")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 12
Differential Diagnosis of Waldenström’s Macroglobulinemia and Early Management: Perspectives from Clinical Practice
Received 5 March 2022
Accepted for publication 28 July 2022
Published 18 August 2022 Volume 2022:12 Pages 107—117
DOI https://doi.org/10.2147/BLCTT.S259860
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wilson Gonsalves
Shashank Cingam,1 Surbhi Sidana2
1Division of Hematology and Oncology, University of New Mexico Comprehensive Cancer Center, Albuquerque, NM, 87102, USA; 2Division of BMT and Cell Therapy, Stanford University School of Medicine, Stanford, CA, 94305, USA
Correspondence: Surbhi Sidana, Division of Blood and Marrow Transplant and Cellular Therapy, Stanford University School of Medicine, 300 Pasteur Drive, Room H0101c, Stanford, CA, 94305, USA, Tel +650 498-6000, Fax +650 725-3321, Email [email protected]
Abstract: Waldenström’s Macroglobulinemia (WM) is a clonal B-lymphocyte neoplasm characterized by the presence of IgM monoclonal protein and ≥ 10% bone marrow involvement with lymphoplasmacytic cells. Several mature B-cell and plasma cell disorders can potentially produce monoclonal IgM immunoglobulin and hence, careful consideration of the differential diagnosis is vital. Clinico-pathological features, immunophenotype, and MYD88 mutation status help distinguish WM from other plasma cell and lymphoproliferative disorders. Treatment is only indicated in patients symptomatic from adenopathy or organomegaly, neuropathy, hyper viscosity, cryoglobulinemia, cold agglutinin disease, cytopenia’s or amyloidosis. Alkylators (cyclophosphamide, bendamustine) in combination with anti-CD20 antibodies and novel targeted agents including Bruton tyrosine kinase (BTK) inhibitors like ibrutinib are the mainstay of frontline treatment in symptomatic WM.
Keywords: Waldenström’s Macroglobulinemia, lymphoplasmacytic lymphoma, WM, LPL
Introduction
Waldenström Macroglobulinemia (WM) is a clonal B-lymphocyte neoplasm characterized by the presence of IgM monoclonal protein and ≥10% bone marrow involvement with lymphoplasmacytic cells. WM is classified as a subset of a broader entity, lymphoplasmacytic lymphoma (LPL), an indolent B-cell lymphoma.1 The distinction between these two entities is controversial, especially in patients with more than one type of monoclonal protein. The typical immunohistochemistry profile of the malignant cells is, surface IgM+, CD5-/+, CD10-, CD19+, CD20+, CD22+, CD23-, CD25+, CD27+, FMC7+, CD103-, CD138-.2
WM is a rare disorder with incidence reported to be approximately 3 per million people per year with 1000–1500 new cases diagnosed in the United States each year.3 WM is a disease primarily seen in older adults, like other non-Hodgkin’s lymphomas, with a median age at the time of diagnosis of 70 years. The incidence is higher in males compared to females and whites compared to blacks.4 The etiology is unknown, but an association has been noted with chronic immune stimulating conditions including chronic Hepatitis C.5 About one-quarter of patients with WM have a family history of lymphoproliferative neoplasms.6 IgM-monoclonal gammopathy of undetermined significance (MGUS) is a precursor condition for WM.7–9
The clonal cells of WM are hypothesized to arise from post-germinal B cells which undergo somatic mutations in a chronically immune activated environment.10 Monoallelic MYD88L265P mutations are detectable in >90% of patients with WM11 Mutated MYD88 leads to a more active “Myddosome” complex, a structure composed of MYD88 dimers, triggering the interleukin-1 receptor-associated kinases (IRAK1/IRAK4) to activate nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) signaling pathway and MAPK pathway through Bruton’s tyrosine kinase (BTK) which leads to chronic activated BCR signaling.10,12 Patients with IgM MGUS with MYD88L265P mutations had an increased rate of progression to WM suggesting that it may be a driver mutation which leads to the initial clonal expansion.13 Mutations in CXCR4, a chemokine receptor are the second most common recurrent mutations in WM and are seen in about 40% of patients with WM.14 CXCR4 mutations in WM lead to impaired CXCR4 desensitization and internalization leading to the prolonged exposure and binding of the chemokine ligand CXCL12, which triggers cell proliferation through the AKT and MAPK signaling pathways.15
Typically, WM has an indolent clinical course with 10-year overall survival rates (OS) rates are around 60%-70%, which continue to improve with recent therapeutic advances.3 Transformation into diffuse large B-cell lymphoma (DLBCL) and acute leukemia has been reported and adversely effects survival.16
Clinical Manifestations
Clinical presentation in WM is highly heterogenous. Patients may present with non-specific constitutional symptoms or may have manifestations related to the abnormal monoclonal IgM protein or organ infiltration.
Manifestations of abnormal IgM protein include hyperviscosity, neuropathy, cryoglobulinemia, cold agglutinin disease and amyloidosis. Bone marrow involvement can result anemia and thrombocytopenia, and organ infiltration results in hepatosplenomegaly or adenopathy. About 25% of patients with WM are asymptomatic at diagnosis.17,18
IgM is a large pentameric protein (920kDa) and when present at higher amounts in the plasma increases the viscosity of the blood and leading to decreased organ perfusion.19 Hyperviscosity, which manifests as blurring or loss of vision, headache, vertigo, nystagmus, dizziness, tinnitus among other neurological symptoms is seen in about 30% of the patients. Higher IgM levels (>4000 mg/dl) and/or serum viscosity >4 centipoise (normal value-1.5) are usually associated with these symptoms.20 A classic fundoscopic finding in WM associated with hyperviscosity is the presence of dilated, segmented, and tortuous retinal veins. Hemorrhages, exudates, and papilledema can also be seen.21
Peripheral neuropathy which usually manifests as a slowly progressive paresthesia’s or weakness is seen in about 20% of the patients with WM, and sometimes the only indication to start treatment.22 It is characteristically a symmetric, demyelinating neuropathy affecting lower extremities more frequently than upper extremities. Anti-myelin-associated glycoprotein (MAG) antibodies are found in about 50% of the patients.22 Other antibodies including those directed against GM1 ganglioside have also been described and are associated with motor neuropathy.22 Patients can also present with rare neurological manifestations such as Bing-Neel syndrome and mononeuropathies caused by the direct infiltration of the malignant cells of the nervous system.23 Bing Neel syndrome is characterized by central nervous involvement and presence of tumor cells in the CSF. The symptoms are often variable and include altered mental status, motor and sensory nerve deficits to headaches and seizures.
Type 1 cryoglobulinemia is seen in approximately 20% of patients with WM, but only a fraction of these patients present with symptoms.24 On exposure to cold, the monoclonal IgM (type 1 cryoglobulin) precipitates and can lead decreased tissue perfusion with minimal inflammation which manifests as purpura, livedo reticularis, Raynaud’s syndrome, acral cyanosis, glomerulonephritis and in severe cases, tissue necrosis. The monoclonal IgM protein can also behave as a cold agglutinin and cause complement mediated intravascular hemolysis at temperatures <37 degree Celsius.19 The presence cryoglobulins or cold agglutinins may give a falsely low IgM level on laboratory testing, which can be overcome by maintaining the serum sample in a warm bath.
Primary amyloidosis due to the deposition of the monoclonal IgM protein in patients with WM can present with nephrotic syndrome with or without renal insufficiency, neuropathy, cardiomyopathy or hepatic dysfunction.25 Renal insufficiency in WM can be also secondary to immune-mediated glomerulonephritis or minimal change disease.26 Fat biopsy and/or evaluation of the bone marrow biopsy with Congo red can help establish the diagnosis. Mass spectrometry should be ideally performed to characterize the amyloid protein. Kidney biopsy may also be needed in patients presenting with renal dysfunction.
Cytopenias are caused by the malignant infiltration of the bone marrow by lymphoplasmacytic cells. Symptomatic anemia is the most common manifestation. Anemia and neutropenia at diagnosis are associated with poor prognosis.27 Patients with WM are also susceptible to recurrent infections due to a decrease in other functional immunoglobulins.
Finally, direct organ infiltration can occur in any organ, but most common manifestations include hepatosplenomegaly and lymphadenopathy. Laboratory workup of suspected Waldenström Macroglobulinemia (WM) is enumerated in Table 1.
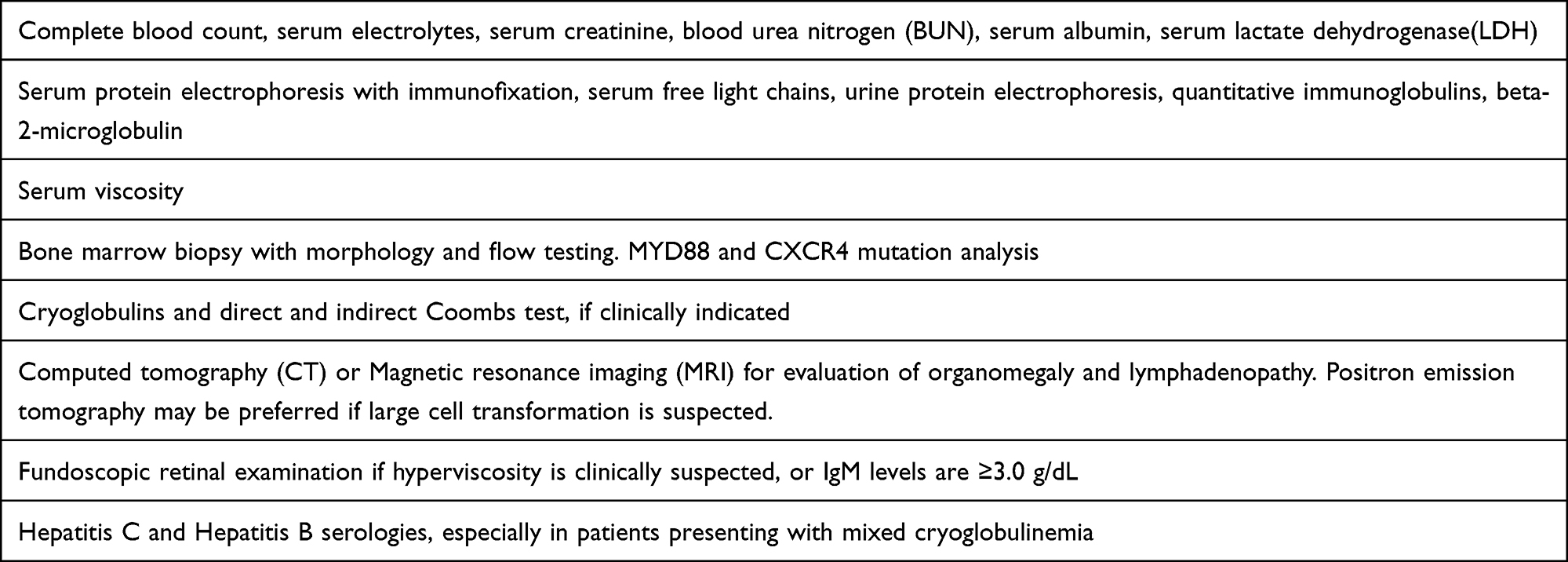 |
Table 1 Clinical Workup for Suspected Waldenstrom’s Macroglobulinemia |
Differential Diagnosis
Any mature B cell or plasma cell malignancy can potentially produce monoclonal IgM and hence, careful consideration of the differential diagnosis is vital.28,29 Clinico-pathological features, immunohistochemistry profile and MYD88 mutation status may help distinguish from other plasma cell and lymphoproliferative disorders (Table 2).
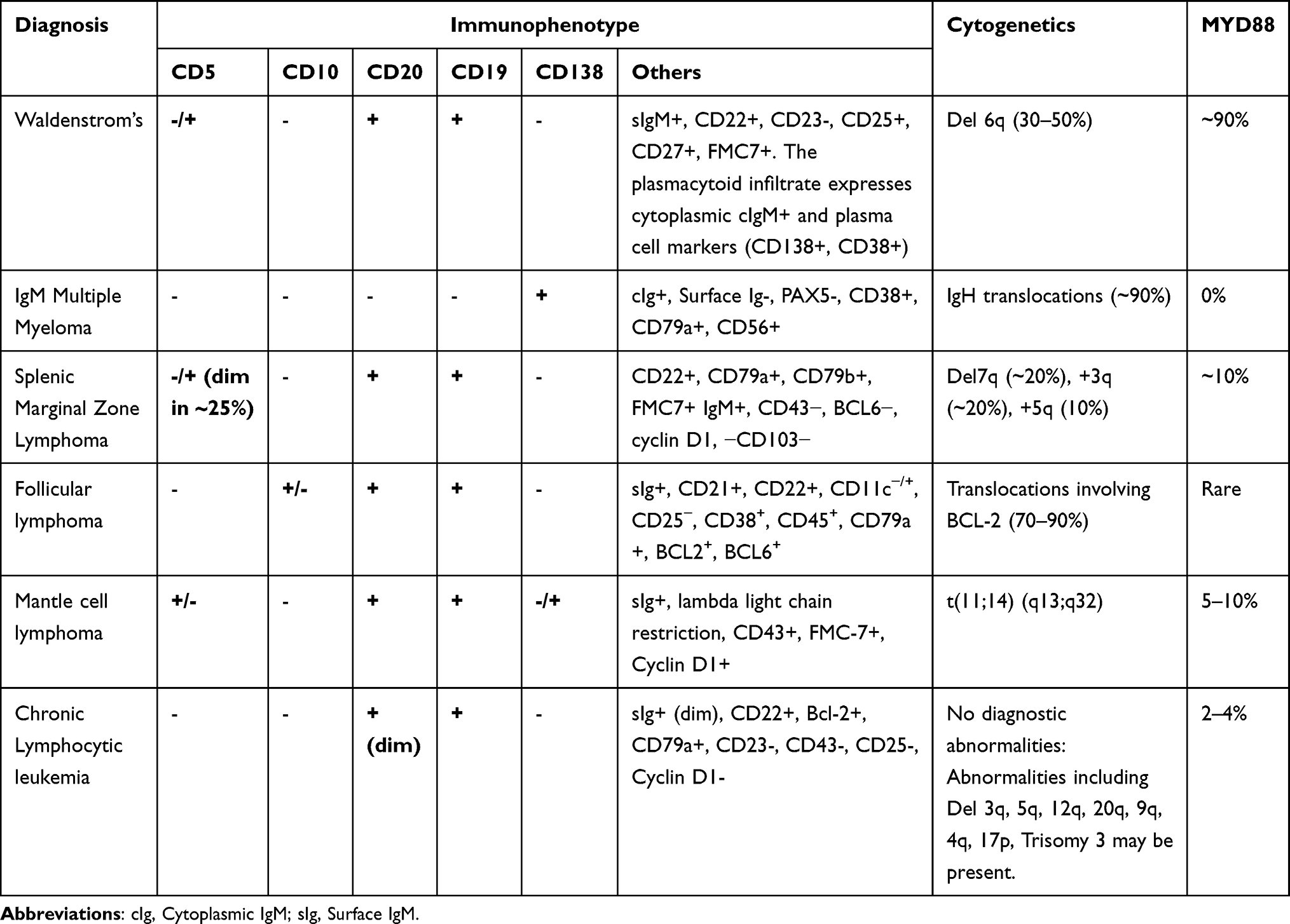 |
Table 2 Differential Diagnosis of IgM Producing Lymphoproliferative Disorders Based on Immunophenotype, Common Cytogenetic Abnormalities and MYD88 Mutation Status ((2, 10, 26, 30–32)) |
Multiple Myeloma and IgM MGUS
IgM MGUS is defined by presence of serum IgM concentration <3.0 g/dL, absence of lymphadenopathy, end organ damage and <10% marrow infiltration. It is important to do the confirmatory bone marrow biopsy to differentiate IgM MGUS from WM. The rate of progression from IgM MGUS to WM is ~ 1.5% per year.8
IgM multiple myeloma (MM) is a rare entity, comprising <1% of MM and often presents with typical features of MM (hypercalcemia, lytic bone lesions, anemia, renal insufficiency). Pathology and IHC profile of MM bone marrow infiltrate is consistent with differentiated plasma cells whereas the WM lymphoplasmacytic B-cell infiltrate is arrested before terminal differentiation to plasma cells. IgH gene translocations are more common in IgM-MM. If the clinical and pathological features do not offer a clear differentiation, a MYD88 mutation panel cab be considered. Typically, IgM MM does not have a MYD88 mutation.
Marginal Zone Lymphoma
IgM-secreting marginal zone lymphoma (MZL) shares IHC profile with WM and furthermore, MYD88 mutations can be seen in 5–10% of patients with MZL. However, MZL tends to present with more extranodal involvement and a lower IgM monoclonal protein. Splenic MZL can be distinguished from WM based on immunophenotypic and molecular cytogenetic studies (Table 2).
Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) with an IgM monoclonal protein may have a similar presentation as WM. In CLL, lymphocytes are typically small and mature, without visible nucleoli with the characteristic smudge cells on a blood smear. The lymphocytes in CLL are positive for CD5 and CD23 and negative for cytoplasmic Ig, whereas CD5+ (95%) and CD23 are negative, and the cells are strongly cytoplasmic Ig positive in WM.
Light Chain Amyloidosis
Light chain amyloidosis can be distinguished from WM with presence of amyloid deposits in the target organs. In addition, the neuropathy in amyloid is usually axonal, unlike WM where it is predominantly demyelinating. Patients with WM can also have overlap with AL amyloidosis.30,31
POEMS Syndrome
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes) is characterized by a λ light chain restricted monoclonal gammopathy and sensorimotor demyelinating neuropathy. The neuropathy in POEMS typically starts with sensory symptoms but progresses to predominantly effect the motor nerves. Patients with WM can also be distinguished from POEMS by the presence of abnormal lymphoplasmacytic infiltrate in the BM and the absence of osteosclerotic bone lesions, elevated levels of vascular endothelial growth factor (VEGF), endocrinopathy, skin changes, and elevated protein in the cerebrospinal fluid.
Risk Stratification
Prognostic Scores
A prognostic system Revised- International Prognostic Scoring System for WM (rIPSSWM) (Table 3) based on age, beta-2-microglobulin, serum LDH, and albumin identifies 5 prognostic groups, ranging from a very low risk group with a 3-year WM related death rate of 0% to a very high risk group with a WM related death rate of 48%.32 It is important note that age is weighted more in these prognostic scores and perhaps, bio-chemical indicators such as B2-microglobulin, LDH and albumin may be more accurate predictors of the biology of the disease. Also, the rIPSSWM and the prior IPSSWM do not consider molecular or genetic characteristics.
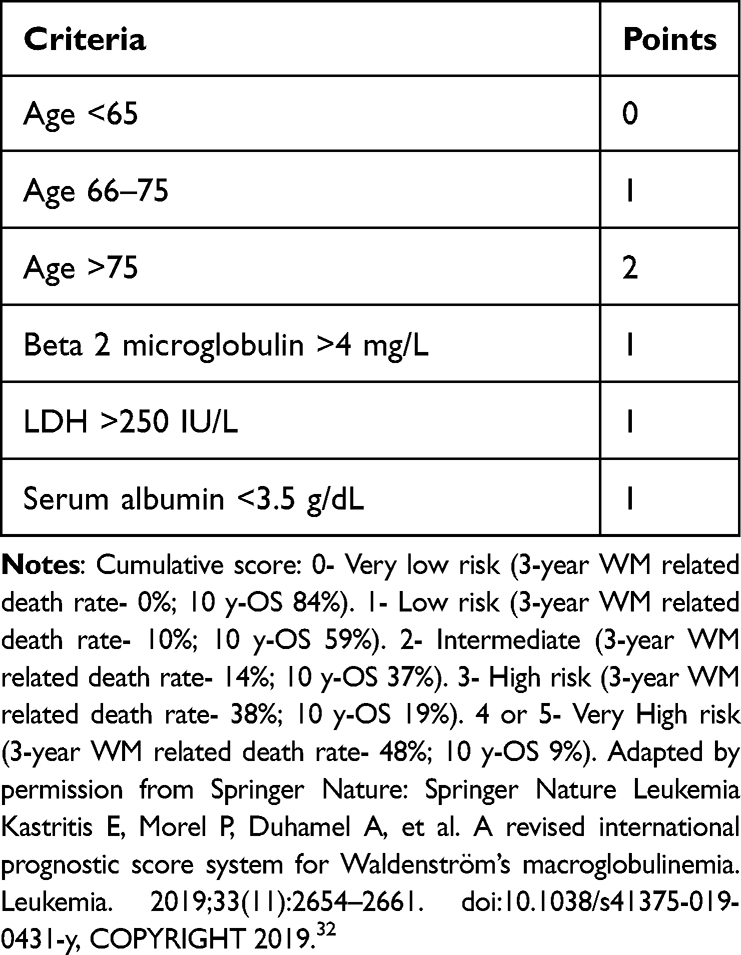 |
Table 3 Revised IPSS Waldenström Macroglobulinemia Scoring System (33) |
Cytogenetics and Molecular Abnormalities
Mutations/deletions of p53 are uncommon but when present in WM are associated with poor prognosis.33 MYD88 mutation predicts for a better response, whereas CXCR4 mutation predicts for decreased response to ibrutinib.34
Early Management
The goal of early management in symptomatic WM is prompt recognition and treatment of complications of the disease. It is important to note that treatment is only indicated in patients symptomatic from adenopathy, organomegaly, neuropathy, hyperviscosity, cryoglobulinemia, cold agglutinin disease, cytopenias (Hb<10 gm/dl, platelets <50 x 103/ μL) or amyloidosis. IgM level alone, in the absence of above signs or symptoms should not trigger initiation of treatment. Fifty percent of the patients who are asymptomatic at diagnosis and who are observed, will not require therapy within almost 3 years.18 The response to treatment should be monitored using the International Working Group on Waldenstrom’s Macroglobulinemia (IWWM) criteria during treatment (Table 4).
 |
Table 4 Response Criteria in Waldenstrom’s Macroglobulinemia |
Plasmapheresis
Plasmapheresis is utilized as a bridge to systemic therapy in patients with symptomatic hyperviscosity, symptomatic cryoglobulinemia or severe hemolysis resulting from cold agglutinin disease.35 Multiple sessions may be required to achieve desired decrease in the IgM levels and resultant symptom control.
Immuno-Chemotherapy
Alkylators (cyclophosphamide, melphalan, bendamustine), nucleoside analogues (fludarabine, cladribine), vinca alkaloids (vincristine), anthracyclines (doxorubicin) and corticosteroids are known to be effective in WM and are used as a backbone of the immunochemotherapy regimens.36 Monoclonal antibodies that target the B-lymphocyte antigen CD20, rituximab and ofatumumab are used in the treatment of WM because CD20 is expressed on lymphoplasmacytic cells in patients with WM. Patient’s treated with anti-CD20 antibodies as a sole therapy may have a slow response to treatment. Treatment with rituximab is also associated with a risk of IgM flare when treatment is started and may exacerbate IgM-related complications.37 Rituximab is often used with other agents as combination and in maintenance strategies. Ofatumumab, a human anti-CD20 monoclonal antibody that binds to an epitope distinct from that recognized by rituximab, is also effective in WM and may be used in patients with intolerance to Rituximab.38
The most common upfront chemoimmunotherapy regimens used in treatment of WM are bendamustine-rituximab (BR) or dexamethasone, rituximab, and cyclophosphamide (DRC). The Study Group Indolent Lymphomas (StiL) examined the activity of BR versus cyclophosphamide, doxorubicin, vincristine, rituximab (R-CHOP) in a large, randomized, multicenter Phase III trial with indolent lymphomas which included 41 patients with WM/LPL. In the 40 patients, who were available for response assessment, at median follow up of 45 months, progression free survival (PFS) was longer in the BR arm (69.5 months) compared to the R-CHOP (28.5 months). BR was also associated with a lower incidence of grade 3 or 4 neutropenia and infectious complications.39 These results suggest that BR is a preferable alternative to CHOP-R as primary therapy for WM. Rituximab with cyclophosphamide and dexamethasone for 6 cycles (DRC, for 6 cycles) is an option in patients who are not candidates for intensive treatment. In a Phase II study of DRC (n=72), objective response rate (ORR) was 83% and the median time to response was 4.1 months. 90% of the patients were progression free at 18 mo. It was well tolerated, with only 9% of patients experiencing grade 3 or 4 neutropenia and approximately 20% of patients experiencing toxicity (any grade) related to rituximab including IgM flare.40 Other class specific adverse events include neuropathy vinca alkaloids and risk of myelodysplastic syndrome/treatment related-acute myeloid leukemia (alkylators and nucleoside analogues), cardiomyopathy (anthracyclines).
Proteosome Inhibitors
Proteasome inhibitors like bortezomib and carfilzomib are active in mature B cell/ plasma cell malignancies including MM, Waldenström and mantle cell lymphoma.41 Bortezomib has been shown to activity in WM in several in vitro and in vivo studies. It can rapidly decrease IgM levels and does not cause IgM flare. In a Phase 2 study, bortezomib, dexamethasone, and rituximab (BDR) combination in previously untreated symptomatic patients with WM (n=55) was associated with an 85% overall response rate including 3% complete response (CR), 7% very good partial response (VGPR), 58% partial response (PR). In 11% of patients, an increase of IgM ≥25% was observed after rituximab; no patient required plasmapheresis. After a minimum follow-up of 32 months, median progression-free survival was 42 months, 3-year duration of response for patients with ≥PR was 70%, and 3-year survival was 81%. Peripheral neuropathy occurred in 46% (grade ≥3 in 7%); only 8% discontinued bortezomib due to neuropathy.42 In a follow up study of BDR, exclusively in treatment naïve patients (n= 23), ORR and major response (greater than or equal to PR) rates (MRR) were 96% and 83% respectively. With a median follow-up of 22.8 months, 18 of 23 patients remained free of disease progression. Peripheral neuropathy was the most common toxicity, with grade 3 peripheral neuropathy was observed in 30% of patients in the study that utilized twice-a-week bortezomib administration. The higher incidence of bortezomib neuropathy in WM compared to MM is attributed to the underlying predisposition to neuropathy in WM due to IgM monoclonal protein, as discussed above. The main limitation with bortezomib which has lead to decreased utilization in the combination regimens is peripheral neuropathy (46–60%) which can last even after discontinuation of the agent. In clinical practice, bortezomib can be administered subcutaneously and in weekly intervals to lower risk of neurotoxicity.
Carfilzomib and ixazomib are potential alternatives to bortezomib with a lower incidence of neuropathy. In a phase II study of Carfilzomib, rituximab, and dexamethasone (CaRD) in symptomatic WM patients (n=26) naïve to bortezomib and rituximab, ORR was 87.1% With a median follow-up of 15.4 months, 20 patients remain progression free. Grade ≥2 toxicities included neutropenia (12.9%), and cardiomyopathy in 1 patient (3.2%).43 Patients with cardiomyopathies or other cardiovascular comorbidities should be carefully monitored while on carfilzomib. Ixazomib, an oral proteasome inhibitor was evaluated in a phase II trial in combination with dexamethasone, and rituximab (IDR) as primary therapy in symptomatic patients with WM. In the 26 patients in the study, ORR was 96% with median time to response was 8 weeks and the median PFS was not reached after a median follow up of 22 months.44
Bruton Tyrosine Kinase (BTK) Inhibitors
BTK, a critical kinase in B-cell receptor (BCR) signaling plays a crucial role in the growth and survival of clonal cells in B-cell malignancies, including WM. Inhibition of BTK induces apoptosis, inhibits DNA replication, and blocks pro-survival signaling pathways in tumor cells. Ibrutinib, acalabrutinib and zanubrutinib have been studied and shown to have activity in WM.45
In a phase II trial of ibrutinib monotherapy in 30 symptomatic, treatment-naïve WM patients treated with ibrutinib, ORR and major (partial or greater than partial) responses for all patients were 100% and 83%, respectively. The 18-month PFS was 92%. Significant grade 2/3 treatment-related toxicities included bruising (7%), neutropenia (7%), upper respiratory tract infection (7%), atrial fibrillation (10%), and hypertension (13%).46
The Phase 3 iNNOVATE trial (n = 150) compared both newly diagnosed and patients with relapsed/refractory WM treated with ibrutinib and rituximab or rituximab plus placebo.47 In this study, the combination significantly prolonged PFS compared to rituximab monotherapy (PFS at 30 months: 82% vs. 28%, p <0.001). The rate of major response was higher with ibrutinib–rituximab than with placebo–rituximab (72% vs. 32%, P<0.001). Atrial fibrillation (any grade (19% vs. 1%), grade ≥ 3 (12% vs. 1%)) and hypertension (13% vs. 4%) were higher in the ibrutinib–rituximab group. In a longer follow up with median follow-up of 5 years, patients treated with the ibrutinib-rituximab continued to have longer PFS (not reached vs. 20.3 months).48 Combination Ibrutinib-Rituximab is approved by the US FDA but not by the European Medicines Agency (EMA).
Zanubrutinib and acalabrutinib are more selective BTK inhibitors that exhibit less off-target inhibition than ibrutinib. In a phase 2 study of acalabrutinib in symptomatic naïve and relapsed or refractory disease (N=106 of which 14 treatment naive), ORR was 93%. Grade 3/4 atrial fibrillation occurred in one (1%) patient and grade 3/4 bleeding occurred in three (3%) patients.45 In a phase 2 study of single agent zanubrutinib in WM (n=77 with 24 treatment naïve patients) ORR was 96% and VGPR/CR rate was 45%. Three-year PFS was 81 months. Significant AEs (any grade) were neutropenia (18.2%), major hemorrhage (4%) and atrial fibrillation/flutter (any grade- 5%, grade ≥ 3–1.3%).49 The phase 3 ASPEN study compared the efficacy and safety of ibrutinib and zanubrutinib in patients with R/R WM (n=201). Major response rates (MRR) were 77% and 78%, respectively. The median PFS were not reached. 84% and 85% of ibrutinib and zanubrutinib patients were progression free at 18 months. Atrial fibrillation (any grade (15% vs. 4%) and grade ≥ 3 (2% vs. none)) and major hemorrhage (6% vs. 9%) was less common in the zanubrutinib arm.50 Neutropenia was more common in the zanubrutinib arm (any grade (25% vs. 12%) and grade ≥3 (20% vs. 8%)). These studies with newer BTK inhibitors suggest comparable efficacy and lower cardiovascular morbidity compared to compared to ibrutinib. The major drawback with BTK inhibitors is the indefinite period of treatment and the efficacy of these agents is substantially effected by poor compliance and treatment disruptions due to adverse events.51
BCL2 Inhibitors
BCL2 expression supports survival of malignant cells by inhibiting apoptosis. BCL2 is expressed in a low level in malignant lymphoplasmacytic cells in WM and BCL2 antagonism has shown invitro activity and potential synergy with BTK inhibitors in WM.52 Venetoclax, a potent BCL2 inhibitor has shown activity in R/R WM in a phase II clinical trial (n=32) with an ORR of 84% and a median PFS of 30 months at a follow up of 33 months.53 The major grade≥ 3 event was neutropenia in 45% of the study participants. A study of venetoclax in addition to ibrutinib in treatment naïve WM patients (ClinicalTrials.gov Identifier: NCT04273139) is ongoing.
Immunomodulatory Agents
Immunomodulatory agents (iMID’s) including thalidomide and lenalidomide are an integral part of therapy in MM. iMIDs are hypothesized to augment antibody dependent cell mediated cytotoxicity of anti-CD20 antibodies. A combination of thalidomide (200 mg for 2 weeks, then 400 mg for 50 weeks) and rituximab was evaluated in a phase 2 study (n=25) of WM, of which a majority (n=20) were treatment naïve. The ORR was 72% with a median PFS of 38 months in responders. The major toxicity was peripheral neuropathy which was attributed to thalidomide and lead to decreased adoption of this agent in the clinical practice.54 A second generation iMID, lenalidomide was evaluated in a phase 2 study in combination with rituximab in patients with WM naïve to either agents, which showed a poor response rate (ORR-50%) and a high rate of anemia (80%) in patients treated with this combination, even with dose reductions.55 Given these adverse events, iMIDs are not recommended in the treatment naïve patients but may be a reasonable approach in relapsed/refractory patients.
Maintenance Therapy
WM is an indolent lymphoma and maintenance therapy has been investigated to improve survival outcomes. In two large observational studies, maintenance rituximab after chemo-immunotherapy induction improved PFS and showed a trend toward better OS.56,57 A large prospective study, StiL NHL7-2008 MAINTAIN trial evaluated maintenance rituximab vs. observation in patients who had partial response or better after BR in patients with WM. Of 218 randomized patients, 2-year rituximab maintenance provided a better disease control (PFS- 101 months vs. 83 months), however this difference was not statistically significant with a hazard ratio of 0.80 (95% CI 0.51–1.25, p=0.32).58
Summary for Upfront Treatment
Several chemoimmunotherapy combinations and novel agents are associated with high ORR in patients with treatment naïve WM. Treatment should only be initiated for symptomatic disease. The choice of the regimen depends on patient and disease characteristics, as well as patient preference. Chemoimmunotherapy with BR or DRC and BTK inhibition with ibrutinib or newer BTK inhibitors are two common treatment options. While chemoimmunotherapy may be associated with increased short-term toxicity, it has the advantage of fixed duration treatment. In addition, patients who are suspected to have lower compliance to oral medications may also benefit from finite upfront strategies. BTK inhibitors may be easier to tolerate in the short term especially in older patients but have unique cardiovascular toxicities and require long-term treatment. Figure 1 describes a treatment algorithm for patients with newly diagnosed WM.
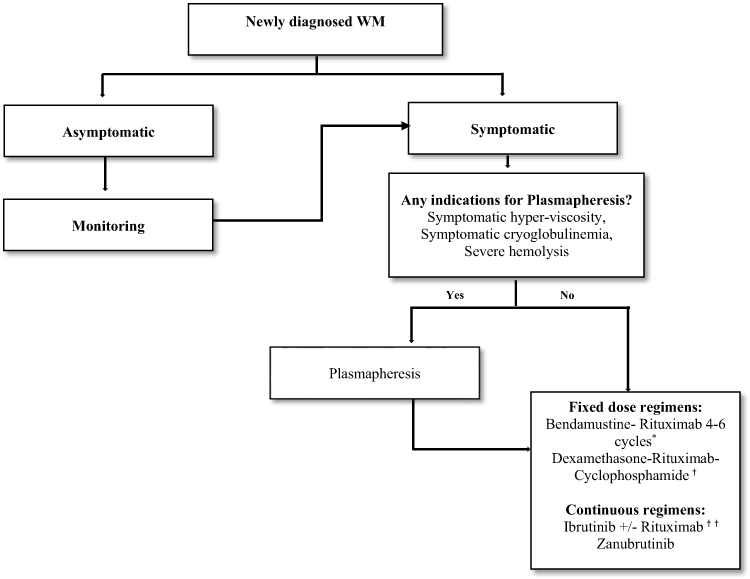 |
Figure 1 Treatment algorithm for newly diagnosed Waldenström. Notes: *BR is preferred in MYD88 WT and CXCR4 mutated due to increased resistance with BTK inhibitor. *BR is preferred in patients where rapid tumor reduction is needed. †DRC is well tolerated in older, less fit patients but decreased PFS compared to BR. ††Combination Ibrutinib+ rituximab only approved in US. |
Conclusion
WM is a rare, indolent, incurable, non-Hodgkin’s B cell lymphoma. The differential diagnosis of WM is broad, and workup including a bone marrow biopsy, cytogenetic and molecular studies are helpful to rule out other mature B cell NHLs. Early recognition and effective treatment of complications, including using plasmapheresis when indicated, is prudent to avoid premature mortality and morbidity in this otherwise indolent lymphoma. Multiple effective therapies with comparable outcomes are now available for the treatment of WM. Patient preference, clinical presentation, co-morbidities, molecular features, and treatment goals should be considered before choosing appropriate initial therapy for newly diagnosed WM. It is also essential to consider the financial toxicities of indefinite treatments such as oral BTK inhibitors and patients’ ability to obtain these medications. Population-based studies have shown considerable improvement in the outcomes of WM over the last two decades, mainly owing to an improved understanding of the pathophysiology of the disease and the introduction of targeted agents.3,4,59
Funding
Surbhi Sidana was supported by: KL2TR003143, KL2 Mentored Career Development Program, Stanford Clinical Translational Science Award Program.
Disclosure
Dr Surbhi Sidana reports personal fees from Magenta Therapeutics, BMS, Janssen, Sanofi, Oncopeptides, other from Magenta Therapeutics, BMS, Allogene, Janssen, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi:10.1182/blood-2016-01-643569
2. Konoplev S, Medeiros LJ, Bueso-Ramos CE, Jorgensen JL, Lin P. Immunophenotypic profile of lymphoplasmacytic lymphoma/Waldenström macroglobulinemia. Am J Clin Pathol. 2005;124(3):414–420. doi:10.1309/3g1x-dx0d-vhbn-vkb4
3. Castillo JJ, Olszewski AJ, Cronin AM, Hunter ZR, Treon SP. Survival trends in Waldenström macroglobulinemia: an analysis of the surveillance, epidemiology and end results database. Blood. 2014;123(25):3999–4000. doi:10.1182/blood-2014-05-574871
4. Yin X, Chen L, Fan F, et al. Trends in incidence and mortality of Waldenström macroglobulinemia: a population-based study. Front Oncol. 2020;10:1712. doi:10.3389/fonc.2020.01712
5. Giordano TP, Henderson L, Landgren O, et al. Risk of non-Hodgkin lymphoma and lymphoproliferative precursor diseases in US veterans with hepatitis C virus. JAMA. 2007;297(18):2010–2017. doi:10.1001/jama.297.18.2010
6. Treon S, Hunter Z, Aggarwal A, et al. Characterization of familial Waldenström’s macroglobulinemia. Ann Oncol. 2006;17(3):488–494. doi:10.1093/annonc/mdj111
7. Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenström macroglobulinemia: long-term results. Blood. 2012;119(19):4462–4466. doi:10.1182/blood-2011-10-384768
8. Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378(3):241–249.
9. Landgren O, Graubard B, Kumar S, et al. Prevalence of myeloma precursor state monoclonal gammopathy of undetermined significance in 12372 individuals 10–49 years old: a population-based study from the National Health and Nutrition Examination Survey. Blood Cancer J. 2017;7(10):e618–e618. doi:10.1038/bcj.2017.97
10. Argyropoulos KV, Vogel R, Ziegler C, et al. Clonal B cells in Waldenström’s macroglobulinemia exhibit functional features of chronic active B-cell receptor signaling. Leukemia. 2016;30(5):1116–1125. doi:10.1038/leu.2016.8
11. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367(9):826–833. doi:10.1056/NEJMoa1200710
12. Rhyasen GW, Starczynowski DT. IRAK signalling in cancer. Br J Cancer. 2015;112(2):232–237. doi:10.1038/bjc.2014.513
13. Varettoni M, Zibellini S, Arcaini L, et al. MYD88 (L265P) mutation is an independent risk factor for progression in patients with IgM monoclonal gammopathy of undetermined significance. Blood. 2013;122(13):2284–2285. doi:10.1182/blood-2013-07-513366
14. Poulain S, Roumier C, Venet-Caillault A, et al. Genomic landscape of CXCR4 mutations in Waldenström macroglobulinemia. Clin Cancer Res. 2016;22(6):1480–1488. doi:10.1158/1078-0432.CCR-15-0646
15. Kaiser LM, Hunter ZR, Treon SP, Buske C. CXCR4 in Waldenström’s macroglobulinema: chances and challenges. Leukemia. 2021;35(2):333–345. doi:10.1038/s41375-020-01102-3
16. Castillo JJ, Gustine J, Meid K, Dubeau T, Hunter ZR, Treon SP. Histological transformation to diffuse large B‐cell lymphoma in patients with Waldenström macroglobulinemia. Am J Hematol. 2016;91(10):1032–1035. doi:10.1002/ajh.24477
17. Ansell SM, Kyle RA, Reeder CB, et al. Diagnosis and management of Waldenström macroglobulinemia: mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc. 2010;85(9):824–833. doi:10.4065/mcp.2010.0304
18. García-Sanz R, Montoto S, Torrequebrada A, et al. Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases. Br J Haematol. 2001;115(3):575–582. doi:10.1046/j.1365-2141.2001.03144.x
19. Stone MJ, Pascual V. Pathophysiology of Waldenström’s macroglobulinemia. Haematologica. 2010;95(3):359–364. doi:10.3324/haematol.2009.017251
20. Weaver A, Rubinstein S, Cornell RF. Hyperviscosity syndrome in paraprotein secreting conditions including Waldenstrom macroglobulinemia. Front Oncol. 2020;10:815. doi:10.3389/fonc.2020.00815
21. Menke MN, Feke GT, McMeel JW, Branagan A, Hunter Z, Treon SP. Hyperviscosity-related retinopathy in Waldenström macroglobulinemia. Arch Ophthalmol. 2006;124(11):1601–1606. doi:10.1001/archopht.124.11.1601
22. Klein CJ, Moon JS, Mauermann ML, et al. The neuropathies of Waldenstrom’s macroglobulinemia (WM) and IgM-MGUS. Can J Neurol Sci. 2011;38(2):289–295. doi:10.1017/S0317167100011483
23. Simon L, Fitsiori A, Lemal R, et al. Bing-Neel syndrome, a rare complication of Waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). haematologica. 2015;100(12):1587. doi:10.3324/haematol.2015.133744
24. Dimopoulos MA, Kyle RA, Anagnostopoulos A, Treon SP. Diagnosis and management of Waldenstrom’s macroglobulinemia. J Clin Oncol. 2005;23(7):1564–1577. doi:10.1200/JCO.2005.03.144
25. Gertz MA, Kyle RA, Noel P. Primary systemic amyloidosis: a rare complication of immunoglobulin M monoclonal gammopathies and Waldenström’s macroglobulinemia. J Clin Oncol. 1993;11(5):914–920. doi:10.1200/JCO.1993.11.5.914
26. Castro H, Valenzuela R, Ruiz P, Lenz O, Monrroy M. Nephrotic syndrome and kidney failure due to immunocomplex-mediated renal damage in a patient with Waldenström’s Macroglobulinemia: a case report. Cases J. 2008;1(1):333. doi:10.1186/1757-1626-1-333
27. Oza A, Rajkumar S. Waldenstrom macroglobulinemia: prognosis and management. Blood Cancer J. 2015;5(3):e394–e394. doi:10.1038/bcj.2015.28
28. Pangalis GA, Kyrtsonis MC, Kontopidou FN, et al. Differential diagnosis of Waldenstrom’s macroglobulinemia and other B-cell disorders. Clin Lymphoma. 2005;5(4):235–240. doi:10.3816/clm.2005.n.006
29. Bustoros M, Sklavenitis-Pistofidis R, Kapoor P, et al. Progression risk stratification of asymptomatic Waldenström macroglobulinemia. J Clin Oncol. 2019;37(16):1403. doi:10.1200/JCO.19.00394
30. Zanwar S, Abeykoon JP, Ansell SM, et al. Primary systemic amyloidosis in patients with Waldenström macroglobulinemia. Leukemia. 2019;33(3):790–794. doi:10.1038/s41375-018-0286-7
31. Sidana S, Larson DP, Greipp PT, et al. IgM AL amyloidosis: delineating disease biology and outcomes with clinical, genomic and bone marrow morphological features. Leukemia. 2020;34(5):1373–1382. doi:10.1038/s41375-019-0667-6
32. Kastritis E, Morel P, Duhamel A, et al. A revised international prognostic score system for Waldenström’s macroglobulinemia. Leukemia. 2019;33(11):2654–2661. doi:10.1038/s41375-019-0431-y
33. Poulain S, Roumier C, Bertrand E, et al. TP53 mutation and its prognostic significance in Waldenstrom’s macroglobulinemia. Clin Cancer Res. 2017;23(20):6325–6335. doi:10.1158/1078-0432.ccr-17-0007
34. Divoka M, Pika T, Krupkova L, et al. Clinical implications of MYD88 and CXCR4 in patients with Waldenström’s macroglobulinemia. Blood. 2018;132:5314. doi:10.1182/blood-2018-99-112855
35. Stone MJ, Bogen SA. Role of plasmapheresis in Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2013;13(2):238–240. doi:10.1016/j.clml.2013.02.013
36. Buske C, Leblond V. How to manage Waldenstrom’s macroglobulinemia. Leukemia. 2013;27(4):762–772. doi:10.1038/leu.2013.36
37. Ghobrial IM, Fonseca R, Greipp PR, et al. Initial immunoglobulin M ‘flare’ after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia: an Eastern Cooperative Oncology Group Study. Cancer. 2004;101(11):2593–2598. doi:10.1002/cncr.20658
38. Furman RR, Eradat HA, DiRienzo CG, et al. Once-weekly ofatumumab in untreated or relapsed Waldenstrom’s macroglobulinaemia: an open-label, single-arm, phase 2 study. Lancet Haematol. 2017;4(1):e24–e34. doi:10.1016/s2352-3026(16)30166-1
39. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–1210. doi:10.1016/S0140-6736(12)61763-2
40. Dimopoulos MA, Anagnostopoulos A, Kyrtsonis M-C, et al. Primary treatment of Waldenstrom macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol. 2007;25(22):3344–3349. doi:10.1200/JCO.2007.10.9926
41. Chen CI, Kouroukis CT, White D, et al. Bortezomib is active in patients with untreated or relapsed Waldenstrom’s macroglobulinemia: a phase II study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(12):1570–1575. doi:10.1200/jco.2006.07.8659
42. Dimopoulos MA, García-Sanz R, Gavriatopoulou M, et al. Primary therapy of Waldenström macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN). Blood. 2013;122(19):3276–3282. doi:10.1182/blood-2013-05-503862
43. Treon SP, Tripsas CK, Meid K, et al. Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström’s macroglobulinemia. Blood. 2014;124(4):503–510. doi:10.1182/blood-2014-03-566273
44. Castillo JJ, Meid K, Gustine JN, et al. Prospective clinical trial of ixazomib, dexamethasone, and rituximab as primary therapy in Waldenström macroglobulinemia. Clin Cancer Res. 2018;24(14):3247–3252. doi:10.1158/1078-0432.CCR-18-0152
45. Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: a single-arm, multicentre, phase 2 study. Lancet Haematol. 2020;7(2):e112–e121. doi:10.1016/S2352-3026(19)30210-8
46. Treon SP, Gustine J, Meid K, et al. Ibrutinib monotherapy in symptomatic, treatment-naïve patients with Waldenström macroglobulinemia. J Clin Oncol. 2018;36(27):2755–2761. doi:10.1200/JCO.2018.78.6426
47. Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenström’s macroglobulinemia. N Engl J Med. 2018;378(25):2399–2410. doi:10.1056/NEJMoa1802917
48. Buske C, Tedeschi A, Trotman J, et al. Five-year follow-up of ibrutinib plus rituximab vs placebo plus rituximab for Waldenstrom’s macroglobulinemia: final analysis from the randomized phase 3 iNNOVATETM study. Blood. 2020;136:24–26. doi:10.1182/blood-2020-134460
49. Tam CSL, Opat S, Marlton P, et al. Three-year follow-up of treatment-naïve and previously treated patients with Waldenström macroglobulinemia (WM) receiving single-agent zanubrutinib. Am Soc Clin Oncol. 2020;38:8051. doi:10.1200/JCO.2020.38.15_suppl.8051
50. Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–2050. doi:10.1182/blood.2020006844
51. Castillo JJ, Gustine JN, Meid K, et al. Impact of ibrutinib dose intensity on patient outcomes in previously treated Waldenström macroglobulinemia. haematologica. 2018;103(10):e466. doi:10.3324/haematol.2018.191999
52. Cao Y, Yang G, Hunter ZR, et al. The BCL2 antagonist ABT-199 triggers apoptosis, and augments ibrutinib and idelalisib mediated cytotoxicity in CXCR4 Wild-type and CXCR4 WHIM mutated Waldenstrom macroglobulinaemia cells. Br J Haematol. 2015;170(1):134–138. doi:10.1111/bjh.13278
53. Castillo JJ, Allan JN, Siddiqi T, et al. Venetoclax in previously treated Waldenström macroglobulinemia. J Clin Oncol. 2022;40(1):63–71. doi:10.1200/jco.21.01194
54. Davies FE, Raje N, Hideshima T, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood. 2001;98(1):210–216. doi:10.1182/blood.v98.1.210
55. Treon SP, Soumerai JD, Branagan AR, et al. Lenalidomide and rituximab in Waldenstrom’s macroglobulinemia. Clin Cancer Res. 2009;15(1):355–360. doi:10.1158/1078-0432.ccr-08-0862
56. Treon SP, Hanzis C, Manning RJ, et al. Maintenance Rituximab is associated with improved clinical outcome in rituximab naive patients with Waldenstrom Macroglobulinaemia who respond to a rituximab‐containing regimen. Br J Haematol. 2011;154(3):357–362. doi:10.1111/j.1365-2141.2011.08750.x
57. Zanwar S, Abeykoon JP, Gertz MA, et al. Rituximab-based maintenance therapy in Waldenström macroglobulinemia: a case control study. Am Soc Clin Oncol. 2019;37:7559. doi:10.1200/JCO.2019.37.15_suppl.7559
58. Rummel MJ, Lerchenmüller C, Hensel M, et al. Two years rituximab maintenance vs. observation after first line treatment with bendamustine plus rituximab (BR) in patients with Waldenström’s macroglobulinemia (MW): results of a prospective, randomized, multicenter phase 3 study (the StiL NHL7-2008 MAINTAIN trial). Blood. 2019;134:343.
59. Madhira BR, Durer S, Ghimire K. Survival trend of Waldenstrom macroglobulinemia over last 3 decade shows health disparities in minority population. Blood. 2021;138:5023. doi:10.1182/blood-2021-154319
60. Owen RG, Kyle RA, Stone MJ, et al. Response assessment in Waldenström macroglobulinaemia: update from the VIth International Workshop. British journal of haematology. 2013 Jan;160(2):171-6.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.