")
Back to Journals » The Application of Clinical Genetics » Volume 8
Diagnostic and treatment strategies in mucopolysaccharidosis VI
Authors Vairo F, Federhen A, Baldo G, Riegel M, Burin M, Leistner-Segal S, Giugliani R
Received 4 July 2015
Accepted for publication 15 September 2015
Published 30 October 2015 Volume 2015:8 Pages 245—255
DOI https://doi.org/10.2147/TACG.S68650
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Filippo Vairo,1–3 Andressa Federhen,1,3,4 Guilherme Baldo,1,2,5–7 Mariluce Riegel,1,6 Maira Burin,1 Sandra Leistner-Segal,1,8 Roberto Giugliani1,5,6,8
1Medical Genetics Service, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil; 2Department of Genetics, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil; 3Clinical Research Group on Medical Genetics, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil; 4Post-Graduate Program in Child and Adolescent Health, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil; 5Gene Therapy Center, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil; 6Post-Graduate Program in Genetics and Molecular Biology, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil; 7Department of Physiology, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil; 8Post-Graduate Program in Medical Sciences, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
Abstract: Mucopolysaccharidosis VI (MPS VI) is a very rare autosomal recessive disorder caused by mutations in the ARSB gene, which lead to deficient activity of the lysosomal enzyme ASB. This enzyme is important for the breakdown of the glycosaminoglycans (GAGs) dermatan sulfate and chondroitin sulfate, which accumulate in body tissues and organs of MPS VI patients. The storage of GAGs (especially dermatan sulfate) causes bone dysplasia, joint restriction, organomegaly, heart disease, and corneal clouding, among several other problems, and reduced life span. Despite the fact that most cases are severe, there is a spectrum of severity and some cases are so attenuated that diagnosis is made late in life. Although the analysis of urinary GAGs and/or the measurement of enzyme activity in dried blood spots are useful screening methods, the diagnosis is based in the demonstration of the enzyme deficiency in leucocytes or fibroblasts, and/or in the identification of pathogenic mutations in the ARSB gene. Specific treatment with enzyme replacement has been available since 2005. It is safe and effective, bringing measurable benefits and increased survival to patients. As several evidences indicate that early initiation of therapy may lead to a better outcome, newborn screening is being considered for this condition, and it is already in place in selected areas where the incidence of MPS VI is increased. However, as enzyme replacement therapy is not curative, associated therapies should be considered, and research on innovative therapies continues. The management of affected patients by a multidisciplinary team with experience in MPS diseases is highly recommended.
Keywords: mucopolysaccharidosis VI, Maroteaux–Lamy syndrome, dermatan sulfate, arylsulfatase b, enzyme replacement therapy, lysosomal storage diseases
Introduction
Mucopolysaccharidosis VI (MPS VI), or Maroteaux–Lamy syndrome (OMIM 253200) is a rare genetic disease first described in 1963 by the French doctors Pierre Maroteaux and Maurice Lamy.1 The disease is inherited as an autosomal recessive trait and is caused by mutations in the ARSB gene, that encodes the lysosomal enzyme E.C.3.1.6.12 or ASB.2
The enzyme deficiency leads to the accumulation of the glycosaminoglycan (GAG) dermatan sulfate (DS) into the lysosome, leading to a variety of signs and symptoms in multiple organs and systems.3 The disorder is clinically heterogeneous. The majority of patients are severely affected (rapidly advancing form). In a minority of patients the disease is more attenuated (slowly-progressing form).4 International studies report that the incidence of MPS VI ranges from one in 43,261 births in a Turkish population in Germany to one in 1,505,160 births in Sweden.5 Table 1 shows the epidemiological studies performed in different countries to date.4,6–17 Some populations seem to have increased frequencies of mutations in the gene ARSB. The prevalence identified in a Turkish population living in Germany, for example, was higher than the non-Turkish population.10 It has also been reported that in Brazil,18 and in Portugal,9 from which many people migrated to Brazil, the frequency of MPS VI is proportionally increased considering the usual distribution of MPS types. In Brazil, for example, in the city of Monte Santo, state of Bahia, the prevalence of MPS VI is estimated as 1:5,000, probably due to a combination of founder effect and endogamy, with a single mutation (p.H178L) present in homozygosis in all cases. A pilot program of newborn screening for MPS VI was implemented in this region, based on measurement of the enzyme activity and the detection of the common mutation.4
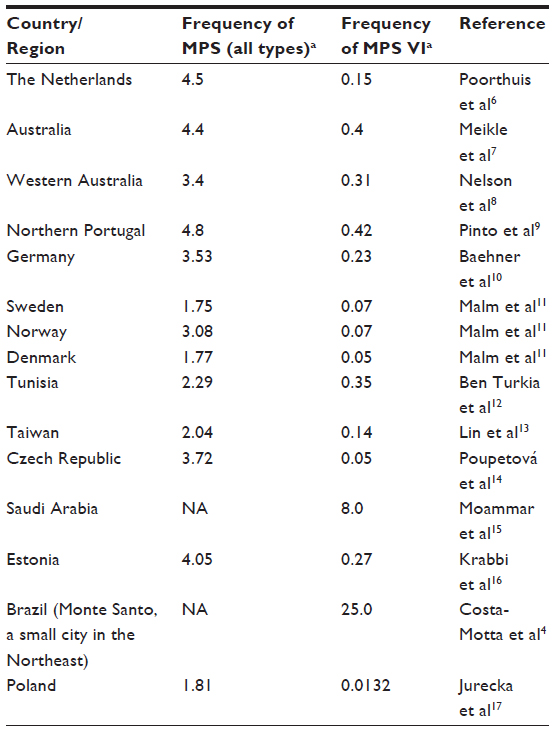 | Table 1 Frequency of MPS (all types) and MPS VI reported in the literature |
Clinical features
The clinical presentation of MPS VI is quite variable regarding the age of onset of symptoms and the disease progression.
The severe form of MPS VI, or rapidly progressive, is characterized by the onset of symptoms between 2 and 3 years of age, restricted joint mobility in infancy, delayed puberty, cervical spine compression, and respiratory failure. Most patients die of cardiac decompensation in the second or third decade of life.2 Although they have no cognitive deficit related directly to the disease, patients may present difficulties due to hearing impairment, visual, and physical limitations. Physical characteristics include joint restriction, pectus carinatum, kyphoscoliosis, macrocephaly, hepatosplenomegaly, umbilical and inguinal hernia, beyond the typical facial features such as low nasal bridge, hirsutism, gingival hypertrophy, dental malocclusion, macroglossia, and corneal opacification (which can lead to glaucoma and papilledema)19,20 (Figure 1). Patients may progress to respiratory failure, heart disease, severe joint involvement, and spinal cord compression (SCC) requiring surgical intervention.3,21 Cholestasis is rare, but has been reported.22
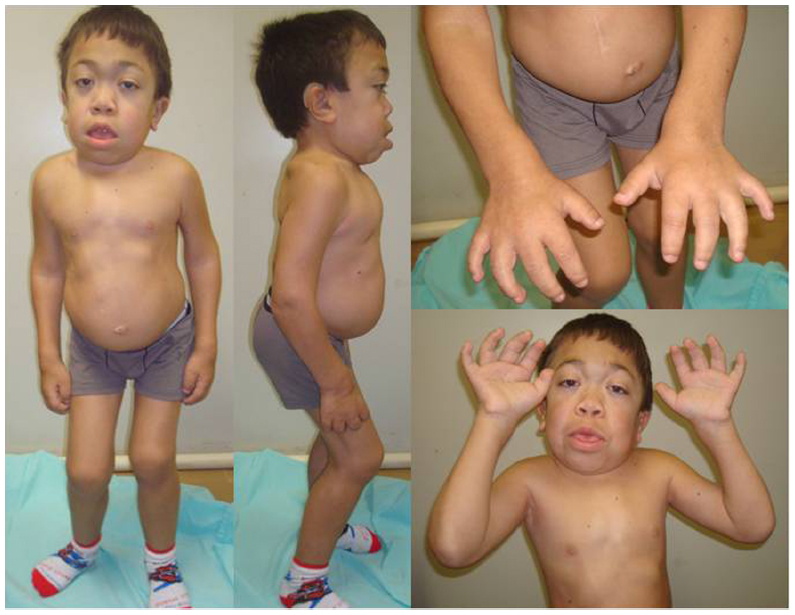 | Figure 1 Male patient (14 years old) with the rapidly advancing form of MPS VI. |
The mild form, or slowly progressive, is characterized by attenuated symptoms, due to lower levels of DS.23 The patients can develop skeletal complications such as short stature, carpal tunnel syndrome, Dupuytren’s contracture, and hip dysplasia. In addition, adult patients may have valve disease, obstructive sleep apnea, and pulmonary complications24 (Figure 2).
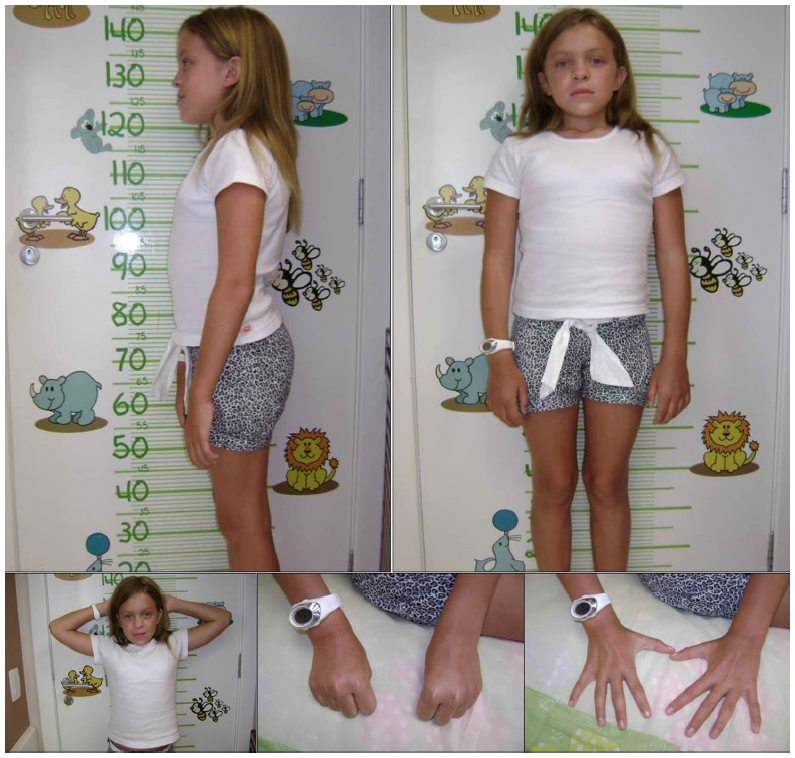 | Figure 2 Female patient (11 years old) with the slowly advancing form of MPS VI. |
Unfortunately, delays to or missed diagnosis are common especially related to patients with mild form since they may present without a typical coarse facies and with less pronounced skeletal disease.
It is believed that the greater the residual activity of ASB, the more attenuated the symptoms25 but this correlation was not confirmed by all groups.26 The correlation with urinary excretion of GAGs is another factor to be considered for the prognosis. A study with 121 patients with untreated MPS VI was performed and a classification into two groups was suggested. Patients with urinary GAG excretion greater than 200 ug/mg creatinine had more severe disease characterized by short stature, low weight, greater difficulty in performing the 6-minute walk test, greater impairment of lung function, and joint restriction. They were characterized as patients with the rapidly progressive form. Patients over 20 years of age rarely showed GAG excretion greater than 100 ug/mg creatinine, suggesting that levels below 100 ug/mg creatinine predict a greater survival.
They were classified as slowly progressive patients. These cases tend to have higher height and levels of GAGs below 100 ug/mg creatinine.23 Although this classification is widespread and frequently used, there are several descriptions of patients with intermediate phenotypes.27–29 In general, MPS VI patients have a chronic progressive course and multisystem especially the musculoskeletal system, cardiopulmonary system, cornea, skin, liver, spleen, brain, and meninges.3
Biochemical diagnosis
In general, the first step in diagnosis of MPS is the assessment of urinary GAGs excretion. GAGs are unbranched polysaccharide chains containing repeated disaccharides with uronic acid, and galactose or hexosamine. Except for hyaluronic acid, sulfate ions are also associated with these substances. Chondroitin sulfate (CS), heparan sulfate (HS), DS, and keratan sulfate are the main GAGs present in body tissues. ASB deficiency in MPS VI causes an important accumulation in tissues of DS and CS, and these compounds are excreted in high concentrations in the urine.2
Using only qualitative screening tests (such as Berry spot test) is not recommended anymore (due to the high proportion of false results) and was replaced by more refined and quantitative colorimetric methods.30 The urinary GAGs may be analyzed quantitatively (to measure the total amount of GAGs) and/or qualitatively (to identify the specific GAG species which are elevated). Performing both analyses simultaneously is strongly recommended, as it reduces the risk of missing an MPS diagnosis. Since quantitative analysis is non-specific, qualitative urinary GAGs analysis is needed to identify the exact GAG types which are elevated, guiding the subsequent investigation needed to identify the specific MPS type. It is important to note that urinary GAGs analyses, quantitative or qualitative, are screening methods and its results should never be considered diagnostic for a specific MPS type. Quantitative GAGs analysis is also important because it allows monitoring a patient’s response to specific treatment.31–33
Quantitative analysis is usually performed by colorimetric analysis of urine combined with a cationic blue dye. The most used is the dimethylmethylene blue based method. Urinary GAGs concentration varies with age, and newborns have higher excretion than adults, therefore there are different reference ranges according to the age of the patients.34
To identify the distinct GAGs excreted in urine, a qualitative pattern can be obtained by thin-layer chromatography (TLC) and/or electrophoresis (ELP). TLC and ELP are qualitative techniques, and a careful interpretation of the results by an experienced technician is required. Generally, these methods provide good results, especially when ELP is used. However, some ambiguous results or even false positives may also occur due to subjective interpretation of the different patterns.32
In MPS VI, the GAGs which are increased are DS and CS. However, DS is also excreted in excess in MPS I, II, and VII, in conjunction with HS (and also with CS, in the case of MPS VII) (Figure 3). Although differentiation of MPS VI from the other MPS disorders causing excess DS excretion seems straightforward, there are several complicating factors. It is not uncommon to observe a strong signal of DS with a faint signal of HS in patients with MPS I or II. Therefore, even if DS is detected, other MPS types should not be immediately ruled out. Conversely, it has been noted that trace amounts of HS may be present in urine of patients with MPS VI or even in the urine of young children (<3 years of age). It is not possible to firmly differentiate MPS I, II, VI, and VII using qualitative urinary GAGs analysis.31 Advantages of urinary GAGs analysis include non-invasive sample collection, relative ease of performance, and availability at many laboratories, including non-specialized ones. Urine collected in the morning is preferred, as it is usually more concentrated, but not mandatory. However, diluted samples should be avoided, as it can lead to false or ambiguous results. In normal neonates the concentration of GAGs may be increased, hindering the detection of MPS patients.31 In an attempt to overcome the difficulties of transportation of liquid urine samples, a new method which uses dried-urine filter paper samples for reliable mucopolysacchariduria detection has been described.35
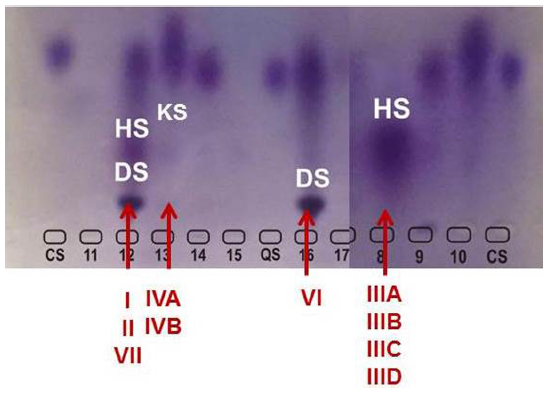 | Figure 3 Electrophoresis of urinary GAGs. |
Quantitative urinary GAGs excretion can be in normal range in some MPS patients, especially in adults with MPS III and MPS IV. So, a negative result in the assay of urinary GAGs should be considered with caution, and the investigation should be continued if clinical suspicion of MPS persists.31,32,36
It is expected that the measurement of GAGs by tandem mass spectrometry would progressively replace colorimetric quantification and TLC/ELP qualitative evaluations,37 but technology may take some time to be incorporated in the routine evaluation due to the high cost of equipment.
A positive result in the evaluation of urinary GAGs should be followed by more specific methods until a conclusion is reached. The usual following step is the measurement of ASB enzyme activity to demonstrate its deficiency and complete the biochemical diagnosis. This can be done using different types of samples, such as dried blood spots (DBS), leukocytes (including lymphocytes), or fibroblasts. Leukocytes (isolated from whole blood) and fibroblasts are considered as the gold standard samples for the measurement of enzyme activity. The enzyme analysis using DBS is a good option, particularly for regions of the world where collecting and/or shipping whole blood is not practical, but should be considered as a screening method. For ASB analysis the activity can be measured using the natural substrate, but in routine practice artificial substrates are used, such as the chromogenic p-nitrocatechol sulfate, or the fluorogenic 4-methylumbelliferyl sulfate. It is always recommended to assay another lysosomal enzyme in the same sample used for ASB analysis to assess the quality of the sample. If the reference enzyme is normal and ASB activity is low, another sulfatase (eg, iduronate-2-sulfatase) should be measured to rule out multiple sulfatase deficiency, a different lysosomal disease in which low levels of all sulfatases are found, including ASB.
It should be pointed out that no clear correlation could be drawn from the level of residual enzyme activity and clinical phenotype, making the prediction of clinical course difficult to be estimated on the basis of the enzyme assay. Although the biochemical tests (urinary GAGs analyses and enzyme assays) are sufficient to confirm the diagnosis of MPS VI, whenever possible the genetic analysis of the ARSB gene should be performed for the characterization of the molecular defect.32
For the biochemical prenatal diagnosis of MPS VI the sample of choice is amniocytes, because there is the presence of high arylsulfatase C activity in chorionic villi which could interfere with the result if this material is used.31
Mutation identification
The gene responsible for MPS VI (ARSB) was identified and its sequence cloned and described in 1990.38 It is mapped at chromosome 5q13-14 and has 2.8 kb organized in eight exons interrupted by seven introns.39 As genetic abnormalities began to be described,25 there was a growing attempt to correlate the genotypes with phenotypes, which has been disappointing most of the time, as a consequence of the broad molecular heterogeneity present in this gene, including disease causing mutations and several polymorphisms.
To date, more than 164 mutations have been described in the ASB gene, most of them being point mutations.40 Also, several polymorphisms have been identified and expression studies have shown than one of them, p.V358M, could reduce the residual activity of ARSB, depending on the concomitant disease causing mutation present. Most mutations are private, and those patients presenting both alleles with mutation predicting synthesis of a non-functional protein such as nonsense, small insertions and/or deletions tend to show a more severe phenotype.25
Generally, protocols for ARSB molecular analysis starts with genomic DNA extraction from several sources such as total blood, leucocytes, saliva, DBS, and others, followed by polymerase chain reaction (PCR) amplification41 of the coding region of the gene, including exons 1–8 and adjacent intronic regions. The amplified product is then analyzed by Sanger sequencing42 in order to identify the alteration which leads to the enzyme defect. When the mutation is identified, it is prudent to confirm it through cDNA analysis obtained from mRNA. This is particularly interesting for mutations within the intron/exon boundaries which can cause the use of alternative splicing sites and consequently deletion or insertion of several nucleotides within the coding sequence.
There is a group of mutations that can be present in more than one unrelated patient (Table 2).40 For those, it is possible to apply a strategy for detection of known recurrent mutations. Nevertheless, this procedure depends on the population studied, since the mutation profile can vary in distinct ethnic groups. A common mutation was described (p.H178L) in a small community in the Northeast of Brazil which was present in homozygosis in all patients tested. In this same sample a haplotype analysis was performed using intragenic polymorphisms and the same haplotype was identified amongst the individuals tested indicating a founder effect.43
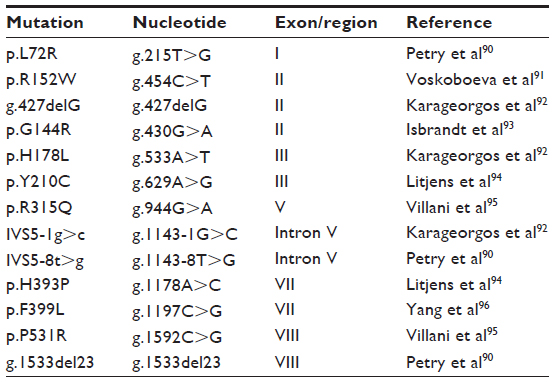 | Table 2 Recurrent mutations observed in specific populations worldwide |
The observation of a high carrier frequency for the p.R152W mutation in the Lithuanian population may indicate a possible founder effect in this region.44 Although its high prevalence in a series consisting of patients from Russia, Kazakhstan, and Central and Eastern Europe suggests that p.R152W may be of Slavic, not Lithuanian origin.45
After the identification of both mutations in the index case, they can be tested in the parents to confirm inheritance and rule out the presence of two “in cis” mutations, as has already been reported for other MPS.46 It is also possible to analyze its presence in other family members for identification of carriers or other affected individuals, using several methodologies according to the lab expertise and equipment available. Simple methods such as restriction fragment length polymorphisms can be used for mutations which lay within an enzyme restriction site which allows the digestion of the PCR product with specific restriction enzymes. This digested product can then be analyzed by ELP in agarose gel and visualization under ultra violet light using double strand DNA intercalants such as ethidium bromide, SYBR Safe, and gel red.
When the mutation found is not within an enzyme restriction site, it is possible to use a technique based on hybridization with a fluorescent labeled probe which is either complementary to the normal or to the mutated sequence. The presence of a positive reaction with one or both probes indicates the homozygous or heterozygous genotype. This method is known as real time PCR and can be very useful for identification of recurrent and frequent mutations in a given community,4 including neonatal screening as it allows for a throughput analysis with quick and sensible results.
Nowadays, modern techniques allow for the identification of a broad number of sequencing reactions in a single assay, allowing for a complete analysis of the whole gene (or even several genes). This technology is based on next generation sequencing (NGS) and can be used as panels of certain genes or even whole exome and genome sequencing and it is becoming an extremely advantageous technique due to its quickness and robustness. Although costs for this kind of analysis are still high, there is a decreasing cost trend as already shown with other novel technologies. NGS can even be useful for diagnosis of patients without a definitive biochemical diagnosis.
Nevertheless, even promising technologies have some pitfalls. For a specific group of mutations such as complete gene deletions including contiguous genes and gene inversions/rearrangements, this strategy would fail to identify the disease causing mutations. In the first example, sequencing or even NGS would not allow for deletion breakpoints mapping and other techniques such as comparative genomic hybridization (CGH) array would be necessary as ancillary techniques.47
If the molecular defect is known in the family, prenatal diagnosis is possible through mutation analysis using several sources of biological material such as chorionic villi, amniocytes, or umbilical cord blood and can be performed concomitantly with the enzyme assay or independently.
DNA sequencing is the primary clinical technique for identifying mutations in human disease, but sequencing often does not detect intragenic or whole-gene deletions/duplications. CGH using oligonucleotide arrays has been implemented in cytogenetic and molecular diagnostic laboratories as a robust, rapid, and sensitive assay for detecting targeted gene deletions.47 Targeted oligonucleotides array can be designed to detect both whole-gene deletions and small intragenic deletions in metabolic disorders and lysosomal storage genes.48,49 This technology may be particularly useful as complementary diagnostic test to the gene sequencing analyses in groups where intragenic deletions, duplications, and rearrangements are causative. Furthermore, genomic positions of the breakpoints cannot be determined by traditional methods. In contrast, the target CGH array is a rapid, highly sensitive, and accurate method that can be useful for detecting single- and multi-exon deletions and duplications in a large set of metabolic and lysosomal disorder genes simultaneously.49 As gene CGH array is a recent technology being used in the metabolic and lysosomal disorder diagnostics and not yet a standard test, the intragenic deletions/duplications detected need to be confirmed. Methods such as breakpoints PCR, Southern blot, real time PCR, or confirmation with dye swap array CGH can be considered to determine the exact breakpoints.
Treatment
Until recently, support therapy and bone marrow transplantation therapies were the only options available for MPS VI. Supportive therapy was focused on nutrition, occupational and physiotherapy, in addition to symptomatic treatment of complications such as the use of oxygen with positive pressure during sleep, tonsillectomy, adenoidectomy, tracheostomy, medical or surgical treatment for heart failure, cervical decompression, corneal transplantation, and ventricular shunt, among others.3
Hematopoietic stem cell transplantation
Bone marrow or hematopoietic cell transplantation has been used for some patients since 198450 with reports of favorable outcome several years after transplantation with.51,52 The rationale is that the hematopoietic system of the receptor is reconstituted with stem cells from an immune-compatible healthy donor, in order to establish a long-term enzyme source.53,54 Although there is an increase in the enzyme ASB and decrease of GAG excretion, skeletal changes already established do not improve, even in cases of successful transplantation. The use of this form of treatment is limited by the high rate of mortality and morbidity related to the procedure, such as graft versus host disease and because of the difficulty of finding donors.55
Enzyme replacement therapy (ERT)
ERT is a specific form of therapy for MPS VI and is safer than hematopoietic cell transplantation. ERT is carried out by a recombinant form of ASB (rhASB) synthesized by genetic engineering from ovarian cells of the Chinese hamster by BioMarin Pharmaceutical Laboratory, Novato, CA, USA.56,57 Galsulfase (Naglazyme®) is administered intravenously in order to provide an active form of the enzyme that lacks or is insufficient in patients with MPS VI. In cats treated from birth, the enzyme prevented or slowed the progression of the skeletal related disease57,58 but was ineffective regarding the GAG accumulation in corneal keratocytes and cartilage.58,59 Some studies reported immune response to the recombinant enzyme, which led to adverse effects such as hypersensitivity reactions and anaphylaxis. The antibody response can inactivate or degrade the enzyme, affecting its efficacy.60,61
A Phase III study was conducted using a weekly dose of 1 mg/kg in 39 patients, for 24 weeks, confirming the Phase II study results, with improvement in overall resistance measured by the 12-minute walk test and stair climbing, reduced GAG excretion in the urine, and good tolerability.62,63 Of the 54 patients who participated in clinical studies to its end, 53 developed specific antibodies to galsulfase without a direct correlation with the excretion of GAGs in the urine.3 A Phase IV study was performed using two different doses, 1 mg/kg and 2 mg/kg, weekly, in patients with MPS VI between 3.3 and 12.7 months, for at least 52 weeks. From this study it was demonstrated that the medication was well tolerated at both doses and no serious adverse events were related to medication. The main measure of improvement was a reduction of approximately 70% of urinary GAG excretion.64 Harmatz et al65 conducted an observational study with 56 patients receiving ERT during Phase I/II, Phase II, Phase III, and extension clinical trials, and reported that the improvement in lung function is a factor that may be contributing to the improved resistance observed in these patients and it was not related to age. Decker et al66 demonstrated that the growth rate in the prepubertal period was higher in patients receiving long-term ERT and that the increase was even greater in patients under the age of 16 years.
Recently, a study with 34 MPS VI patients under 5 years of age showed that ERT with galsulfase is safe and effective in this population67 and has a similar profile to that reported in clinical trials with older patients.62,68 The results of this study also showed a significant negative correlation between the ERT and levels of urinary GAGs. In addition, patients maintained or increased the growth rate after treatment, and from the nine patients who underwent sleep studies at the beginning of this study, one showed improvement in sleep apnea after 56 months of ERT.67
Clinical trials have shown that ERT with galsulfase has been well tolerated by patients with MPS VI.69 However, there are a few reports of adverse reactions.70,71
Di Natale et al72 conducted a study with four patients with the severe form of MPS VI who were on ERT with galsulfase to analyze genes that could be potential biomarkers of response to therapy. The results of this study showed that the expression of TNF-α, a cytokine produced by cells such as macrophages, chondrocytes, and synoviocytes significantly reduced after treatment, suggesting TNF-α as a potential biomarker of response to treatment.
A major complication: SCC
SCC is a frequent complication of MPS VI and may lead to important morbidity. Surgery for spinal decompression usually results in a significant neurological improvement73,74 and must be performed in experienced and specialized MPS treatment centers. ERT cannot cross the blood–brain barrier and has no access to the central nervous system including spinal cord by intravenous infusion. Horovitz et al75 described six MPS VI children with SCC who presented increased joint mobility after ERT. As ERT increases neck mobility and reduces stiffness by GAG clearance from paravertebral structures, patients can experience an increased range of vertebral motion. In MPS VI animal models, intrathecal (IT) ERT prevents and removes dural storage76 with no adverse reactions. Furthermore, a pioneer report of IT ERT to treat SCC in a single case of MPS VI has been described in the literature.77 The patient had pachymeningitis cervicalis hypertrophica and received IT infusions of rhASB administered monthly by lumbar puncture. Despite significant urodynamic and neurological improvement, the patient had worsened walking capacity. The authors concluded that IT ERT can be used as an alternative approach for MPS VI patients with pachymeningitis cervicalis hypertrophica, as long as neck stability is previously obtained, but further studies are ongoing to support these findings.
Perspectives
Based on the limitations observed with the current available treatments, new approaches have been tested for MPS VI. In that scenario, mainly the rat and cat animal models have been particularly useful for studies of disease pathogenesis and effectiveness of new treatments.78,79
Using MPS VI rats, and based on previous studies in other animal models that suggested an important inflammatory component on the pathogenesis of this disease,80 Eliyahu et al81 demonstrated that, when an anti-TNF-α drug is used in combination with ERT, benefits in organs such as the trachea and even the bones can be achieved. Further studies using pentosan polysulfate, a US Food and Drug Administration approved medication with anti-inflammatory effects have shown reduction in eye and nasal secretions and improvement in bone and tracheal deformities as well.82 Currently, Phase I clinical trials are under development, and anti-inflammatory therapy for MPS VI seems to be a promising approach at least to alleviate some of the disease symptoms.
Gene therapy has also been tested in animal models of MPS VI. Different types of viral vectors have shown promising results in delivering a correct version of the ARSB gene and achieving high levels of enzyme activity. A study with an adeno-associated virus 2/8 suggests that this approach could be used as good therapy for MPS VI. A study in cats has shown improvement in visceral organs, joint mobility, skeletal abnormalities, and survival.83 A second study used a retroviral vector in neonatal MPS VI cats. This approach was able to produce sustained high ASB serum levels for 8 years, with improvements in visceral organs, increased bone length as well as improvements in joints, heart valves, and aorta, which are known to be organs not easily corrected by ERT.84 Major problems with gene therapy approaches are still the pre- existing immunity against adeno-associated viruses and the risk of insertional oncogenesis.83,84
One point that several reports seem to agree on is that treatment is more effective when started earlier in life.85 Newborn screening may play a role in the early detection of cases, with high-throughput methods based on tandem mass spectrometry already in development.86 While no formal massive screening for MPS VI is in place so far, an interesting approach is being used in an area with high incidence of MPS VI. In this selected area, a community-based neonatal screening will enable early diagnosis and more effective treatments. Also, it opens up the possibility of more effective genetic counseling and development of comprehensive prevention programs for these families.4,87
Final remarks
Ten years after a specific ERT for MPS VI was approved, it still remains a challenging disease. Despite the clear benefits of ERT in terms of reduced morbidity and even mortality88 (Figure 4), the affected patients still present significant disease manifestations. Early start of treatment, as already elegantly demonstrated in a sibling study89 should bring additional benefits. Present methods for biochemical and genetic diagnosis are accurate, but depend on clinical suspicion, which could be delayed. The detection of affected babies by newborn screening is being considered as a potential diagnostic tool, and pilot screening programs are already in place in selected areas. Additional treatment options (such as pentosan polysulfate) to be used jointly with ERT, or gene therapy, in combination with early diagnosis, will probably positively change the course of the disease in affected patients.
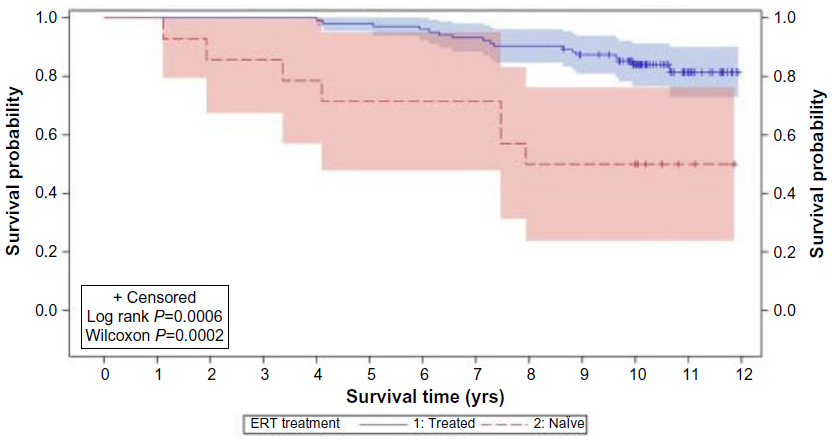 | Figure 4 Product-limit survival estimates of MPS VI patients who received ERT and who were treatment naïve. |
Acknowledgments
We thank Dr Carolina Souza for providing the pictures of the MPS VI patients. Legal guardians gave the consent to these pictures being published.
Disclosure
The authors have no conflicts of interest to disclose.
References
Maroteaux P, Leveque B, Marie J, Lamy M. Une nouvelle dysostose avec elimination urinaire de chondroitine sulfate B [A new dysostosis with urinary elimination of chondroitin sulfate B]. Presse Med. 1963;71:1849–1852. French. | |
Neufeld E, Muenzer J. The mucopolysaccharidosis. In: Scriver C, Beandet A, Sly S, et al, editors. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 2001:3421–3452. | |
Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120(2):405–418. | |
Costa-Motta FM, Bender F, Acosta A, et al. A community-based study of mucopolysaccharidosis type VI in Brazil: the influence of founder effect, endogamy and consanguinity. Hum Hered. 2014;77(1–4):189–196. | |
Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. | |
Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105(1–2):151–156. | |
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249–254. | |
Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A. 2003;123A(3):310–313. | |
Pinto R, Caseiro C, Lemos M, et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. 2004;12(2):87–92. | |
Baehner F, Schmiedeskamp C, Krummenauer F, et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28(6):1011–1017. | |
Malm G, Lund AM, Mansson JE, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta Paediatr. 2008;97(11):1577–1581. | |
Ben Turkia H, Tebib N, Azzouz H, et al. Incidence of mucopolysaccharidoses in Tunisia. Tunis Med. 2009;87(11):782–785. | |
Lin HY, Lin SP, Chuang CK, et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984–2004. Am J Med Genet A. 2009;149A(5):960–964. | |
Poupetová H, Ledvinová J, Berná L, Dvoráková L, Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis. 2010;33(4):387–396. | |
Moammar H, Cheriyan G, Mathew R, Al-Sannaa N. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Ann Saudi Med. 2010;30(4):271–277. | |
Krabbi K, Joost K, Zordania R, et al. The live-birth prevalence of mucopolysaccharidoses in Estonia. Genet Test Mol Biomarkers. 2012; 16(8):846–849. | |
Jurecka A, Ługowska A, Golda A, Czartoryska B, Tylki-Szymanska A. Prevalence rates of mucopolysaccharidoses in Poland. J Appl Genet. 2015;56(2):205–210. | |
Coelho JC, Wajner M, Burin MG, Vargas CR, Giugliani R. Selective screening of 10,000 high-risk Brazilian patients for the detection of inborn errors of metabolism. Eur J Pediatr. 1997;156(8):650–654. | |
Koseoglu ST, Harmatz P, Turbeville S, Nicely H. Reversed papilledema in an MPS VI patient with galsulfase (Naglazyme) therapy. Int Ophthalmol. 2009;29(4):267–269. | |
Gomes BD, Souza Gda S, Viana GM, et al. Visual Dysfunction of Type I and VI Mucopolysaccharidosis Patients Evaluated with Visual Evoked Cortical Potential. Case Rep Ophthalmol. 2012;3(1):104–112. | |
Cardoso-Santos A, Azevedo AC, Fagondes S, Burin MG, Giugliani R, Schwartz IV. Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome): assessment of joint mobility and grip and pinch strength. J Pediatr (Rio J). 2008;84(2):130–135. | |
Arslan N, Mavi A, Kalkan S, et al. Findings of hepatobiliary scintigraphy and liver biopsy in Maroteaux-Lamy syndrome presenting as neonatal cholestasis. Pediatr Int. 2006;48(5):498–500. | |
Swiedler SJ, Beck M, Bajbouj M, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). Am J Med Genet A. 2005;134A(2):144–150. | |
Thumler A, Miebach E, Lampe C, et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis. 2012;35(6):1071–1079. | |
Litjens T, Hopwood JJ. Mucopolysaccharidosis type VI: Structural and clinical implications of mutations in N-acetylgalactosamine-4-sulfatase. Hum Mutat. 2001;18(4):282–295. | |
Azevedo AC, Schwartz IV, Kalakun L, et al. Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin Genet. 2004;66(3):208–213. | |
Pilz H, von Figura K, Goebel HH. Deficiency of arylsulfatase B in 2 brothers aged 40 and 38 years (Maroteaux-Lamy syndrome, type B). Ann Neurol. 1979;6(4):315–325. | |
Paterson DE, Harper G, Weston HJ, Mattingley J. Maroteaux-Lamy syndrome, mild form – MPS vib. Br J Radiol. 1982;55(659):805–812. | |
Wicker G, Prill V, Brooks D, et al. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). An intermediate clinical phenotype caused by substitution of valine for glycine at position 137 of arylsulfatase B. J Biol Chem. 1991;266(32):21386–21391. | |
de Jong JG, Wevers RA, Liebrand-van Sambeek R. Measuring urinary glycosaminoglycans in the presence of protein: an improved screening procedure for mucopolysaccharidoses based on dimethylmethylene blue. Clin Chem. 1992;38(6):803–807. | |
Wood T, Bodamer OA, Burin MG, et al. Expert recommendations for the laboratory diagnosis of MPS VI. Mol Genet Metab. 2012;106(1):73–82. | |
Giugliani R, Brusius-Facchin A, Souza C, et al. Diagnosis and therapy options in mucopolysaccharidosis II (Hunter syndrome). Expert Opinion on Orphan Drugs. 2015;3(2):10. | |
Kircher S, Bajbouj M, Beck M. Mucopolysaccharidoses: A Guide for Physicians and Parents (Uni-Med Science). UNI-MED Verlag AG; 2007. | |
Whitley CB, Draper KA, Dutton CM, Brown PA, Severson SL, France LA. Diagnostic test for mucopolysaccharidosis. II. Rapid quantification of glycosaminoglycan in urine samples collected on a paper matrix. Clin Chem. 1989;35(10):2074–2081. | |
Civallero G, Bender F, Gomes A, et al. Reliable detection of mucopolysacchariduria in dried-urine filter paper samples. Clin Chim Acta. 2013;415:334–336. | |
Lampe C, Bellettato CM, Karabul N, Scarpa M. Mucopolysaccharidoses and other lysosomal storage diseases. Rheum Dis Clin North Am. 2013; 39(2):431–455. | |
Auray-Blais C, Bherer P, Gagnon R, et al. Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I, II and VI. Mol Genet Metab. 2011;102(1):49–56. | |
Peters C, Schmidt B, Rommerskirch W, et al. Phylogenetic conservation of arylsulfatases. cDNA cloning and expression of human arylsulfatase B. J Biol Chem. 1990;265(6):3374–3381. | |
Modaressi S, Rupp K, von Figura K, Peters C. Structure of the human arylsulfatase B gene. Biol Chem Hoppe Seyler. 1993;374(5):327–335. | |
hgmd.cf.ac.uk [homepage on the Internet]. The Human Gene Mutation Database. Available from: http://www.hgmd.cf.ac.uk/ac/index.php. Accessed September 20, 2015. | |
Erlich HA. PCR Technology: Principles and applications for DNA amplification. Macmillan Publishers; 1989. | |
Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–5467. | |
Costa-Motta FM, Acosta AX, Abe-Sandes K, et al. Genetic studies in a cluster of mucopolysaccharidosis type VI patients in Northeast Brazil. Mol Genet Metab. 2011;104(4):603–607. | |
Jurecka A, Piotrowska E, Cimbalistiene L, et al. Molecular analysis of mucopolysaccharidosis type VI in Poland, Belarus, Lithuania and Estonia. Mol Genet Metab. 2012;105(2):237–243. | |
Jurecka A, Zakharova E, Cimbalistiene L, et al. Mucopolysaccharidosis type VI in Russia, Kazakhstan, and Central and Eastern Europe. Pediatr Int. 2014;56(4):520–525. | |
Schwartz IV, Lima LC, Tylee K, et al. Further cases of “neighbor” mutations inmucopolysaccharidosis type II. Am J Med Genet A. 2006;140(15):1684–1686. | |
Brusius-Facchin AC, De Souza CF, Schwartz IV, et al. Severe phenotype in MPS II patients associated with a large deletion including contiguous genes. Am J Med Genet A. 2012;158A(5):1055–1059. | |
Wong LJ, Dimmock D, Geraghty MT, et al. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin Chem. 2008;54(7):1141–1148. | |
Tayeh MK, Chin EL, Miller VR, Bean LJ, Coffee B, Hegde M. Targeted comparative genomic hybridization array for the detection of single- and multiexon gene deletions and duplications. Genet Med. 2009;11(4):232–240. | |
Krivit W, Aubourg P, Shapiro E, Peters C. Bone marrow transplantation for globoid cellleukodystrophy, adrenoleukodystrophy, metachromatic leukodystrophy, and Hurler syndrome. Curr Opin Hematol. 1999;6(6):377–382. | |
Wang CC, Hwu WL, Lin KH. Long-term follow-up of a girl with Maroteaux-Lamy syndrome after bone marrow transplantation. World J Pediatr. 2008;4(2):152–154. | |
Herskhovitz E, Young E, Rainer J, et al. Bone marrow transplantation for Maroteaux-Lamysyndrome (MPS VI): long-term follow-up. J Inherit Metab Dis. 1999;22(1):50–62. | |
Krivit W. Allogeneic stem cell transplantation for the treatment of lysosomal and peroxisomal metabolic diseases. Springer Semin Immunopathol. 2004;26(1–2):119–132. | |
Giugliani R, Carvalho CG, Herber S, de Camargo Pinto LL. Recent Advances in Treatment Approaches of Mucopolysaccharidosis VI. Curr Pharm Biotechnol. 2011;12(6):956–962. | |
Rovelli AM. The controversial and changing role of haematopoietic cell transplantation for lysosomal storage disorders: an update. Bone Marrow Transplant. 2008;41 Suppl 2:S87–S89. | |
Fuller M, Hopwood JJ, Anson DS. Receptor mediated binding of two glycosylation forms of N-acetylgalactosamine-4-sulphatase. Biochim Biophys Acta. 1998;1406(3):283–290. | |
Auclair D, Hopwood JJ, Brooks DA, Lemontt JF, Crawley AC. Replacement therapy in Mucopolysaccharidosis type VI: advantages of early onset of therapy. Mol Genet Metab. 2003;78(3):163–174. | |
Yogalingam G, Crawley A, Hopwood JJ, Anson DS. Evaluation of fibroblast-mediated gene therapy in a feline model of mucopolysaccharidosis type VI. Biochim Biophys Acta. 1999;1453(2):284–296. | |
Bielicki J, Crawley AC, Davey RC, Varnai JC, Hopwood JJ. Advantages of using same species enzyme for replacement therapy in a feline model of mucopolysaccharidosis type VI. J Biol Chem. 1999;274(51):36335–36343. | |
Turner CT, Hopwood JJ, Bond CS, Brooks DA. Immune response to enzyme replacement therapy: 4-sulfatase epitope reactivity of plasma antibodies from MPS VI cats. Mol Genet Metab. 1999;67(3):194–205. | |
Brooks DA, Gibson GJ, Karageorgos L, Hein LK, Robertson EF, Hopwood JJ. An index case for the attenuated end of the mucopolysaccharidosis type VI clinical spectrum. Mol Genet Metab. 2005;85(3):236–238. | |
Harmatz P, Giugliani R, Schwartz I, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr. 2006;148(4):533–539. | |
Harmatz P. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis VI: clinical facts and figures. Turk J Pediatr. 2010;52(5):443–449. | |
Harmatz PR, Garcia P, Guffon N, et al. Galsulfase (Naglazyme®) therapy in infants with mucopolysaccharidosis VI. J Inherit Metab Dis. 2014;37(2):277–287. | |
Harmatz P, Giugliani R, Schwartz IV, et al. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine 4- sulfatase. Mol Genet Metab. 2008;94(4):469–475. | |
Decker C, Yu ZF, Giugliani R, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: Growth and pubertal development in patients treated with recombinant human N- acetylgalactosamine 4-sulfatase. J Pediatr Rehabil Med. 2010;3(2):89–100. | |
Horovitz DD, Magalhaes TS, Acosta A, et al. Enzyme replacement therapy with galsulfase in 34 children younger than five years of age with MPS VI. Mol Genet Metab. 2013;109(1):62–69. | |
Harmatz P, Ketteridge D, Giugliani R, et al. Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4- sulfatase. Pediatrics. 2005;115(6):e681–e689. | |
Hendriksz CJ, Giugliani R, Harmatz P, et al. Design, baseline characteristics, and early findings of the MPS VI (mucopolysaccharidosis VI) Clinical Surveillance Program (CSP). J Inherit Metab Dis. 2013;36(2):373–384. | |
Dogan M, Cesur Y, Peker E, Oner AF, Dogan SZ. Thrombocytopenia associated with galsulfase treatment. Hum Exp Toxicol. 2011;30(7):768–771. | |
Begin P, Chapdelaine H, Lemyre E, Paradis L, Des Roches A. Successful desensitization in a type VI mucopolysaccharidosis patient with probable IgE-mediated allergy to galsulfase [Naglazyme]. Ann Allergy Asthma Immunol. 2013;110(1):55–56. | |
Di Natale P, Villani GR, Parini R, et al. Molecular markers for the follow-up of enzyme-replacement therapy in mucopolysaccharidosis type VI disease. Biotechnol Appl Biochem. 2008;49(Pt 3):219–223. | |
So anki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012;107(1–2):15–24. | |
Lampe C, Schwarz M, Muller-Forell W, Harmatz P, Mengel E. Craniocervical decompression in patients with mucopolysaccharidosis VI: development of a scoring system to determine indication and outcome of surgery. J Inherit Metab Dis. 2013;36(6):1005–1013. | |
Horovitz DD, Magalhaes Tde S, Pena e Costa A, et al. Spinal cord compression in young children with type VI mucopolysaccharidosis. Mol Genet Metab. 2011;104(3):295–300. | |
Auclair D, Finnie J, White J, et al. Repeated intrathecal injections of recombinant human 4-sulphatase remove dural storage in mature mucopolysaccharidosis VI cats primed with a short-course tolerisation regimen. Mol Genet Metab. 2010;99(2):132–141. | |
Munoz-Rojas MV, Horovitz DD, Jardim LB, et al. Intrathecal administration of recombinant human N-acetylgalactosamine 4-sulfatase to a MPS VI patient with pachymeningitis cervicalis. Mol Genet Metab. 2010;99(4):346–350. | |
Yoshida M, Noguchi J, Ikadai H, Takahashi M, Nagase S. Arylsulfatase B-deficient mucopolysaccharidosis in rats. J Clin Invest. 1993;91(3):1099–1104. | |
Bradbury AM, Gurda BL, Casal ML, Ponder KP, Vite CH, Haskins ME. A review of gene therapy in canine and feline models of lysosomal storage disorders. Hum Gene Ther Clin Dev. 2015;26(1):27–37. | |
Simonaro CM, Ge Y, Eliyahu E, He X, Jepsen KJ, Schuchman EH. Involvement of the Toll- like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc Natl Acad Sci U S A. 2010;107(1):222–227. | |
Eliyahu E, Wolfson T, Ge Y, Jepsen KJ, Schuchman EH, Simonaro CM. Anti-TNF-alpha therapy enhances the effects of enzyme replacement therapy in rats with mucopolysaccharidosis type VI. PLoS One. 2011;6(8):e22447. | |
Schuchman EH, Ge Y, Lai A, et al. Pentosan polysulfate: a novel therapy for the mucopolysaccharidoses. PLoS One. 2013;8(1):e54459. | |
Ferla R, O’Malley T, Calcedo R, et al. Gene therapy for mucopolysaccharidosis type VI is effective in cats without pre-existing immunity to AAV8. Hum Gene Ther. 2013;24(2):163–169. | |
Ponder KP, O’Malley TM, Wang P, et al. Neonatal gene therapy with a gamma retroviral vector in mucopolysaccharidosis VI cats. Mol Ther. 2012;20(5):898–907. | |
Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63–72. | |
Gelb MH, Scott CR, Turecek F. Newborn screening for lysosomal storage diseases. Clin Chem. 2015;61(2):335–346. | |
Acosta AX, Abe-Sandes K, Giugliani R, Bittles AH. Delivering genetic education and genetic counseling for rare diseases in rural Brazil. J Genet Couns. 2013;22(6):830–834. | |
Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome) – 10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953–1964. | |
McGill JJ, Inwood AC, Coman DJ, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age – a sibling control study. Clin Genet. 2010;77(5):492–498. | |
Petry MF, Nonemacher K, Sebben JC, et al. Mucopolysaccharidosis type VI: Identification of novel mutations on the arylsulphatase B gene in South American patients. J Inherit Metab Dis. 2005;28(6):1027–1034. | |
Voskoboeva E, Krasnopol’skaia KD, Peters K, von Figura K. [Identification of mutations in the arylsulfatase B gene in Russian mucopolysaccharidosis type VI patients]. Genetika. 2000;36(6):837–843. | |
Karageorgos L, Brooks DA, Pollard A, et al. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Hum Mutat. 2007;28(9):897–903. | |
Isbrandt D, Arlt G, Brooks DA, Hopwood JJ, von Figura K, Peters C. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): six unique arylsulfatase B gene alleles causing variable disease phenotypes. Am J Hum Genet. 1994;54(3):454–463. | |
Litjens T, Brooks DA, Peters C, Gibson GJ, Hopwood JJ. Identification, expression, and biochemical characterization of N-acetylgalactosamine-4-sulfatase mutations and relationship with clinical phenotype in MPS-VI patients. Am J Hum Genet. 1996;58(6):1127–1134. | |
Villani GR, Balzano N, Vitale D, Saviano M, Pavone V, Di Natale P. Maroteaux-lamy syndrome: five novel mutations and their structural localization. Biochim Biophys Acta. 1999;1453(2):185–192. | |
Yang CF, Wu JY, Lin SP, Tsai FJ. Mucopolysaccharidosis type VI: Report of two Taiwanese patients and identification of one novel mutation. J Formos Med Assoc. 2001;100(12):820–823. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.