")
Back to Journals » OncoTargets and Therapy » Volume 12
Dicerandrol B: a natural xanthone dimer induces apoptosis in cervical cancer HeLa cells through the endoplasmic reticulum stress and mitochondrial damage
Authors Gao DD, Guo ZM, Wang JB, Hu GF, Su YQ, Chen LJ, Lv QW, Yu HM, Qin JC, Xu W
Received 17 October 2018
Accepted for publication 3 January 2019
Published 13 February 2019 Volume 2019:12 Pages 1185—1193
DOI https://doi.org/10.2147/OTT.S191204
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjay Singh
Dandan Gao,1 Zhimin Guo,1 Jiabin Wang,2 Gaofeng Hu,1 Yuqiao Su,3 Lijun Chen,1 Qianwen Lv,1 Huimei Yu,2 Jianchun Qin,3 Wei Xu1
1Department of Clinical Laboratory, The First Hospital of Jilin University, Changchun 130021, China; 2Department of Pathology and Pathophysiology, School of Basic Medical Sciences, Jilin University, Changchun 130021, China; 3Department of Biotechnology, College of Plant Sciences, Jilin University, Changchun, Jilin 130062, China
Background: Dicerandrol B is a natural antitumor agent that can be isolated from the endophytic fungus, Phomopsis sp. The present study investigated the effects of dicerandrol B on human cervical cancer HeLa cells.
Materials and methods: In this study, dicerandrol B was identified by electrospray ionization mass spectrometry and nuclear magnetic resonance spectroscopy. We used MTT to detect the cell viability. Flow cytometry was used to analyze the apoptosis and cell cycle. Western blot was used to examine the expression of related proteins.
Results: Dicerandrol B was isolated from the endophytic fungus Phomopsis sp. The MTT assay and flow cytometry showed that dicerandrol B significantly inhibited HeLa cell viability and induced G2/M cell cycle arrest. Western blot analysis demonstrated that dicerandrol B increased the levels of GRP78, ubiquitin, cleaved PARP, and Bax protein, decreased the levels of PARP and Bcl-2 protein, and caused an increase in the Bax/Bcl-2 ratio in HeLa cells. Dicerandrol B increased the production of ROS in HeLa cells, which was attenuated by the antioxidant N-acetyl-l-cysteine.
Conclusion: These findings suggest that dicerandrol B induces apoptosis in human HeLa cells, possibly through the endoplasmic reticulum stress and mitochondrial apoptotic pathways. This suggests that dicerandrol B possesses strong anticancer activity in cervical cancer and provides insight into the underlying mechanisms.
Keywords: apoptosis, cervical cancer, endoplasmic reticulum stress, mitochondrial damage
Introduction
Cervical cancer is the fourth most common type of cancer among women and the seventh most common cancer globally.1 In China, the 5-year relative survival rate for cervical cancer is estimated at 45.4%.2 Management of cervical cancer includes surgery, radiotherapy, and chemotherapy, either as a single agent or in combination with radiotherapy (concurrent chemotherapy) to sensitize cancer cells to radiation.3 Although chemotherapy is effective in the treatment of cervical cancer, it is associated with toxicity,4 and some cervical tumors are chemotherapy resistant. Currently, there is an unmet need for novel therapies in cervical cancer that have low toxicity and achieve good response rates.
Natural products represent effective therapeutic options for the prevention and treatment of many cancers.5 In particular, the xanthone dimer metabolite dicerandrol B, isolated from the endophytic fungus Phomopsis species,6–8 displays significant antitumor activity in vitro. Dicerandrol B was first isolated in 2001 from Phomopsis longicolla, an endophytic fungus of the mint species Dicerandra frutescens.9,10 Dicerandrol B demonstrated antimicrobial activities against Bacillus subtilis and Staphylococcus aureus and cytotoxic activity against the human cancer cell lines colon HCT-116, lung A549 and Calu-3, and breast MDA-MB-435.9,10 However, the specific mechanisms underlying the antitumor effect of dicerandrol B remain to be elucidated.
Apoptosis is mainly activated by cell surface death receptors or mitochondria-mediated apoptosis signaling pathways.11 The death receptor apoptotic pathway (extrinsic) pathway involves Fas and tumor necrosis factor receptor family. The mitochondrial (intrinsic) pathway is triggered by the release of mitochondrial apoptotic factors.7 The release of cytochrome c, together with Apaf-1, activates caspase-9, which cleaves downstream caspase-3. Caspase-3 is capable of hydrolyzing substrate proteins, such as PARP, leading to PARP dysfunction,12 all of which ultimately leads to cell death. In addition, endoplasmic reticulum (ER) stress (ERS) can also cause apoptosis. The ER is the principal organelle responsible for multiple cellular functions including protein folding and maturation and the maintenance of cellular homeostasis. ERS is activated by a variety of factors and triggers the unfolded protein response (UPR), which restores homeostasis or activates cell death.13
In this study, we investigated the effects of dicerandrol B on human cervical cancer HeLa cells. Findings showed that this compound can affect the cell cycle and induce apoptosis in HeLa cells through the ERS and mitochondrial apoptotic pathways. These data advocate for the use of natural metabolites in antitumor therapies.
Materials and methods
Cell line and cell culture
The cervical cancer HeLa cell line, which was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), was obtained from the Institute of Virology and AIDS Research of the First Hospital of Jilin University (Changchun, China). The human gastric cancer cell line MGC803 and the human mammary epithelial cell line MCF10A were obtained from the First Hospital of Jilin University. HeLa cells and MGC803 cells were cultured in DMEM and Roswell Park Memorial Institute Medium 1640, respectively, supplemented with 10% FBS and 1% antibiotics. MCF10A cells were cultured in DMEM/F-12 supplemented with 5% horse serum, 20 ng/mL epidermal growth factor, 0.01 mg/mL insulin, 500 ng/mL hydrocortisone, and 100 ng/mL cholera toxin. Cells were maintained in 5% CO2 and 100% humidity at 37°C.
Dicerandrol B preparation
The endophytic fungus Phomopsis sp. was isolated from Polygonatum cyrtonema Hua. Fungal isolates were grown in an incubator on potato dextrose agar for 5 days at 26°C and inoculated into 500 mL Erlenmeyer flasks containing 200 mL sterile solid rice medium (prepared by soaking 100 g of commercially available rice in 100 mL distilled water overnight) under static culture conditions at room temperature. After 40 days of culture, the solid fugal culture was overlaid with a cellophane film and dicerandrol B was extracted with ethyl acetate. Crude extract (108.60 g) was prepared by removing the solvent by evaporation under reduced pressure. Five fractions, A–F, were prepared by subjecting the extract to silica gel column chromatography using CH2Cl2:MeOH (CH2Cl2, 50:1, 30:1, 20:1, 10:1, MeOH) as the eluent. Fraction B (13.52 g) was purified to a yellow amorphous compound (526.85 mg) with Sephadex LH-20 with CH2Cl2:MeOH (6:4) and repeated silica gel column chromatography. The yellow amorphous compound was identified as dicerandrol B by electrospray ionization mass spectrometry (ESI-MS) and nuclear magnetic resonance (NMR) spectroscopy.
Cell viability assay
Cell viability was determined with the MTT assay. Briefly, HeLa cells were seeded at 1×104 cells/well in 96-well flat-bottom microtiter plates. After 24 hours, the medium was replaced with fresh DMEM containing 3 or 5 μg/mL dicerandrol B or dimethyl sulfoxide (DMSO; untreated control), and cells were incubated for 24, 48, or 96 hours. Subsequently, 20 μL of 5 mg/mL MTT was added to each well, and cells were incubated for 4 hours. Formazan was solubilized in 150 μL DMSO, and the OD at 490 nm was detected with a 96-well microplate reader (BioTek, Winooski, VT, USA). Cell viability was evaluated according to the formula: cell viability (%) = [1− (OD of the samples/OD of the control)] ×100%.
Colony formation assay
About 200 cells/well were added into a 24-well culture plate, with three wells per sample. After 2 weeks of incubation with different concentrations of dicerandrol B, the cells were washed three times with PBS and stained with the Giemsa solution. The plate clone formation efficiency was calculated as: (number of colonies/number of cells inoculated) ×100%.
Propidium iodide (PI) staining for cell cycle analysis
Cultures of HeLa cells were treated with dicerandrol B (3 or 5 μg/mL) or DMSO (untreated control) in DMEM and incubated for 24 hours. Cells were detached by treating with 0.25% trypsin for 2–3 minutes, washed, centrifuged, fixed in 70% cold ethanol (10 mL) at 4°C overnight, and incubated with PI buffer (50 mg/mL PI, 20 mg/mL RNase A; BD Biosciences, San Jose, CA, USA). After 30 minutes in the dark, cell cycle distribution was analyzed with flow cytometry (BD FACSAria II; BD Biosciences) and the MultiCycle software (Phoenix Flow Systems, San Diego, CA, USA).
Apoptosis assay
HeLa cells were treated with dicerandrol B (3 or 5 μg/mL) or DMSO (untreated control) in DMEM and incubated for 24 hours. Cells (1×106) were detached by treating with 0.25% trypsin and washed twice with cold PBS. Cells were resuspended in 500 μL binding buffer and stained with 5 μL Annexin V-fluorescein isothiocyanate (FITC) and 5 μL PI (Annexin V-FITC/PI Apoptosis Detection kit; BD Biosciences) in the dark at room temperature for 15 minutes. Apoptosis was measured with the FACSVerse™ flow cytometer (BD biosciences). Apoptotic cells were counted by the total percentage of Annexin V-positive cells, including the early apoptotic and late apoptotic cells.
Measurement of intracellular ROS level
Intracellular ROS levels were measured using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Beyotime, Nanjing, China), according to the manufacturer’s recommendations. Briefly, HeLa cells in six-well tissue culture plates were treated with dicerandrol B (3 or 5 μg/mL) or DMSO (untreated control) in DMEM for 24 hours; some cells were pretreated with 100 μmol/L N-acetyl-L-cysteine (NAC; antioxidant) for 60 minutes. After treatment, cells were incubated with 10 μM DCFH-DA diluted in serum-free culture medium in the dark at 37°C for 30 minutes, washed twice with PBS, and micrographs were obtained using a conventional fluorescent microscope (Olympus).
Western blot analysis
Cultured HeLa cells were detached by treating with 0.25% trypsin, washed, and pelleted by centrifugation at 12,000 rpm for 1 minute. Protein concentrations were quantified with the BCA protein assay kit using bicinchoninic acid (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. Cells were lysed in RIPA buffer and boiled for 10 minutes. Protein samples (30 μg/sample) were subjected to 10% SDS-PAGE and electrotransferred to polyvinylidene difluoride membranes (Bio-Rad). Membranes were blocked with 5% nonfat dry milk in TBS buffer (20 mM Tris-HCl, pH 7.4, 500 mM NaCl) and incubated with primary antibody at 4°C overnight. Primary antibodies included rabbit polyclonal anti-Bcl-2(1:1,000), Bax (1:1,000), PARP-1 (1:1,000), cleaved PARP (1:1,000), ubiquitin (Ub; 1:1,000), and GRP78 (1:1,000) (Cell Signaling Technology, Beverly, MA, USA). Immunoblots were washed with TBS-T and incubated with HRP-conjugated secondary antibodies at 37°C for 1–2 hours. Secondary antibodies were goat anti-rabbit IgG (1:5,000) or rabbit anti-mouse IgG (1:5,000). Chemiluminescence signals were detected using the BioSpectrum Imaging System (BioSpectrum 410, UVP; Analytik Jena US LLC, Upland, CA, USA). β-Actin served as the internal control.
Quantitative real-time RT-PCR
RNA was extracted from cultured HeLa cells using Trizol reagent (Thermo Fisher Scientific) and isolated, according to the manufacturer’s instructions. RNA was treated with DNAse (DNase I, RNase-Free; Ambion), and 200 ng of total RNA was reverse-transcribed with oligo dT primers using the High-Capacity cDNA RT Kit (Thermo Fisher Scientific) in a 20 μL cDNA reaction, according to the manufacturer’s instructions. For quantitative PCR, template cDNA was added to a 20 μL reaction mixture with SYBR GREEN PCR Master Mix (Thermo Fisher Scientific) and 0.2 mM of primer. The marker of proliferation Ki67 (MKi-67) primers were: forward, 5′-GCCTGCTCGACCCTACAGA-3′ and reverse, 5′-GCTTGTCAACTGCGGTTGC-3′. The UBE2S primers were: forward, 5′-CCGACACGTACTGCTGACC-3′ and reverse, 5′-GCCGCATACTCCTCGTAGTTC-3′. The Gpx primers were: forward, 5′-CAGTCGGTGTATGCCTTCTCG-3′ and reverse, 5′-GAGGGACGCCACATTCTCG-3′. The GAPDH primers were: forward, 5′-CGG AGT CA A CGG ATT TGG TCG TAT-3′ and reverse, 5′-AGC CTT CTC CAT GGT GGT GAA GAC-3′. Amplification was performed using real-time quantitative PCR instrument (the LightCycler®480; Roche) for 40 cycles under the following conditions: initial denaturation at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Changes relative to GAPDH were calculated using the ΔΔ Ct method.
Statistical analysis
Statistical analyses were conducted with SPSS 17.0 software. All experiments were repeated three times. Data are presented as mean ± SD. Differences between test and control groups were compared with one-way ANOVA and Student’s t-test. P<0.05 was considered as a significant difference.
Results
Identification of dicerandrol B
ESI-MS (positive mode): m/z=709 [M+H]+, 731 [M+Na]; HR-ESI-MS: m/z 709.2116 [M+H]+ (calcd for C36H376O15, 709.2127), 731.1977 [M+Na] (calcd for C36H36NaO15, 731.1946), 1H NMR (acetone-d6, 300 MHz), and 13C NMR (acetone-d6, 125 MHz) data were in accordance with a previous report (Figure 1).9
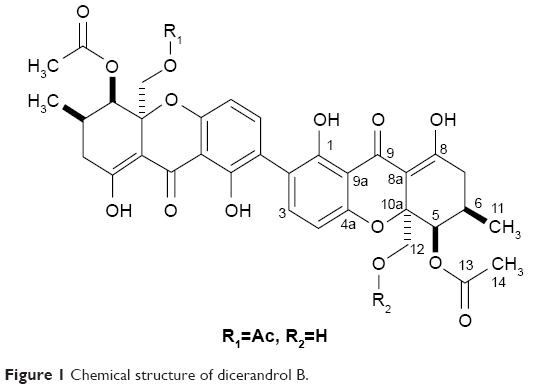 | Figure 1 Chemical structure of dicerandrol B. |
Effect of dicerandrol B on the growth of HeLa cells
In our experiment, we detected the cell viability of dicerandrol B on different human cells, including human gastric cancer cell MGC803, human mammary epithelial cell MCF10A, and human cervical cancer HeLa cells. We used MCF10A as a normal cell. The results showed that dicerandrol B could inhibit the cell viability of all three cells, but the inhibition of MGC803 and HeLa cells was more significant than that of MCF10A (Figure 2A). MTT assay indicated that dicerandrol B inhibited the viability of HeLa cells in a dose- and time-dependant manner. IC50 values for dicerandrol B were 7.13, 3.00, and 1.84 μg/mL after 24, 48, and 96 hours of incubation, respectively (Figure 2B). We also performed a clone formation experiment. The results were similar to the MTT results (Figure 2C). The apoptosis assay indicated that dicerandrol B (0, 3, and 5 μg/mL for 24 hours) induced apoptosis in HeLa cells in a dose-dependent manner; 30.9% HeLa cells were apoptotic following incubation with 3 μg/mL dicerandrol B, and 45% HeLa cells were apoptotic following incubation with 5 μg/mL dicerandrol B (Figure 2D). Western blot analysis showed a significant dose-dependent increase in the expression levels of cleaved PARP in HeLa cells incubated with dicerandrol B for 24 hours compared to control (Figure 2E and F). These findings suggest that dicerandrol B inhibits HeLa cell proliferation and promotes apoptosis by increasing the expression of cleaved PARP.
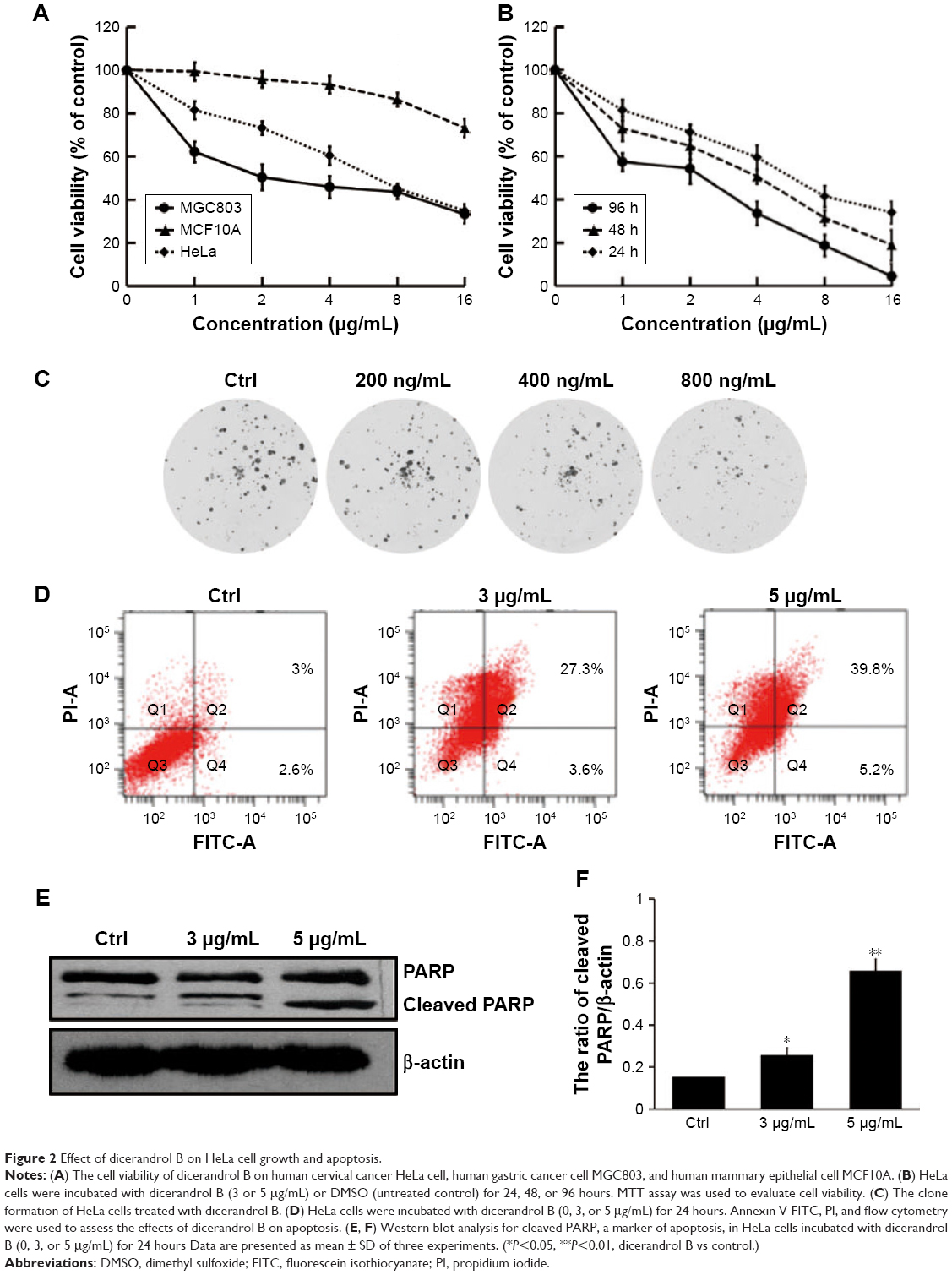 | Figure 2 Effect of dicerandrol B on HeLa cell growth and apoptosis. |
Effect of dicerandrol B on the HeLa cell cycle
Flow cytometry revealed a significant (67.24%) increase in the number of HeLa cells in the G2/M phase following incubation with 3 μg/mL dicerandrol B for 24 hours compared to control and a significant (17.91%) decrease in the number of HeLa cells in the S phase. These findings suggest that dicerandrol B significantly increases the number of HeLa cells in the G2/M phase of the cell cycle (Figure 3A and B).
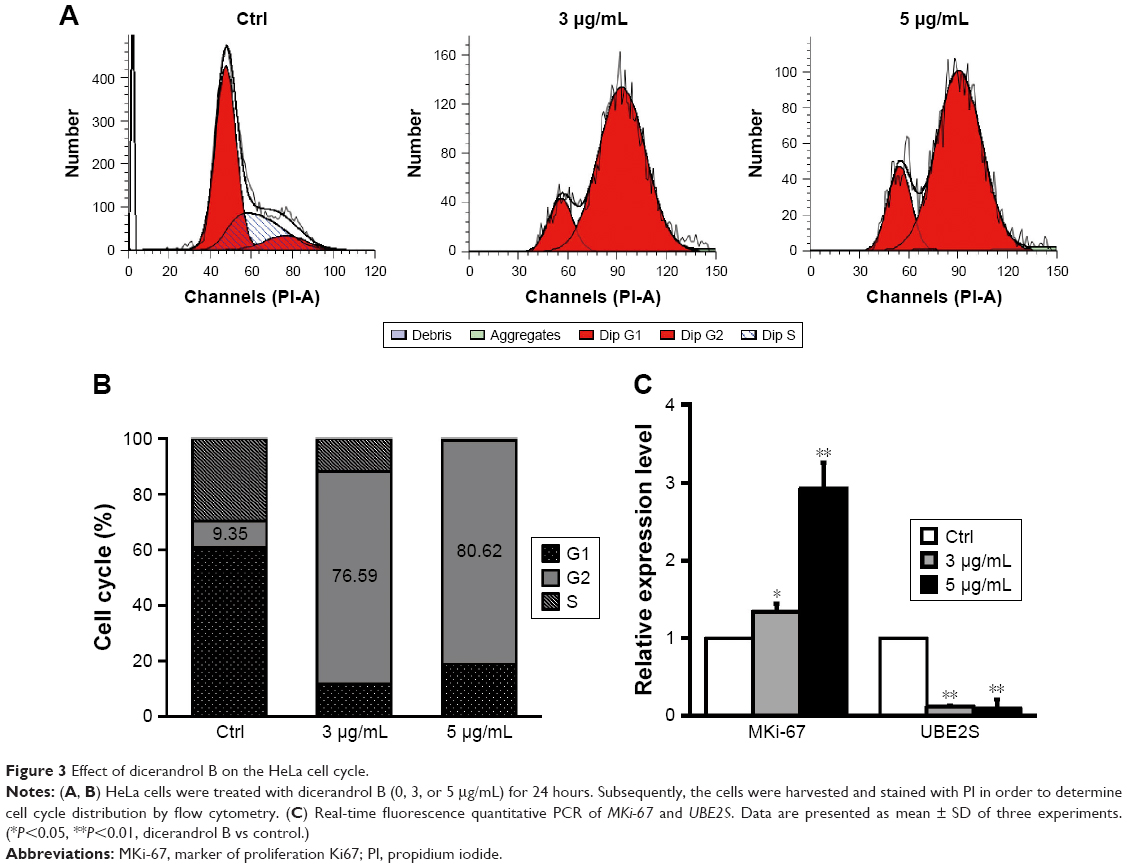 | Figure 3 Effect of dicerandrol B on the HeLa cell cycle. |
Real-time fluorescence quantitative PCR was used to investigate the expression levels of the cell cycle-related gene MKi-67 and ubiquitin conjugating enzyme E2S (UBE2S/E2-EPF) in HeLa cells incubated with dicerandrol B (0, 3, or 5 μg/mL for 24 hours). There was a significant dose-dependent increase in the expression level of MKi-67 and a significant dose-dependent decrease in the expression level of UBE2S in HeLa cells incubated with dicerandrol B compared to control (Figure 3C). These findings suggest that dicerandrol B induces G2/M cell cycle arrest in HeLa cells.
Effect of dicerandrol B on ERS in HeLa cells
Western blot analysis was used to investigate GRP78 and Ub protein levels in HeLa cells incubated with dicerandrol B (0, 3, or 5 μg/mL for 12 hours). GRP78 and Ub proteins are markers of ERS-associated apoptosis in HeLa cells (Figure 4A–C). Western blot analysis showed a significant dose-dependent increase in the expression levels of GRP78 and Ub in HeLa cells incubated with dicerandrol B compared to control. These findings suggest that dicerandrol B induces apoptosis in HeLa cells by the ERS signaling pathway.
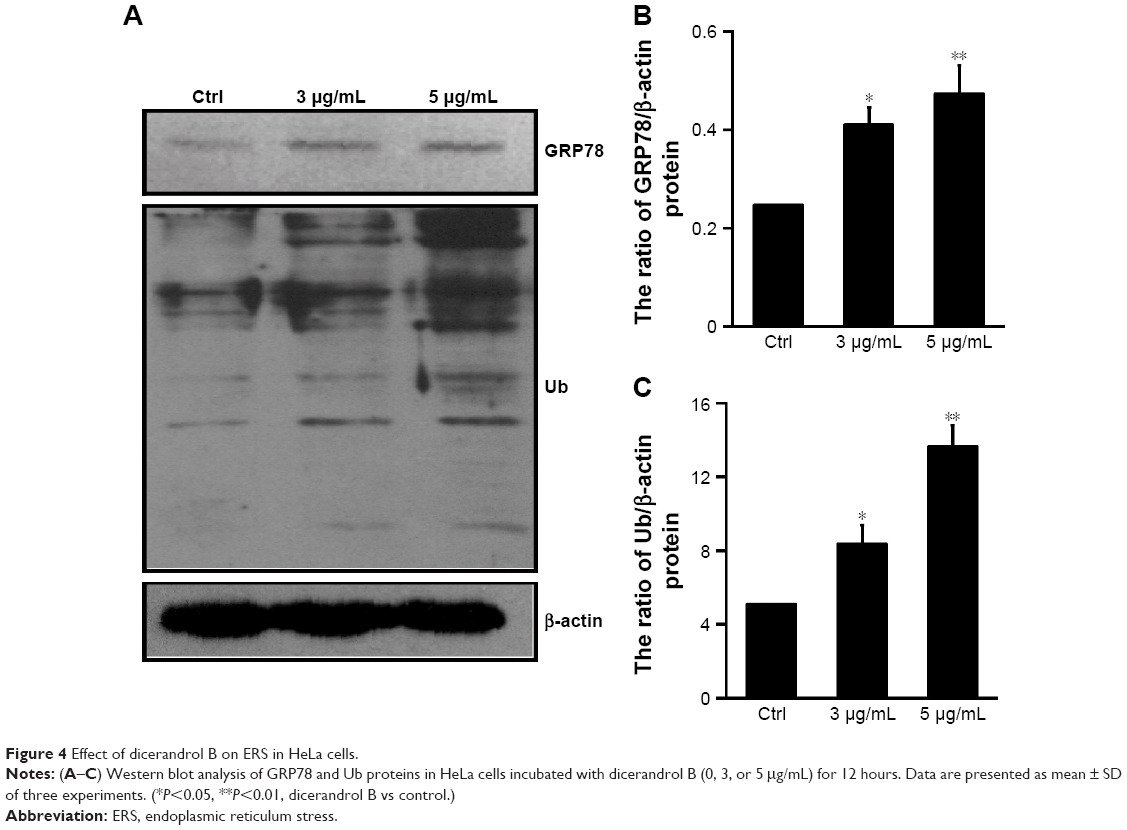 | Figure 4 Effect of dicerandrol B on ERS in HeLa cells. |
Effect of dicerandrol B on mitochondrial dysfunction of HeLa cells
The ROS-sensitive fluorescent probe DCFH-DA was used to monitor cellular oxidative stress in HeLa cells incubated with dicerandrol B (0, 3, or 5 μg/mL for 24 hours) in the presence or absence of 100 μmol/L NAC. Dicerandrol B considerably increased ROS production in HeLa cells, and NAC significantly attenuated the dicerandrol B-induced increase in ROS production (Figure 5A). Western blot analysis demonstrated that dicerandrol B significantly increased the Bax/Bcl-2 ratio (determinant of apoptotic potential in cells) in HeLa cells (Figure 5B and C). Real-time fluorescence quantitative PCR showed a significant dose-dependent increase in the expression of the Gpx gene, which encodes an antioxidant enzyme, in HeLa cells incubated with dicerandrol B compared to control (Figure 5D). These findings suggest that dicerandrol B induces ROS production in HeLa cells, disturbs the balance between cell death and cell growth, and promotes the production of Gpx to avoid the mitochondrial dysfunction caused by the ROS.
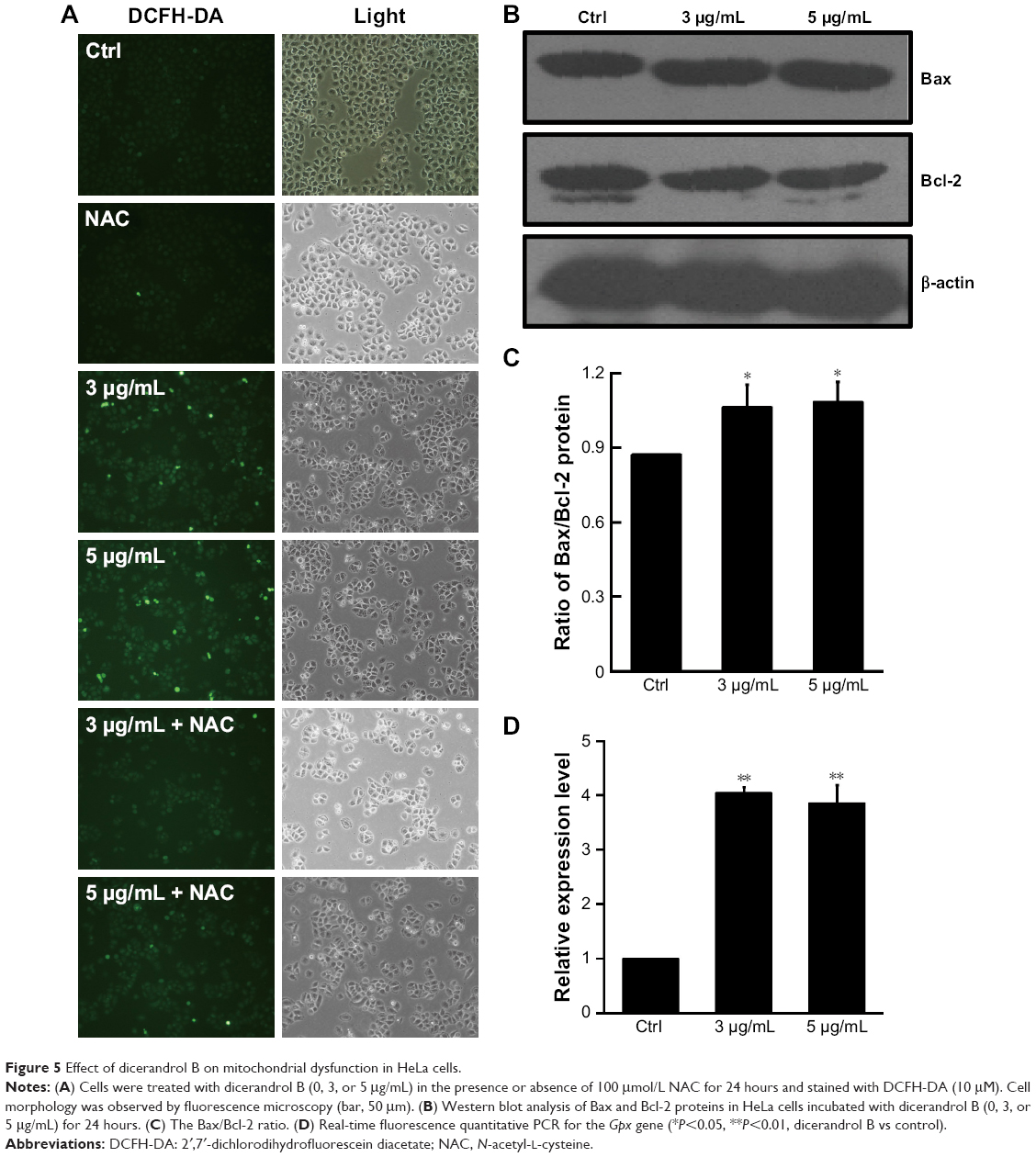 | Figure 5 Effect of dicerandrol B on mitochondrial dysfunction in HeLa cells. |
Discussion
Cancer remains a major global health problem. All available anticancer agents currently have disadvantages such as resistance and side effects.14 Therefore, it is necessary to introduce a new anticancer agent. The utilization of natural products and/or their novel structures, in order to discover and develop the final drug entity, is still alive and well.15 The xanthone dimer metabolite dicerandrol B, isolated from the endophytic fungus Phomopsis sp., displays marked cyctotoxic activity against some tumor cells, but less-marked cytotoxic effect on the immortalized human breast epithelial cell line.10 Dicerandrol B was found to be the most promising candidate for further investigations among the tested xanthones.16
This study investigated the effects of dicerandrol B on human cervical cancer HeLa cells. Dicerandrol B significantly inhibited HeLa cell viability, increased the number of HeLa cells in the G2/M phase of the cell cycle, and increased the expression MKi-67 and decreased the expression of UBE2S in a dose-dependent manner. MKi-67 and UBE2S are cell cycle-related genes. MKi-67 is a nuclear protein that is present during all cell cycle phases, including G1, S, G2, and mitosis; it is not expressed in resting cells.17 UBE2S promotes efficient anaphase-promoting complex (APC/C) substrate degradation, spindle assembly, and progression through mitosis.18,19 UBE2S depletion prolongs drug-induced mitotic arrest and suppresses mitotic slippage.20 Taken together, these data indicate that dicerandrol B induces G2/M arrest and apoptosis in HeLa cells.
Apoptosis is programmed cell death. Apoptosis is essential for controlling the number of cells in a multicellular organism.21 Modification of apoptotic pathways can lead to disease, including cancer.22 The three key apoptotic pathways include mitochondrial, ER, and death receptor-activated apoptotic pathways.11
The ER apoptotic pathway is initiated by ERS, which is caused by factors such as cytotoxicity and nutrient limitation and is characterized by the accumulation of unfolded or misfolded proteins.23 ERS activates the UPR to restore ER homeostasis. In mild ERS, the UPR triggers and promotes the ER-associated protein degradation pathway, which removes misfolded proteins and restores normal ER function. In prolonged or severe ERS, the UPR triggers ERS-induced apoptosis.24 Findings from the present study suggest that dicerandrol B caused ERS-mediated apoptosis in HeLa cells, evidenced by the accumulation of substantial amounts of ubiquitinated protein and changes in GRP78 expression levels.25,26 Ubiquitin is a key modulator of cell survival. The accumulation of ubiquitinated proteins in the cells after treatment with dicerandrol B suggests increase of the misfolded proteins in the ER, which can induce ERS-mediated apoptosis. GRP78 is a major ER chaperone that exerts antiapoptotic effects and regulates the ERS response.27,28
The mitochondrial pathway is another important apoptotic pathway. Mitochondria are the major ATP producer and a producer and target of ROS.28 ROS interact with Bcl-2 proteins to activate diverse redox-sensitive signaling cascades, including the mitochondrial intrinsic apoptotic cascade.29,30 Bcl-2 proteins are proapoptotic (Bax, Bak, Bad, Bim) or antiapoptotic (Bcl-2, Bcl-dicerandrol BL, Bcl-w).31,32 ROS activate and translocate Bax to the outer mitochondrial membrane, where it forms oligomers that alter the mitochondrial membrane permeability.29,31 This facilitates the release of apoptogenic factors such as cyt c from the mitochondrial inner membrane space and activates caspase-9 and caspase-3, which are crucial mediators of apoptosis.11 Findings from the present study suggest that dicerandrol B activates the mitochondrial apoptotic pathway in HeLa cells, evidenced by the dose-dependent increase in ROS production, Bax/Bcl-2 ratio (a key determinant of a cell’s apoptotic potential), and cleaved PARP expression.
In summary, findings from this study demonstrate that dicerandrol B isolated from a medicinal plant induces apoptosis in human HeLa cells possibly through the ERS and mitochondrial apoptotic pathways. This suggests that dicerandrol B possesses strong anticancer activity in cervical cancer, and provides insight into the mechanisms underlying dicerandrol B’s antitumor effects.
Acknowledgments
This study was funded by the National Major Increase or Decrease Project-Construction of the sustainable utilization capacity of famous traditional Chinese medicine resources (2060302), Natural Science Foundation of China (81672106, 31870332, 31470414), the 13th Five-year Science and Technology Project of Jilin Provincial Education Department (JJKH20180190KJ), and the Youth Foundation of Health and Family Planning Commission of Jilin Province (2017Q025).
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Zeng H, Zheng R, Guo Y, et al. Cancer survival in China, 2003–2005: a population-based study. Int J Cancer. 2015;136(8):1921–1930. | ||
China. NHaFPCotPsRo. Guidelines for the standardized diagnosis and treatment of cervical cancer and precancerous lesions. Chin Med J (Engl). 2013;5(8):40–49. | ||
Fu ZZ, Li K, Peng Y, et al. Efficacy and toxicity of different concurrent chemoradiotherapy regimens in the treatment of advanced cervical cancer: a network meta-analysis. Medicine. 2017;96(2):e5853. | ||
Yu ZY, Liang YG, Xiao H, et al. Melissoidesin G, a diterpenoid purified from Isodon melissoides, induces leukemic-cell apoptosis through induction of redox imbalance and exhibits synergy with other anticancer agents. Int J Cancer. 2007;121(9):2084–2094. | ||
Gao JM, Yang SX, Qin JC. Azaphilones: chemistry and biology. Chem Rev. 2013;113(7):4755–4811. | ||
Zhou L, Qin J, Ma L, et al. Rosoloactone: a natural diterpenoid inducing apoptosis in human cervical cancer cells through endoplasmic reticulum stress and mitochondrial damage. Biomed Pharmacother. 2017;95:355–362. | ||
Li X, Tian Y, Yang SX, Zhang YM, Qin JC, Yang SX. Cytotoxic azaphilone alkaloids from Chaetomium globosum Ty1. Bioorg Med Chem Lett. 2013;23(10):2945–2947. | ||
Wagenaar MM, Clardy J. Dicerandrols, new antibiotic and cytotoxic dimers produced by the fungus Phomopsis longicolla isolated from an endangered mint. J Nat Prod. 2001;64(8):1006–1009. | ||
Ding B, Yuan J, Huang X, et al. New dimeric members of the phomoxanthone family: phomolactonexanthones a, B and deacetylphomoxanthone C isolated from the fungus Phomopsis sp. Mar Drugs. 2013;11(12):4961–4972. | ||
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30(1):87. | ||
Oliver FJ, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase in the cellular response to DNA damage, apoptosis, and disease. Am J Hum Genet. 1999;64(5):1282–1288. | ||
Yadav RK, Chae SW, Kim HR, Chae HJ. Endoplasmic reticulum stress and cancer. J Cancer Prev. 2014;19(2):75–88. | ||
Hassan AHE, Choi E, Yoon YM, et al. Natural products hybrids: 3,5,4′-trimethoxystilbene–5,6,7-trimethoxyflavone chimeric analogs as potential cytotoxic agents against diverse human cancer cells. Eur J Med Chem. 2019;161:559–580. | ||
Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79(3):629–661. | ||
Cao S, Mcmillin DW, Tamayo G, Delmore J, Mitsiades CS, Clardy J. Inhibition of tumor cells interacting with stromal cells by xanthones isolated from a Costa Rican Penicillium sp. J Nat Prod. 2012;75(4):793–797. | ||
Martinez-Useros J, Garcia-Foncillas J. Can molecular biomarkers change the paradigm of pancreatic cancer prognosis? Biomed Res Int. 2016;2016(3):1–13. | ||
Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, Rape M. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci U S A. 2009;106(43):18213–18218. | ||
Ayesha AK, Hyodo T, Asano E, et al. UBE2S is associated with malignant characteristics of breast cancer cells. Tumour Biol. 2016;37(1):763–772. | ||
Garnett MJ, Mansfeld J, Godwin C, et al. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol. 2009;11(11):1363–1369. | ||
Alberts BJA, Lewis J. Molecular Biology of the Cell. 4th ed. New York: Garland Science Programmed Cell Death; 2002. | ||
Hassan M, Watari H, AbuAlmaaty A, Onba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014;2014:150845. | ||
Yuan S, Wu B, Yu Z, et al. The mitochondrial and endoplasmic reticulum pathways involved in the apoptosis of bursa of Fabricius cells in broilers exposed to dietary aflatoxin B1. Oncotarget. 2016;7(40):65295–65306. | ||
Sophonnithiprasert T, Mahabusarakam W, Nakamura Y, Watanapokasin R. Goniothalamin induces mitochondria-mediated apoptosis associated with endoplasmic reticulum stress-induced activation of JNK in HeLa cells. Oncol Lett. 2017;13(1):119–128. | ||
Zhao CQ, Zhang YH, Jiang SD, Jiang LS, Dai LY. Both endoplasmic reticulum and mitochondria are involved in disc cell apoptosis and intervertebral disc degeneration in rats. Age (Dordr). 2010;32(2):161–177. | ||
Ariyasu D, Yoshida H, Hasegawa Y. Endoplasmic reticulum (ER) stress and endocrine disorders. Int J Mol Sci. 2017;18(2):382. | ||
Broemer M, Meier P. Ubiquitin-mediated regulation of apoptosis. Trends Cell Biol. 2009;19(3):130–140. | ||
Dai C, Li J, Tang S, Li J, Xiao X. Colistin-induced nephrotoxicity in mice involves the mitochondrial, death receptor, and endoplasmic reticulum pathways. Antimicrob Agents Chemother. 2014;58(7):4075–4085. | ||
Khan M, Li T, Ahmad Khan MK, et al. Alantolactone induces apoptosis in HepG2 cells through GSH depletion, inhibition of STAT3 activation, and mitochondrial dysfunction. Biomed Res Int. 2013;2013(9):1–11. | ||
Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48(6):749–762. | ||
Ryter SW, Kim HP, Hoetzel A, et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9(1):49–89. | ||
Mohammad RM, Muqbil I, Lowe L, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35 Suppl:S78–S103. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.