")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
Dietary Advanced Glycation End Products Shift the Gut Microbiota Composition and Induce Insulin Resistance in Mice
Authors Wang J, Cai W, Yu J, Liu H, He S, Zhu L, Xu J
Received 27 October 2021
Accepted for publication 22 January 2022
Published 15 February 2022 Volume 2022:15 Pages 427—437
DOI https://doi.org/10.2147/DMSO.S346411
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Jiao Wang,1– 3,* Wei Cai,4,* Jiao Yu,1 Honghong Liu,1 Shasha He,1 Lingyan Zhu,1– 3 Jixiong Xu1– 3
1Department of Endocrinology and Metabolism, First Affiliated Hospital of Nanchang University, Nanchang, 330006, People’s Republic of China; 2Jiangxi Clinical Research Center for Endocrine and Metabolic Disease, Nanchang, 330006, People’s Republic of China; 3Jiangxi Branch of National Clinical Research Center for Metabolic Disease, Nanchang, 330006, People’s Republic of China; 4Department of Medical Genetics and Cell biology, Medical College of Nanchang University, Nanchang, 330006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jixiong Xu, Email [email protected]
Objective: This study aimed to explore the associations between gut microbiota characteristics and glycometabolic profiles in mice fed diets high in advanced glycation end products (AGEs).
Methods: C57BL/6 mice were exposed to a heat-treated diet or exogenous AGEs for 24 weeks, and glucose metabolism was assessed via the intraperitoneal glucose-tolerance test (IPGTT). Serum AGE and lipopolysaccharide-binding protein (LBP) levels were quantified using ELISA kits. 16S rDNA sequencing was performed to analyze the changes in gut microbiota according to α- and β-diversity. Key operational taxonomic units (OTUs) were evaluated, and co-abundance groups (CAGs) were delineated using weighted correlation network analysis. Associations between CAGs and clinical parameters were analyzed using Spearman correlation; predictive functional analysis of gut microbiota was performed using Kyoto Encyclopedia of Genes and Genomes data.
Results: We identified significant increases in fasting blood glucose (FBG) and fasting insulin levels, as well as homeostatic model assessment insulin resistance (HOMA-IR) and glucose area under the receiver operating characteristic curve from IPGTT, in the high-AGE diet groups relative to controls at week 24. Serum AGE and LBP levels were elevated, and the α- and β-diversity of gut microbiota reduced in high-AGE diet groups. We identified 92 key OTUs that clustered into six CAGs, revealing positive correlations between CAG2/3/5 and insulin levels and mice weight and negative correlations between CAG1/3/4/5 and AGE, FBG, and LBP levels and HOMA-IR in mice fed high-AGE diets. We observed a reduced abundance of butyrate-producing bacteria, including Bacteroidales_S24-7, Ruminococcaceae, and Lachnospiraceae, in mice fed high-AGE diets, with pathway analysis of gut microbiota revealing significantly enriched fructose and mannose metabolism.
Conclusion: High-AGE diets altered the gut microbiota composition and structure, and induced insulin resistance in mice. In the pathogenesis of insulin resistance, the loss of butyrate-producing bacteria might impair the colonic epithelial barrier, thereby triggering chronic low-grade inflammation.
Keywords: diet, advanced glycation end products, gut microbiota, insulin resistance
Background
Advanced glycation end products (AGEs) are a group of heterogeneous compounds formed by non-enzymatic glycation of proteins and lipids.1 AGEs were first recognized as endogenous compounds during circumstances of hyperglycemia in diabetes.2,3 Endogenous AGEs accumulate in the body with aging, and their formation is accelerated by hyperglycemia and oxidative stress.4 In addition to endogenous AGEs, exogenous sources, such as diet and smoking, substantially contribute to the AGE pool.5–7 The major factors determining dietary AGEs include nutrient composition (protein > fat > carbohydrate), high temperature (grilling, broiling, roasting, barbecuing, and frying), and long cooking times.8,9 The processes of globalization and urbanization have influenced dietary patterns worldwide. Among the most profound changes is the worldwide prevalence of the modern western diet that is rich in heat-processed food, which lead to the formation of large amounts of AGEs.10
Excessive AGEs are closely associated with the risk of chronic diseases, such as metabolic syndrome,11 atherosclerosis,12 cancer,13 and kidney disease.14 AGEs exert deleterious effects by binding to their receptor (RAGE).15 Early evidence has shown that only ~10–30% of dietary AGEs are intestinally absorbed into circulation,16 and that ~20–50% of ingested AGEs are excreted in urine and feces,17 suggesting that a portion of dietary AGEs not absorbed and excreted might be metabolized by the gut microbiota. Subsequently, elucidation of the role of unabsorbed dietary AGEs has gained increasing attention, as AGEs carry a possible correlation with gut microbiota composition.
The gut microbiota comprises a complex and dynamic ecosystem, and maintaining its homeostasis is important for human health. Evidence from animal studies has suggested that high-AGE diets are associated with alterations in gut microbiota composition, including decreases in the abundance of Bacteroidetes, Bifidobacteria, and Lactobacilli.18,19 However, another study reported an increased abundance of Bifidobacteria in rats fed bread crusts or bread crumbs.20 These disparities in microbial signatures between studies may be due to the intrinsic flaw of taxon phylogenetic analysis, which ignores variations in bacterial strains belonging to the same species. Therefore, more research is required to understand how members of the gut bacteria interact with one another as functional groups when exposed to high-AGE diets as a new environmental resource.21,22
In this study, we fed C57BL/6 mice a heat-treated diet or exogenous AGEs for 24 weeks, and subsequently analyzed their gut microbial characteristics using 16S rDNA gene sequencing to determine the associations between bacterial co-abundance groups (CAGs) and glycometabolic profiles. The findings provide novel insights into the effect of dietary AGEs on insulin resistance in mice.
Methods
Methylglyoxal-Bovine Serum Albumin (MG-BSA) Preparation
The glycation of BSA by MG was performed as described by Lee et al.,23 with slight modifications. Briefly, BSA (0.1 mM) was incubated in sodium phosphate buffer (0.1 M) with MG (100 mM) for 72 h, dialyzed against ammonium bicarbonate buffer (pH 8.0, 4 °C), and then lyophilized and stored at −80 °C.
Animals and Diets
Male C57BL/6 mice (12-weeks old; ~25–28 g) were purchased from Slake Jingda Experimental Animal Co., Ltd (Hunan, China) and housed under controlled environmental conditions at 23 ± 1 °C with a relative humidity of 45–55% and a 12-h dark-light cycle. Animal experiments were performed in accordance with the Guidelines on the Ethical Treatment of Experimental Animals (2006, No. 398) and issued by the Ministry of Science and Technology of China.
After adaptive feeding for 2 weeks, 40 mice were randomly assigned to three groups fed regular chow diet (control group; n = 10), regular chow with MG-BSA (1 mg/g; exogenous AGE group; n = 15), or a heat-treated diet (dietary AGE group; n = 15) for 24 weeks. The heat-treated diet consisted of regular feed exposed to heat treatment of 120 °C for 15 min. The nutrient composition of the diet in the 3 groups is shown in Supplementary Table S1. AGE content was measured using an ELISA kit for AGEs (Cloud-Clone Corp., Houston, TX, USA) according to the manufacturer’s instructions. The AGE contents in the exogenous AGE and dietary AGE groups was ~4- and ~2-fold that of the control group, respectively (Supplementary Figure S1). Body weights of mice were monitored weekly throughout the study; fasting blood glucose (FBG) was measured from blood collected from the tail vein, and fecal samples were collected at baseline and at the end of the 12th and 24th weeks.
Intraperitoneal Glucose-Tolerance Test (IPGTT)
Glucose tolerance was assessed by IPGTT after animals fasted for 12 h. Mice received an intraperitoneal injection of glucose (1 g/kg body weight), and blood samples were collected sequentially from the tail vein at various time points (0, 15, 30, 60, and 120 min), followed by determination of glucose levels using a glucometer (Abbott, Alameda, CA, USA). The area under the receiver operating characteristic (ROC) curve (AUC) was calculated.
Measurements of Serum Insulin, AGEs, and Lipopolysaccharide-Binding Protein (LBP)
At the end of the 24th week, mice were euthanized under CO2 anesthesia, and blood samples collected retro-orbitally were centrifuged for serum separation and stored at −80 °C until further use. Serum insulin, AGEs, and LBP levels were quantified using commercially available ELISA kits (Cloud-Clone Corp.) according to the manufacturer’s instructions. The homeostatic model assessment insulin resistance (HOMA-IR) was calculated using the following formula: HOMA-IR = fasting plasma glucose (mM) × fasting insulin (mIU/L)/22.5.
Fecal DNA Extraction and 16S rDNA Sequencing
Fecal DNA was extracted using the QIAamp DNA stool mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The integrity and size of the extracted DNA samples were assessed via gel electrophoresis, and the concentration and purity were determined by spectrophotometry. V4 regions of the 16S rDNA gene were amplified using the 515F (GTGCCAGCMGCCGCGGTAA) and 806R (GGACTACHVGGGTWTCTAAT) primers with dual-index barcodes used to tag each sample. The PCR products were purified with AmpureXPbeads (Agencourt Bioscience Corp., Beverly, MA, USA) to remove non-specific products. Purified amplicons were pooled in equimolar amounts and paired-end sequenced on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) according to standard protocols.
Sequencing Data Analysis
The sequences were clustered into operational taxonomic units (OTUs) by setting a 97% threshold in USEARCH (v.7.0).24 A representative sequence for each OTU was aligned against the Silva reference database for taxonomy analysis (https://www.arb-silva.de/) using the RDP Classifier (v.2.2; https://rdp.cme.msu.edu/classifier/classifier.jsp;jsessionid=1A02A869E05F11CEBB89409DE3440CB6.10.0.0.9) with a bootstrap cut-off of 70%. For α-diversity analysis, Simpson’s diversity and observed species indices were used to assess the species diversity and richness, respectively, and differences among groups were analyzed using the Kruskal–Wallis test. The β-diversity was analyzed using principal coordinate analysis (PCoA) plots based on weighted UniFrac distances. Statistical significance between groups was tested using permutational multivariate analysis of variance (ANOVA) with 999 permutations.
Microbial Cluster Analysis Using Weighted Correlation Network Analysis (WGCNA)
OTUs were selected at the end of week 24 from the three groups using the random forest package in R (https://www.r-project.org/), and the core OTUs were determined as those shared by at least 70% of all samples.25 WGCNA was performed to delineate CAGs based on the abundance of their key OTUs.26 A weighted network was constructed by computing the Pearson correlation coefficient between any pair of nodes by setting a soft-thresholding power equal to 7 (Supplementary Figure S2). Module clusters (labeled by color) represented different CAGs, each containing a set of OTUs. The gray module represents a grouping of nodes with outlying profiles and was not analyzed further. Module membership was computed for every node in each module and represents the correlation between module eigengene and OTU expression profile. Computations were carried out using the R package WGCNA.
Spearman Correlation Analysis
Spearman correlations between CAGs and clinical indices were analyzed using R packages. Differential abundances of CAGs among the three groups were tested using the Kruskal–Wallis test, and ROC curves were applied to examine the predictive discriminating values. The AUC was >0.8, indicating good performance of the CAGs.
Functional Analysis of Gut Microbiota
Predictive functional analysis of the gut microbiota associated with the various dietary treatments was performed using PICRUSt (https://picrust.github.io/picrust/), based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.27 Differences between the control group and the high-AGE diet groups (exogenous and dietary AGE groups) were determined using the STAMP software (v2.1.3; https://beikolab.cs.dal.ca/software/STAMP).
Statistical Analysis
Clinical data were statistically analyzed using the GraphPad Prism software (v.8.0; GraphPad Software, La Jolla, CA, USA). Data with normal distributions were expressed as the mean ± standard deviation, and those with non-normal distributions were expressed as the medians (interquartile ranges). Two-way ANOVA was used for the intra- and inter-group comparisons of the bioclinical parameters. P < 0.05 was considered statistically significant.
Results
Effects of High-AGE Diets on Bioclinical Parameters in Mice
We observed no significant changes in body weight increase among the three groups during the experimental period (Figure 1A). However, FBG levels were significantly higher in the exogenous and dietary AGE groups relative to the control group at week 12, with a similar trend observed at week 24 (Figure 1B). At the end of the intervention period, the fasting insulin level and HOMA-IR were significantly elevated in the exogenous and dietary AGE groups relative to the control group, whereas no evident difference was observed between the exogenous and dietary AGE groups (Figure 1C and D). Additionally, the glucose AUC during the IPGTT test was significantly increased in the exogenous and dietary AGE groups relative to the control group, indicating glucose intolerance in mice fed high-AGE diets (Figure 1E and F). Moreover, serum AGE levels were elevated in the exogenous and dietary AGE groups relative to the control group, with a more evident increase observed in the exogenous AGE group as compared with the dietary AGE group (Figure 1G). Furthermore, levels of LBP, an indicator of host intestinal permeability and inflammation,28 were notably elevated in the exogenous AGE group relative to those in the control and dietary AGE groups, although no evident difference could be observed between the dietary AGE group and control groups (Figure 1H). These results indicate that high-AGE diets induced insulin resistance and increased intestinal permeability in mice.
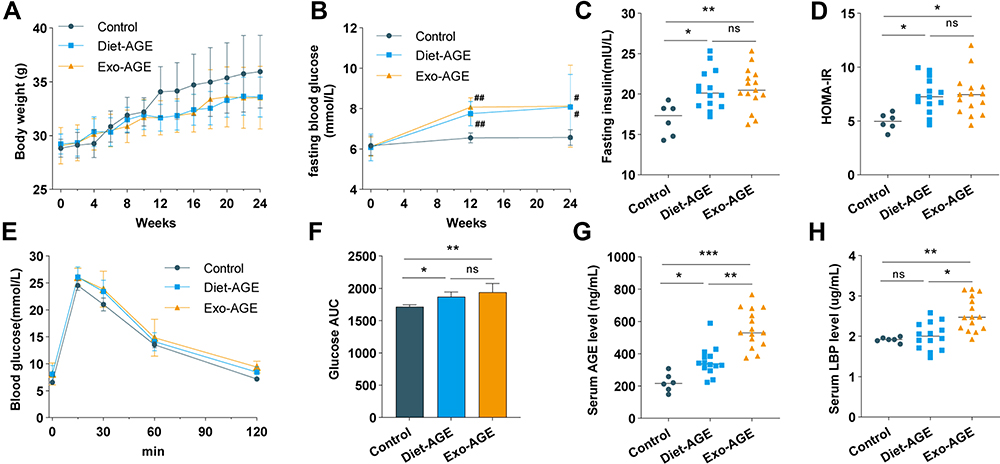 |
Figure 1 High-AGE diets induce insulin resistance and increase intestinal permeability in mice. (A) Changes in body weight among the control and dietary and exogenous AGE groups during the 24-week feeding period. (B) Changes in serum fasting blood glucose levels among the three groups at baseline and weeks 12 and 24. (C) Fasting insulin levels among the three groups at week 24. (D) Homeostatic model assessment insulin resistance (HOMA-IR) among the three groups at week 24. (E) Glucose tolerance was assessed by the intraperitoneal glucose-tolerance test (IPGTT) in the three groups at week 24. (F) Glucose area under the curve (AUC) during IPGTT. (G) Serum AGE levels among the three groups at week 24. (H) Serum lipopolysaccharide-binding protein (LBP) levels among the three groups at week 24. *p < 0.05, **p < 0.01, and ***p < 0.001 for intergroup comparisons; #p < 0.05 and ##p < 0.01 for comparison with the control group at the same time point. Abbreviations: AGE, advanced glycation end product; Diet-AGE, dietary AGE group; Exo-AGE, exogenous AGE group. |
Effects of High-AGE Diets on Gut Microbiota Structure and Composition in Mice
To compare the gut microbiota structures of mice fed with various diets, we performed bacterial 16S rDNA sequencing in fecal samples, and a total of 830 OTUs were clustered with a cut-off of 97% identity. There were no differences in the richness and diversity of the initial gut microbiota in mice from the three groups before high-AGE diet intervention. At the end of week 24, the diversity and richness of the gut microbiota were significantly decreased in the dietary AGE group relative to the control and exogenous AGE groups (Figure 2A and B). Prior to high-AGE diet intervention, PCoA based on weighted UniFrac distance showed no difference in the structure of the gut microbiota among the three groups. During intervention, only the microbiota structure in the exogenous AGE diet group changed significantly relative to baseline (p = 0.012), and at the end of intervention, the gut microbiota structure in the exogenous AGE diet group significantly diverged from that of the other two groups (p = 0.008) (Figure 2C).
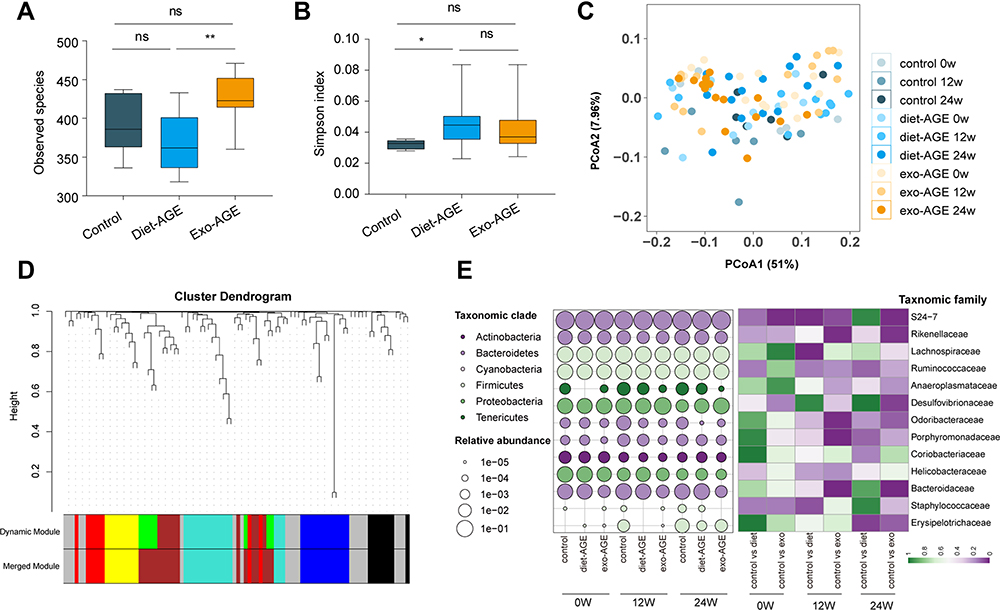 |
Figure 2 High-AGE diets alter gut microbiota structure and composition in mice. (A) Evaluation of α-diversity according to Observed species. (B) Evaluation of α-diversity according to Simpson’s diversity index. (C) Evaluation of β-diversity via principal coordinate analysis (PCoA) based on weighted UniFrac distance. (D) Key operational taxonomic units (OTUs) were clustered into six co-abundance group (CAGs) using weighted correlation network analysis (WGCNA), labeled by various colors. (E) Time-course changes in the abundance of gut bacteria at the family level among the three groups. Circle size and color indicate the relative abundance and phylum level of gut bacteria, respectively. The heatmap in the right panel was generated based on p-values. The Wilcoxon rank-sum test was used to compare two groups at the same time point. *p < 0.05; **p < 0.01 for intergroup comparisons. Abbreviations: AGE, advanced glycation end product; Diet-AGE, dietary AGE group; Exo-AGE, exogenous AGE group. |
Effects of High-AGE Diets on Bacterial CAGs in Mice
Because bacteria act as functional groups (guilds) in the gut ecosystem,29 we established a co-abundance weighted network containing 92 core OTUs, as determined by WGCNA correlation coefficients. These OTUs were clustered into six CAGs (labeled by color), ranging in size from eight to 18 members (Figure 2D and Supplementary Table S2). The results showed that bacterial species of the same genus exhibited variable responses to environment perturbations. For example, OTU589, OTU760, OTU45, OTU46, OTU72, and OTU383 belonging to the genus Bacteroides were clustered into CAG2 (OTU589 and 760), CAG3, CAG4 (OTU46 and 72), and CAG5, respectively. The changes in the relative abundance of key OTUs is shown in Supplementary Figure S3, with these OTUs mainly belonging to Ruminococcus, Bacteroides, Oscillospira, and Clostridium at the genus level (Supplementary Table S2). Prior to high-AGE diets intervention, there were no differences in the relative abundance of bacterial species among the three groups. During intervention, the abundance of Bacteroidales_S24-7, Bacteroidaceae, Porphyromonadaceae, Odoribacteraceae, Lachnospiraceae, Rikenellaceae, and Erysipelotrichaceae decreased, whereas that of Desulfovibrionaceae increased in mice fed high-AGE diets (Figure 2E).
Compared with the control group at the same time point, the relative abundance of CAG1 (OTU137, 180, 190, 439, 508, 525, 672, and 81) decreased in the dietary AGE group and markedly increased in the exogenous AGE group. Conversely, the relative abundance of CAG2 (OTU1, 216, 28, 313, 352, 38, 456, 589, 606, 741, 760, 93, and 97) and CAG3 (OTU128, 130, 154, 156, 166, 226, 244, 260, 270, 280, 337, 361, 45, 455, 463, 544, 576, and 88) significantly decreased in only the exogenous AGE group. Additionally, the relative abundance of CAG4 (OTU118, 163, 316, 403, 409, 46, 48, and 72) decreased in the exogenous AGE group, but increased in the dietary AGE group. Moreover, the relative abundance of CAG5 (OTU102, 145, 164, 201, 205, 212, 245, 27, 31, 339, 378, 383, 386, 710, 77, and 824) significantly decreased in the exogenous and dietary AGE groups, and the relative abundance of CAG6 (OTU135, 168, 282, 309, 499, 51, 554, 60, and 649) markedly increased in only the dietary AGE group (Figure 3).
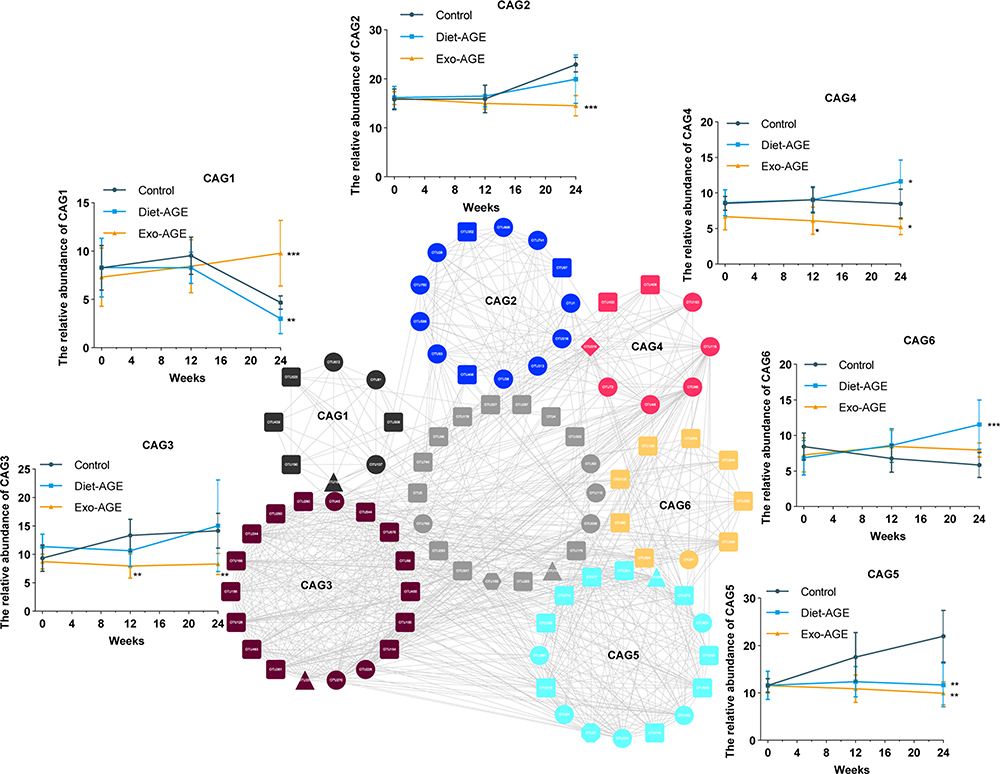 |
Figure 3 Changes among the six co-abundance groups (CAGs) in the gut microbiota of mice fed high-AGE diets for 24 weeks. Different node colors and shapes represent different CAGs and phyla, respectively. Round, square, triangle, diamond, hexagon, and octagon represent Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, Tenericutes, and Cyanobacteria, respectively. The relative abundance of each CAG was summed from the relative abundance of the included operational taxonomic units (OTUs) in all samples of each group. *p < 0.05; **p < 0.01; ***p < 0.001 for comparison with the control group at the same time point. Abbreviations: AGE, advanced glycation end product; Diet-AGE, dietary AGE group; Exo-AGE, exogenous AGE group. |
Relationship Between CAGs and Clinical Phenotype in Mice
We found that all CAG modules helped distinguish between the exogenous AGE and control groups (AUC > 0.8), whereas CAG1/5/6 helped distinguish between the dietary AGE and control groups (Figure 4A). We then analyzed the correlations between the CAGs and clinical phenotype to further explore the features of the gut microbiota in mice fed high-AGE diets. CAG1/3 were negatively correlated with levels of LBP and weight in control mice. CAG2/3/5 were positively correlated with levels of insulin and weight, while CAG1/3/4/5 were negatively correlated with AGE, FBG, and LBP levels and HOMA-IR in mice fed exogenous AGE diets. Furthermore, CAG3 was negatively correlated with AGE levels in mice fed dietary AGE diets (Figure 4B).
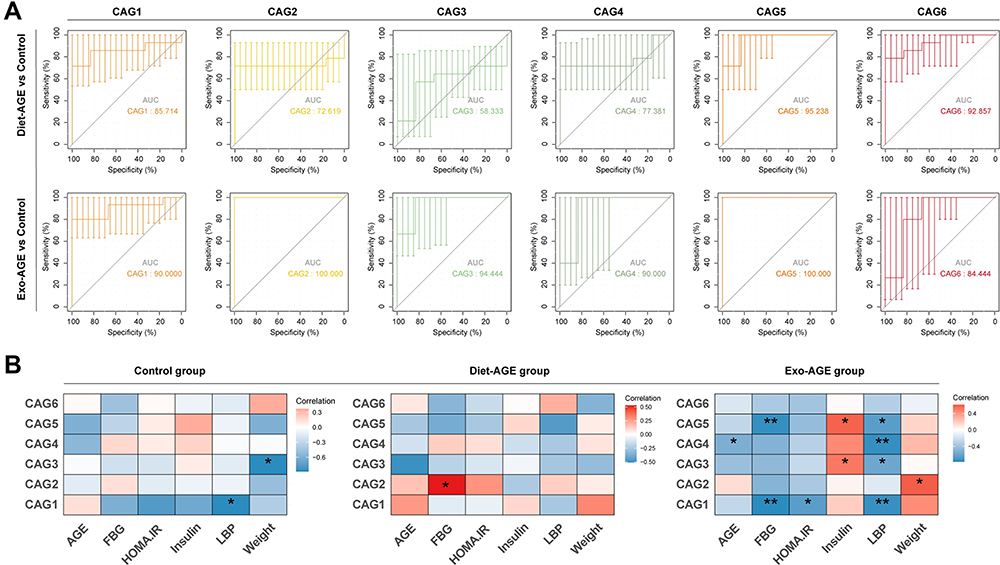 |
Figure 4 Correlation analysis between co-abundance groups (CAGs) and clinical phenotype in the three groups. (A) Receiver operating characteristic (ROC) curves were applied to assess the ability of the six CAGs to discriminate between the high-AGE diet and control groups. (B) Heatmap of correlations between CAGs and clinical phenotype in the three groups. The color represents the level of correlation: red, positive correlation; blue, negative correlation. *p < 0.05; **p < 0.01. Abbreviations: AGE, advanced glycation end product; FBG, fasting blood glucose; HOMA-IR, homeostatic model assessment insulin resistance; LBP, lipopolysaccharide-binding protein; Diet-AGE, dietary AGE group; Exo-AGE, exogenous AGE group. |
Predicted Functional Changes in Gut Microbiota
To further understand the effects of high-AGE diets on gut microbial metabolism in mice, we analyzed a set of significant KEGG pathways using a non-parametric t-test. Compared with the control group, pathways associated with fructose and mannose metabolism, amino sugar and nucleotide sugar metabolism, methane metabolism, peptidoglycan biosynthesis, lysine biosynthesis, starch and sucrose metabolism, glycolysis/gluconeogenesis, and biosynthesis of various secondary metabolites were enhanced in mice fed high-AGE diets. Additionally, those related to propanoate metabolism, amino sugar and nucleotide sugar metabolism, arginine and proline metabolism, glutathione metabolism, ubiquinone and other terpenoid-quinone biosynthesis, and nicotinate and nicotinamide metabolism were significantly impaired (Figure 5).
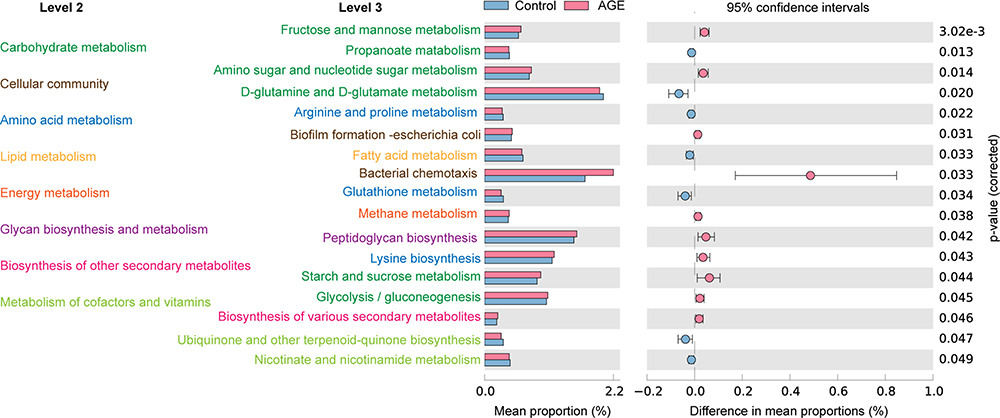 |
Figure 5 Functional changes in gut microbiota, as predicted by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Hierarchical clusters of metabolic pathways are denoted with different colors. Abbreviation: AGE, advanced glycation end product. |
Discussion
This study showed that high-AGE diets induced insulin resistance and altered gut microbiota composition in mice, and that these changes were associated with an altered glycometabolic phenotype and host intestinal inflammation. These results are consistent with those of several animal and human studies showing that high-AGE diets can induce insulin resistance and exert detrimental effects on metabolic diseases,30,31 and that low-AGE intervention ameliorates insulin resistance in obese individuals with metabolic syndrome, and patients with type 2 diabetes.32,33 Furthermore, investigation of mechanistic links between AGEs and insulin resistance identified 92 key OTUs that clustered into six CAGs, revealing key OTUs altered in response to high-AGE diets, as determined by 16S rDNA sequencing of fecal samples.
We confirmed that the α- and β-diversities of the gut microbiota were reduced in mice fed high-AGE diets at the end of week 24, which was consistent with a previous report.34 Ecologically, core members of a CAG should thrive or decline together in response to environmental perturbations.35 Compared with taxon-based analysis, the CAG-based analysis performed in the present study provides a more ecologically reasonable method to explore the members of the gut bacteria with functional importance in insulin resistance. We found that the composition of the gut bacteria was altered in mice fed high-AGE diets relative to that observed in controls. Bacterial abundance in CAG1/2/3/4/5 was markedly decreased in mice fed exogenous or dietary AGE diets, and these CAGs were negatively correlated with AGE, FBG, LBP levels, and HOMA-IR and positively correlated with insulin levels. These CAGs mainly contained OTUs from Bacteroidales_S24-7, Ruminococcaceae, and Lachnospiraceae, and are primary butyrate producers with anti-inflammatory properties.36 However, we observed no significant correlations between CAG6 and high-AGE diet-associated metabolic phenotypes. Additionally, these CAGs could discriminate between high-AGE diets (especially the exogenous AGE diet) and the control group, suggesting the gut microbiota phenotype as a novel and potentially promising biomarker.
Butyrate is the preferred energy source for the colonic epithelium and is important for colonic homeostasis.37,38 Ruminococcaceae is reportedly negatively correlated with insulin resistance,39 which is consistent with the present results. Additionally, our results agree with those of a previous study reporting reduced abundance of Lachnospiraceae in mice fed high-AGE diets.34 Notably, we found that the abundance of Bacteroidales_S24-7 was also decreased by dietary AGE intervention, with this genus reportedly associated with alleviation of intestinal inflammation, and negatively correlated with obesity and inflammation.40,41 Overall, the presence of glycated proteins may have hampered butyrate formation through the loss of their producers, such as Bacteroidales_S24-7, Ruminococcaceae, and Lachnospiraceae, leading to the impairment of the colonic epithelial barrier (as evidenced by elevated serum LBP levels), and subsequent triggering of chronic low-grade systemic inflammation involved in the pathogenesis of insulin resistance.
To better understand the relationship between key gut bacterial functions and insulin resistance, we predicted the microbiota functional composition using the PICRUSt software. Differential functional pathway analysis of the gut microbiota revealed that fructose and mannose metabolism was significantly enriched in mice fed high-AGE diets. Excess fructose can lead to insulin resistance and metabolic syndrome,42 and Jaiswal et al.43 reported that fructose-induced generation of reactive oxygen species impairs glucose utilization and insulin signaling in skeletal muscle cells. In the present study, fructose metabolism may have altered glucose homeostasis, resulting in insulin resistance.
This study has several limitations. First, we confirmed the association between gut bacteria (especially butyrate-producing bacteria) and insulin resistance, exclusively in mice fed high-AGE diets. Further experiments on germ-free or pseudo-germ-free animal models are required to determine causality between gut microbiota and insulin resistance related to dietary AGE intervention. Second, the heat-treated diet used in this study was exposed to 120 °C for 15 min, leading to underrepresented AGE contents. AGEs as complex compounds demonstrate differences in digestion, absorption, and distribution. Seiquer et al.44 reported that the putative effects of dietary AGEs on gut bacterial composition depend on the chemical structure and amounts of the different compounds. Therefore, further research is needed to pursue the influence of specific AGEs on gut bacteria.
Conclusions
In this study, we confirmed that high-AGE diets altered gut microbiota composition and structure and induced insulin resistance in mice. Moreover, CAG-based analysis revealed that the abundance of butyrate-producing bacteria, such as Bacteroidales_S24-7, Ruminococcaceae, and Lachnospiraceae was reduced following high-AGE diet intervention. Mechanistically, we speculate that loss of butyrate-producing bacteria leads to the impairment of the colonic epithelial barrier, triggering the chronic low-grade systemic inflammation involved in the pathogenesis of insulin resistance. Therefore, application of low-AGE-producing culinary techniques (boiling, poaching, or stewing) may present an attractive prevention alternative in managing diabetes and potentially other metabolic diseases.
Abbreviations
AGE, advanced glycation end product; ANOVA, analysis of variance; AUC, area under the receiver operating characteristic curve; BSA, bovine serum albumin; CAG, co-abundance group; FBG, fasting blood glucose; HOMA-IR, homeostatic model assessment insulin resistance; IPGTT, intraperitoneal glucose-tolerance test; KEGG, Kyoto Encyclopedia of Genes and Genomes; LBP, lipopolysaccharide-binding protein; MG, methylglyoxal; OTU, operational taxonomic unit; PCoA, principal coordinate analysis; ROC, receiver operating characteristic; WGCNA, weighted correlation network analysis.
Data Sharing Statement
The 16S rDNA sequencing set of this article has been deposited in the Genome Sequence Archive (GSA) under the BioProject accession code PRJCA006137.
Ethics Approval and Consent to Participate
Animal experiments were performed in accordance with the Guidelines on the Ethical Treatment of Experimental Animals issued by the Ministry of Science and Technology of China and approved by the Ethics Committee of the First Affiliated Hospital of Nanchang University (No. 2021-5-073).
Acknowledgments
The authors would like to thank BGI Company (Shenzhen, China) for performing the fecal microbiota 16S rDNA sequencing.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by grants from the National Natural Science Funds of China [grant no. 81760168] and Natural Science Foundation of Jiangxi Province [grant no. 20192BAB205031].
Disclosure
The authors declare that they have no competing interests.
References
1. Rabbani G, Ahn SN. Structure, enzymatic activities, glycation and therapeutic potential of human serum albumin: a natural cargo. Int J Biol Macromol. 2019;123:979–990. doi:10.1016/j.ijbiomac.2018.11.053
2. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi:10.1038/414813a
3. Baig MH, Jan AT, Rabbani G, et al. Methylglyoxal and advanced glycation end products: insight of the regulatory machinery affecting the myogenic program and of its modulation by natural compounds. Sci Rep. 2017;7(1):5916. doi:10.1038/s41598-017-06067-5
4. Moldogazieva NT, Mokhosoev IM, Mel’nikova TI, Porozov YB, Terentiev AA. Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxid Med Cell Longev. 2019;2019:3085756. doi:10.1155/2019/3085756
5. Ashraf JM, Ahmad S, Rabbani G, et al. Physicochemical analysis of structural alteration and advanced glycation end products generation during glycation of H2A histone by 3-deoxyglucosone. IUBMB Life. 2014;66(10):686–693. doi:10.1002/iub.1318
6. Ashraf JM, Ahmad S, Rabbani G, et al. 3-Deoxyglucosone: a potential glycating agent accountable for structural alteration in H3 histone protein through generation of different AGEs. PLoS One. 2015;10(2):e0116804. doi:10.1371/journal.pone.0116804
7. Ashraf JM, Rabbani G, Ahmad S, et al. Glycation of H1 histone by 3-deoxyglucosone: effects on protein structure and generation of different advanced glycation end products. PLoS One. 2015;10(6):e0130630. doi:10.1371/journal.pone.0130630
8. Goldberg T, Cai W, Peppa M, et al. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc. 2004;104(8):1287–1291. doi:10.1016/j.jada.2004.05.214
9. Uribarri J, Woodruff S, Goodman S, et al. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. 2010;110(6):911–916.e12. doi:10.1016/j.jada.2010.03.018
10. Bettiga A, Fiorio F, Di Marco F, et al. The modern western diet rich in advanced glycation end-products (AGEs): an overview of its impact on obesity and early progression of renal pathology. Nutrients. 2019;11(8):1748. doi:10.3390/nu11081748
11. Gugliucci A. Formation of fructose-mediated advanced glycation end products and their roles in metabolic and inflammatory diseases. Adv Nutr. 2017;8(1):54–62. doi:10.3945/an.116.013912
12. Hartog JW, Voors AA, Bakker SJ, Smit AJ, van Veldhuisen DJ. Advanced glycation end-products (AGEs) and heart failure: pathophysiology and clinical implications. Eur J Heart Fail. 2007;9(12):1146–1155. doi:10.1016/j.ejheart.2007.09.009
13. van Heijst JW, Niessen HW, Hoekman K, Schalkwijk CG. Advanced glycation end products in human cancer tissues: detection of Nepsilon-(carboxymethyl)lysine and argpyrimidine. Ann N Y Acad Sci. 2005;1043:725–733. doi:10.1196/annals.1333.084
14. Uribarri J, Peppa M, Cai W, et al. Dietary glycotoxins correlate with circulating advanced glycation end product levels in renal failure patients. Am J Kidney Dis. 2003;42(3):532–538. doi:10.1016/s0272-6386(03)00779-0
15. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014;2:411–429. doi:10.1016/j.redox.2013.12.016
16. Koschinsky T, He CJ, Mitsuhashi T, et al. Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci U S A. 1997;94(12):6474–6479. doi:10.1073/pnas.94.12.6474
17. Delgado-Andrade C, Tessier FJ, Niquet-Leridon C, Seiquer I, Pilar Navarro M. Study of the urinary and faecal excretion of Nε-carboxymethyllysine in young human volunteers. Amino Acids. 2012;43(2):595–602. doi:10.1007/s00726-011-1107-8
18. Zhang Z, Li D. Thermal processing of food reduces gut microbiota diversity of the host and triggers adaptation of the microbiota: evidence from two vertebrates. Microbiome. 2018;6(1):99. doi:10.1186/s40168-018-0471-y
19. Marungruang N, Fåk F, Tareke E. Heat-treated high-fat diet modifies gut microbiota and metabolic markers in apoe-/- mice. Nutr Metab. 2016;13:22. doi:10.1186/s12986-016-0083-0
20. Helou C, Anton PM, Niquet-Léridon C, Spatz M, Tessier FJ, Gadonna-Widehem P. Fecal excretion of Maillard reaction products and the gut microbiota composition of rats fed with bread crust or bread crumb. Food Funct. 2017;8(8):2722–2730. doi:10.1039/c7fo00430c
21. Rabbani G, Choi I. Roles of osmolytes in protein folding and aggregation in cells and their biotechnological applications. Int J Biol Macromol. 2018;109:483–491. doi:10.1016/j.ijbiomac.2017.12.100
22. Haq SK, Rabbani G, Ahmad E, Atif SM, Khan RH. Protease inhibitors: a panacea? J Biochem Mol Toxicol. 2010;24(4):270–277. doi:10.1002/jbt.20335
23. Lee C, Yim MB, Chock PB, Yim HS, Kang SO. Oxidation-reduction properties of methylglyoxal-modified protein in relation to free radical generation. J Biol Chem. 1998;273(39):25272–25278. doi:10.1074/jbc.273.39.25272
24. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998. doi:10.1038/nmeth.2604
25. Berry D, Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol. 2014;5:219. doi:10.3389/fmicb.2014.00219
26. Guidi L, Chaffron S, Bittner L, et al. Plankton networks driving carbon export in the oligotrophic ocean. Nature. 2016;532(7600):465–470. doi:10.1038/nature16942
27. Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi:10.1038/nbt.2676
28. Weiss J. Bactericidal/permeability-increasing protein (BPI) and lipopolysaccharide-binding protein (LBP): structure, function and regulation in host defence against Gram-negative bacteria. Biochem Soc Trans. 2003;31(Pt 4):785–790. doi:10.1042/bst0310785
29. Zhang C, Yin A, Li H, et al. Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. EBioMedicine. 2015;2(8):968–984. doi:10.1016/j.ebiom.2015.07.007
30. Angoorani P, Ejtahed HS, Mirmiran P, Mirzaei S, Azizi F. Dietary consumption of advanced glycation end products and risk of metabolic syndrome. Int J Food Sci Nutr. 2016;67(2):170–176. doi:10.3109/09637486.2015.1137889
31. Mastrocola R, Collotta D, Gaudioso G, et al. Effects of exogenous dietary advanced glycation end products on the cross-talk mechanisms linking microbiota to metabolic inflammation. Nutrients. 2020;12(9):2497. doi:10.3390/nu12092497
32. Vlassara H, Cai W, Tripp E, et al. Oral AGE restriction ameliorates insulin resistance in obese individuals with the metabolic syndrome: a randomised controlled trial. Diabetologia. 2016;59(10):2181–2192. doi:10.1007/s00125-016-4053-x
33. Luévano-Contreras C, Garay-Sevilla ME, Wrobel K, Malacara JM, Wrobel K. Dietary advanced glycation end products restriction diminishes inflammation markers and oxidative stress in patients with type 2 diabetes mellitus. J Clin Biochem Nutr. 2013;52(1):22–26. doi:10.3164/jcbn.12-40
34. Qu W, Yuan X, Zhao J, et al. Dietary advanced glycation end products modify gut microbial composition and partially increase colon permeability in rats. Mol Nutr Food Res. 2017;61(10). doi:10.1002/mnfr.201700118
35. Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10(8):538–550. doi:10.1038/nrmicro2832
36. Jangi S, Gandhi R, Cox LM, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. 2016;7:12015. doi:10.1038/ncomms12015
37. Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27(2):104–119. doi:10.1111/j.1365-2036.2007.03562.x
38. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273. doi:10.1126/science.1223490
39. Khorraminezhad L, Leclercq M, O’Connor S, et al. Dairy product intake modifies gut microbiota composition among hyperinsulinemic individuals. Eur J Nutr. 2021;60(1):159–167. doi:10.1007/s00394-020-02226-z
40. Rooks MG, Veiga P, Wardwell-Scott LH, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. Isme j. 2014;8(7):1403–1417. doi:10.1038/ismej.2014.3
41. Ma L, Hu L, Jin L, et al. Rebalancing glucolipid metabolism and gut microbiome dysbiosis by nitrate-dependent alleviation of high-fat diet-induced obesity. BMJ Open Diabetes Res Care. 2020;8(1):e001255. doi:10.1136/bmjdrc-2020-001255
42. Tappy L. Metabolism of sugars: a window to the regulation of glucose and lipid homeostasis by splanchnic organs. Clin Nutr. 2021;40(4):1691–1698. doi:10.1016/j.clnu.2020.12.022
43. Jaiswal N, Maurya CK, Pandey J, Rai AK, Tamrakar AK. Fructose-induced ROS generation impairs glucose utilization in L6 skeletal muscle cells. Free Radic Res. 2015;49(9):1055–1068. doi:10.3109/10715762.2015.1031662
44. Seiquer I, Rubio LA, Peinado MJ, Delgado-Andrade C, Navarro MP. Maillard reaction products modulate gut microbiota composition in adolescents. Mol Nutr Food Res. 2014;58(7):1552–1560. doi:10.1002/mnfr.201300847
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.