB, et al. Protein phosphatase 2Cδ/Wip1 regulates phospho-p90RSK2 activity in lesional psoriatic skin. J Inflamm Res. 2017;10:169–180. doi:10.2147/JIR.S152869
21. Romeo Y, Zhang X, Roux PP. Regulation and function of the RSK family of protein kinases. Biochem J. 2012;441(2):553–569. doi:10.1042/BJ20110289
22. Schouten GJ, Vertegaal ACO, Whiteside ST, et al. IκBα is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. EMBO J. 1997;16(11):3133–3144. doi:10.1093/emboj/16.11.3133
23. Peng C, Cho YY, Zhu F, et al. RSK2 mediates NF-κB activity through the phosphorylation of IκBα in the TNF-R1 pathway. FASEB J. 2010;24(9):3490–3499. doi:10.1096/fj.09-151290
24. Peng C, Cho YY, Zhu F, et al. Phosphorylation of caspase-8 (Thr-263) by ribosomal S6 Kinase 2 (RSK2) mediates caspase-8 ubiquitination and stability. J Biol Chem. 2011;286(9):6946–6954. doi:10.1074/jbc.M110.172338
25. Steinhoff M, Meinhardt A, Steinhoff A, et al. Evidence for a role of macrophage migration inhibitory factor in psoriatic skin diseases. Br J Dermatol. 1999;141:1061–1066. doi:10.1046/j.1365-2133.1999.03206.x
26. Shimizu T, Nishihira J, Mizue Y, et al. High macrophage migration inhibitory factor (MIF) serum levels associated with extended psoriasis. J Invest Dermatol. 2001;116:989–990. doi:10.1046/j.0022-202x.2001.01366.x
27. Gesser B, Rasmussen MK, Raaby L, et al. Dimethyl fumarate inhibits MIF-induced proliferation of keratinocytes by inhibiting MSK1 and RSK1 activation and by inducing nuclear p-c-Jun (c63) and p-p53 (S15) expression. Inflamm Res. 2011;60(7):643–653. doi:10.1007/s00011-011-0316-7
28. Anderson KS, Petersson S, Wong J, et al. Elevation of serum epidermal growth factor and interleukin 1 receptor antagonist in active psoriasis vulgaris. Br J Dermatol. 2010;163(5):1085–1089. doi:10.1111/j.1365-2133.2010.09990.x
29. Perkins ND. Post–translational modifications regulate the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25(51):6717–6730. doi:10.1038/sj.onc.1209937
30. Lee FS, Peters RT, Dang LC, Maniatis T. MEKK1 activates both IκB kinase α and IκB kinase β. Proc Natl Acad Sci. 1998;95(16):9319–9324. doi:10.1073/pnas.95.16.9319
31. Perkins ND. Integrating cell-signaling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49–62. doi:10.1038/nrm2083
32. Sakurai H, Chiba H, Miyoshi H, et al. IκB kinases phosphorylate NF-κB subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274(43):3053–3056. doi:10.1074/jbc.274.43.30353
33. Lawrence T, Bebien M, Liu G, Nizet V, Karin M. IKKalpha limits macrophage NF-κB activation and contributes to the resolution of Inflammation. Nature. 2005;434(7037):1138–1143. doi:10.1038/nature03491
34. Sasaki CY, Barberi TJ, Ghosh P, Longo DJ. Phosphorylation of RelA/p65 on Serine 536 defines an IκBα-independent NF-κB pathway. J Biol Chem. 2005;280(41):34538–34547. doi:10.1074/jbc.M504943200
35. Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science. 1993;259(5103):1912–1915. doi:10.1126/science.8096091
36. Nicolay JP, Müller-Decke K, Schroeder A, et al. Dimethyl fumarate restores apoptosis sensitivity and inhibits tumor growth and metastases in CTCL by targeting NF-κB activity. Blood. 2016;128(6):805–815. doi:10.1182/blood-2016-01-694117
37. Bunell TM, Burbach BJ, Shimizu Y, Ervasti JM. β-Actin specifically controls cell growth, migration and the G-actin pool. Mol Biol Cell. 2011;22(21):4047–5835.
38. Rhee I, Zhong MC, Reizis B, et al. Control of dendritic cell migration, T cell-dependent immunity, and autoimmunity by protein tyrosine phosphatase PTPN12 expressed in dendritic cells. Mol Cell Biol. 2014;34(5):888–899.
39. Krishnan N, Bencze G, Cohen P, Tonks NK. The anti-inflammatory compound BAY 11-7082 is a potent inhibitor of protein tyrosine phosphatases. FEBS J. 2013;280(12):2830–2841.
40. Andersen JL, Gesser B, Funder ED, et al. Dimethyl fumarate is an allosteric covalent inhibitor of the p90 ribosomal S6 kinases. Nat Commun. 2018;9(1):4344.
41. Mrowietz U, Morrision PJ, Suhrkamp I, Kumanova M, Clement B. The pharmacokinetics of fumaric acid esters reveal their in vivo effects. Trends Pharmacol Sci. 2018;39(1):1–12.
42. Walker F, Adamczyk A, Kellerer C, et al. Fumaderm® in daily practice for psoriasis: dosing, efficacy and quality of life. Br J Dermatol. 2014;171(5):1197–1205.
43. Treumer F, Zhu K, Gläser R, Mrowietz U. Dimethylfumarate is a potent inducer of Apoptosis in human T cells. J Invest Dermatol. 2003;121(6):1383–1388.
44. Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signaling. Nat Rev Mol Cell Biol. 2008;10:747–758.
45. Bohuslav J, Chen LF, Kwon H, et al. P53 induces NF-κB kinase independent mechanism involving RSK1 phosphorylation of p65. J Biol Chem. 2004;279(25):26115–26125.
46. Harden JL, Krueger JG, Bowcock A. The immunogenetics of psoriasis: a comprehensive review. J Autoimmun. 2016;64:66–73.
47. Chew J, Biswas S, Shreeram S, et al. WiP1 phosphatase is negative regulator of NF-κB signaling. Nat Cell Biol. 2009;11(5):659–666.
48. Yu J, Zhang L. The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Comm. 2005;331(3):851–858.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.