")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Diagnosis and Treatment of Hereditary Transthyretin Amyloidosis (hATTR) Polyneuropathy: Current Perspectives on Improving Patient Care
Authors Luigetti M , Romano A , Di Paolantonio A, Bisogni G, Sabatelli M
Received 22 November 2019
Accepted for publication 27 January 2020
Published 21 February 2020 Volume 2020:16 Pages 109—123
DOI https://doi.org/10.2147/TCRM.S219979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Video abstract presented by Marco Luigetti.
Views: 778
Marco Luigetti,1,2 Angela Romano,2 Andrea Di Paolantonio,2 Giulia Bisogni,3 Mario Sabatelli2,3
1Neurology Unit, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy; 2Università Cattolica del Sacro Cuore, Rome, Italy; 3Centro Clinico NEMO Adulti, Rome, Italy
Correspondence: Marco Luigetti
Neurology Unit, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Largo A Gemelli 8, Rome 00168, Italy
Tel +39-06-3015 4435
Fax +39-06-3550 1909
Email [email protected]
Abstract: Hereditary transthyretin amyloidosis (hATTR) with polyneuropathy (formerly known as Familial Amyloid Polyneuropathy) is a rare disease due to mutations in the gene encoding transthyretin (TTR) and characterized by multisystem extracellular deposition of amyloid, leading to dysfunction of different organs and tissues. hATTR amyloidosis represents a diagnostic challenge for neurologists considering the great variability in clinical presentation and multiorgan involvement. Generally, patients present with polyneuropathy, but clinicians should consider the frequent cardiac, ocular and renal impairment. Especially a hypertrophic cardiomyopathy, even if usually latent, is identifiable in at least 50% of the patients. Therapeutically, current available options act at different stages of TTR production, including synthesis inhibition (liver transplantation and/or gene-silencing drugs) or tetramer TTR stabilization (TTR stabilizers), increasing survival at different disease stages.
Keywords: amyloid, polyneuropathy, clinical care, therapy, transthyretin
Introduction
Hereditary transthyretin amyloidosis (hATTR) is a severe, heterogeneous multisystem condition with prevalent peripheral (both somatic and autonomic) nervous system impairment, due to mutations in the transthyretin (TTR) gene.1,2 The condition, presenting as an adult-onset, autosomal-dominant disease with variable penetrance, is characterized by extracellular deposition of amyloid fibrils in different organs. Besides the peripheral nerves, heart, kidney and ocular vitreous may also be involved (Figure 1), leading to a life-threatening, multisystem disease with a great variability in clinical presentation and course, and death within 10 years on average.1
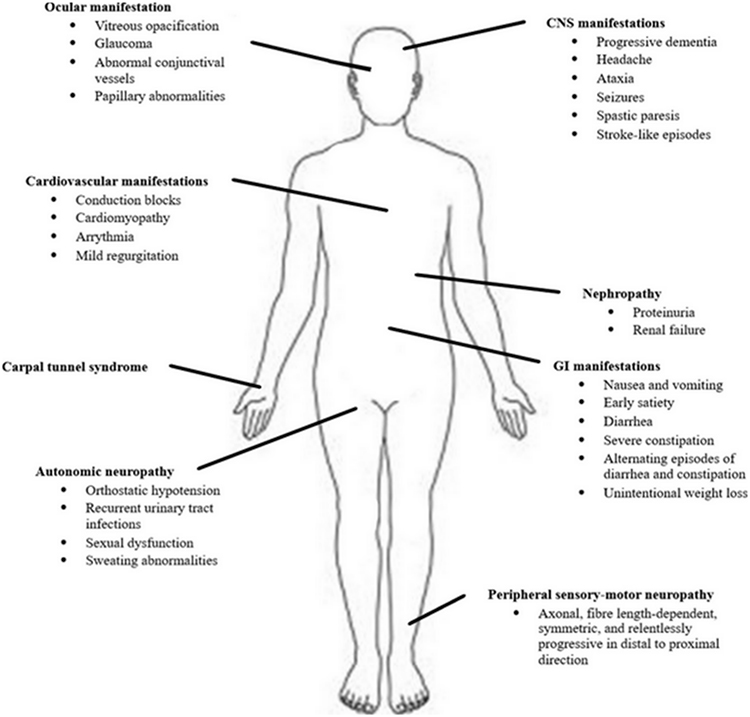 |
Figure 1 Multisystem involvement in hATTR. |
hATTR has traditionally been described according to the predominant clinical features, typically either a polyneuropathy (hATTR-PN), formerly referred to as Familial Amyloid Polyneuropathy (FAP),1 or a cardiomyopathy (hATTR-CM), termed Familial Amyloid Cardiomyopathy (FAC),3 although most patients show symptoms and signs of both nerve and heart involvement. A cardiomyopathy is also characteristic of a wild-type form of the disease (ATTRwt), previously known as senile cardiac amyloidosis.4
To date, over 120 TTR variants have been identified as a cause of hATTR, the most common being the p.Val30Met mutation.5 The majority of TTR mutations cause a “neuropathic” or a “mixed” phenotype; yet, some variants typically manifest with a predominant or isolated cardiomyopathy.
The classical clinical presentation of the p.Val30Met mutation, causing the common “Portuguese phenotype”, is characterized by a length-dependent neuropathy mainly involving the small fibres. When fully developed, the disease generally discloses a sensory-motor polyneuropathy with autonomic involvement (postural hypotension, and gastrointestinal, bladder and erectile dysfunction). However, neurological manifestations may vary according to the geographic area. In endemic areas, such as Portugal, patients usually show an early-onset disease (usually in the third or fourth decade), presenting as a small-fibre predominant neuropathy, with frequent autonomic dysfunction. Conversely, in non-endemic areas, many patients present with a late-onset FAP (in the sixth to eighth decade) with a slowly progressive polyneuropathy, with more severe motor impairment than early-onset cases, often with cardiac involvement but with less degree of autonomic symptoms.1
In this review, we focus on hATTR amyloidosis: an increasing interest has been observed in this field over the last few years, especially due to a great advance in the therapeutic management of these patients with the possibility to change the natural course of the disease. Hence, with the availability of new therapeutic agents acting either as kinetic TTR stabilizers or gene-silencing drugs,6–9 liver transplantation10 is no longer the only treatment option.
Epidemiology
In the past, hATTR amyloidosis has classically been considered a rare disease with three major endemic clusters in Portugal (where it was first described),11 Sweden, and Japan;1,2 smaller endemic foci have been subsequently identified in Cyprus12 and Majorca.13 Within Europe, the incidence of hATTR amyloidosis is highly variable, and cases are mainly sporadic.
Nowadays, given the increasing awareness of the disease among physicians and the current widespread availability of genetic testing, the incidence of hATTR amyloidosis is probably expected to rise, particularly in non-endemic regions.2 Consistently, hATTR has now been reported in at least 29 countries around the world.2
Genetics
The TTR gene, composed of four exons, is located on chromosome 18.14 Over 120 TTR variants have so far been identified, the great majority of which are pathogenic. The most common pathogenic variant consists of a point mutation leading to replacement of valine by methionine at position 30 of the mature protein.5 The Val30Met mutation is responsible for hATTR amyloidosis in the endemic regions,12,13,15,16 and is still the most frequent amyloidogenic mutation worldwide, accounting for about 50% of TTR variants all around the world.1
The disease is inherited as an autosomal dominant trait. However, patients carrying homozygous pathogenic mutations have also been reported, as well as compound heterozygous patients.2
Penetrance of hATTR may vary depending on the geographic region. A complete penetrance has often been described in multi-generation pedigrees from endemic areas.17 Conversely, outside endemic areas, incomplete penetrance that constantly increases with age is commonly seen, reaching nearly 100% in the ninth decade of life.17–19
Factors influencing the age at hATTR onset and phenotypic expression in families with Val30Met mutations remain unknown.1 Both genetic and non-genetic modifying factors,20 such as genotype, geographic origin and the gender of the parent from whom the mutation is inherited,17–19,21 can influence the variability in disease onset and in phenotypic presentation.
Regarding the age at onset, anticipation seems to be more prominent in male offspring inheriting the mutation from the mother, as reported by three different studies conducted in Portugal, Japan and Sweden. Conversely, father-to-daughter inheritance appears to be a protective factor for this phenomenon.2
Pathophysiology
TTR, originally known as prealbumin, is a 55-kDa transport protein for both thyroxine (T4) and retinol-binding protein, that circulates in soluble form in the serum and cerebrospinal fluid (CSF) of healthy humans.22 The liver represents the main source of TTR synthesis, although a small amount of protein is produced by the choroid plexus and by the retinal and ciliary pigment epithelia. Under normal conditions, TTR circulates as a homotetramer with a central channel containing two T4-binding sites, only one of which is physiologically occupied.22
hATTR amyloidosis is a protein misfolding disease: most pathogenic mutations reduce the stability of TTR tetramers as compared with the wild-type protein and enhance their dissociation into monomers, resulting in misfolding, aggregation and subsequent extracellular deposition of TTR amyloid fibrils in different sites, leading to progressive multisystem dysfunction.1 Amyloid deposition begins years before clinical onset, as in other neurodegenerative conditions, such as Alzheimer’s and Parkinson’s disease.
TTR amyloid fibrils can also originate from deposition of wild-type transthyretin protein in ATTRwt amyloidosis: this condition is an age-related disease, thus called senile amyloidosis,4 in which the wild-type TTR protein gets structurally dissociated and misassembled into amyloid fibrils.
Clinical Features
The clinical picture of hATTR classically shows a great heterogeneity, varying from an almost exclusive neurologic phenotype within an endemic cohort to a strictly cardiologic presentation in sporadic cases (Table 1).23
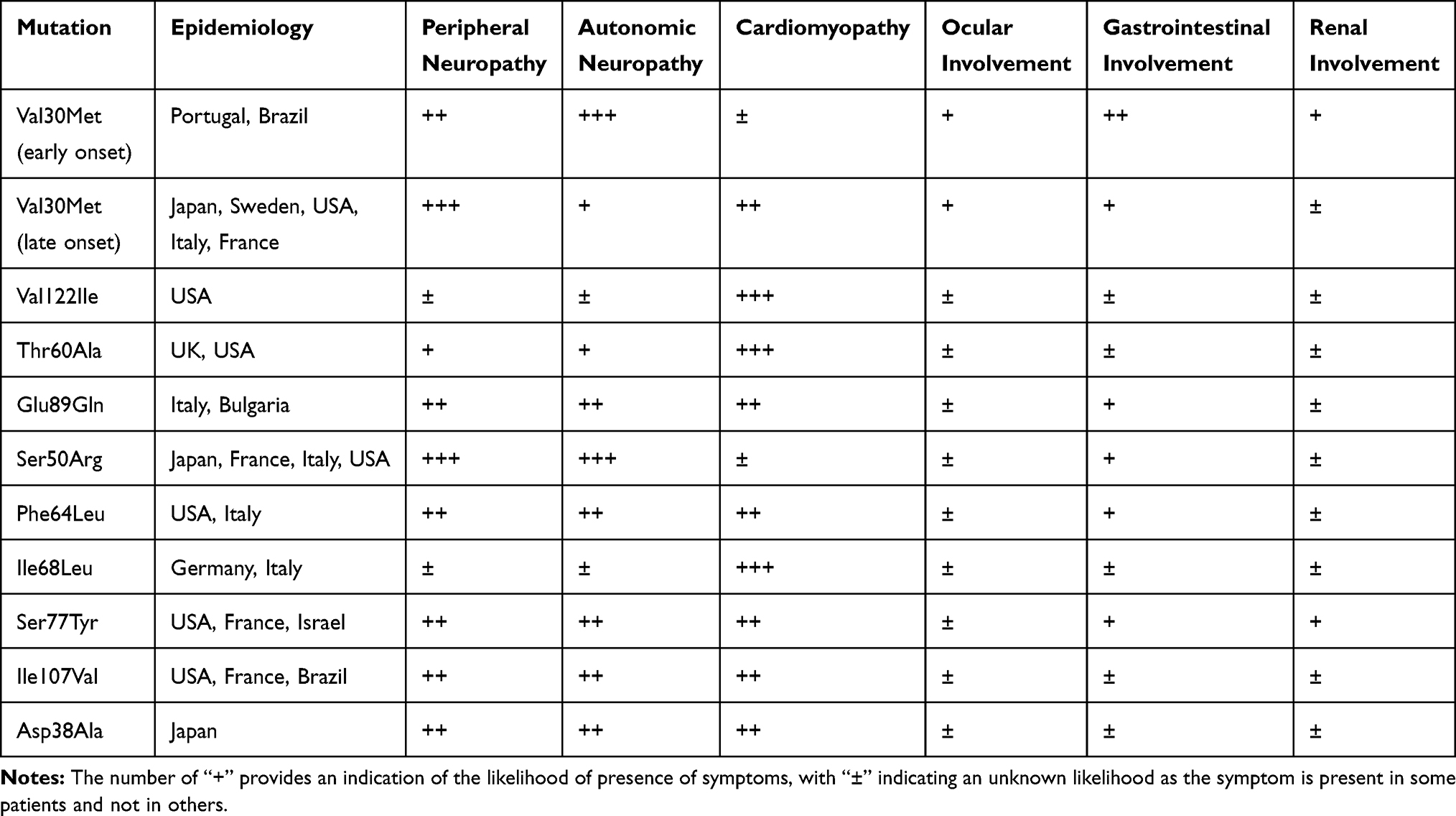 |
Table 1 Clinical Manifestations and Epidemiology of the Most Common hATTR Mutations |
Firstly, clinical phenotype appears to be related to the specific TTR mutation: some variants (such as Val122Ile, Ile68Leu, Thr60Ala, and Leu111Met) induce a cardiomyopathy as the predominant feature,24,25 while others (such as Ala97Ser and Ser50Arg) cause a pure neurological phenotype.26,27
Also, age at onset has a great variability and seems to influence the phenotype and the clinical course, along with the geographic origin. Patients from endemic areas have traditionally been described as having an early-onset (age <50 years) or late-onset disease (age ≥50 years).15,28,29 These two groups, even if sharing the same genotype, differ in pathological and clinical features.30
Early-onset patients in endemic regions usually show a progressive sensory-motor and autonomic neuropathy, with an expected survival of approximately 10–20 years from onset.23 By contrast, late-onset Val30Met patients, as well as non-Val30Met cases, have usually a more rapid and severe progression, with common cardiac involvement, and are associated with a median survival of nearly 7 years since the disease onset.26,31,32
Prognosis is, by far, poorer for the exclusively cardiac phenotypes (FAC), with a life expectancy of 2–5 years from symptom onset.33,34
Symptoms
Some patients, especially those with an early-onset hATTR-Val30Met amyloidosis, can complain of systemic unspecific symptoms, such as fatigue or unintentional weight loss, as the initial manifestations of the disease.
The specific clinical picture of the somatic neuropathy is influenced by the causal TTR mutation and the age of onset. Early-onset hATTR-Val30Met amyloidosis presents with “discomfort” (numbness and neuropathic pain) in the feet, which gradually extends proximally, associated with impaired pain and/or temperature sensation, related to the involvement of unmyelinated and small myelinated fibres; impairment of light touch and deep sensibility and of motor fibres usually occurs late in the course of the disease.1 Clinical presentation of late-onset hATTR-Val30Met amyloidosis and other non-Val30Met variants is, by far, more variable, as it may manifest with sensory or sensorimotor symptoms, starting either distally in lower or all four limbs or even exclusively in the upper limbs, mimicking a carpal tunnel syndrome.31,35
Autonomic impairment is frequently observed, especially in early-onset cases, and may present with gastrointestinal symptoms (including constipation, daily diarrhoea or alternating constipation/diarrhoea, early satiety, nausea, and vomiting), orthostatic hypotension, bladder and erectile dysfunction.
Cardiac amyloidosis, remaining latent for a long time, results in progressive biventricular wall thickening, diastolic dysfunction deriving from loss of compliance, conduction disorders, and symptoms of congestive heart failure with a preserved ejection fraction. Cardiac arrhythmias and/or conduction blocks are frequent in early-onset Val30Met or in other non-Val30Met variants (ie, Glu89Gln).31,33
Kidney involvement, usually presenting as nephrotic syndrome and/or progressive renal failure, occurs in about one-third of Portuguese Val30Met patients, and in only 6% of sporadic hATTR cases.1,36
Ocular involvement is quite frequent, and its prevalence seems to increase with disease duration. Ocular disorders include dry eye syndrome (nearly 70%), amyloid deposition on the iris (38%) or on the anterior capsule of the lens (33%), pupillary disorders (as scalloped iris in about 28%), glaucoma (20%), vitreous opacity (17%), abnormal conjunctiva vessels (14%), and amyloidotic retinal angiopathy (4%).37,38
Finally, central nervous system (CNS) involvement, resulting from leptomeningeal amyloid angiopathy, has also been described, sometimes in combination with eye impairment (oculoleptomeningeal amyloidosis). Leptomeningeal and meningovascular amyloidosis are rare forms of amyloidosis caused by a limited spectrum of TTR variants, such as Ala25Thr39 or Tyr69His,40 but CNS impairment can also be found in advanced stages of hATTR-Val30Met amyloidosis, causing stroke, subarachnoid haemorrhage, hydrocephalus, cerebellar ataxia, spastic paresis, seizures, hearing loss, and dementia.40,41
Diagnosis
In endemic areas, where a positive family history is frequent, the diagnosis of hATTR is usually easy and often made within 1 year from disease onset. In these areas, once a mutation has been identified within a family, genetic counselling for asymptomatic carriers is fundamental. Regular follow-up of carriers is necessary to detect early symptoms or signs of the disease, allowing a prompt initiation of anti-amyloid therapy.
Diagnosis is clearly more challenging in non-endemic regions, where it can be delayed by 4–5 years as a result of several factors, especially a common negative family history.42–44 Furthermore, clinical presentation, given its heterogeneity, may mimic various peripheral neuropathies, explaining the high frequency of misdiagnosis (Table 2).35,42,45–47 Common misdiagnoses include idiopathic axonal polyneuropathy, chronic inflammatory demyelinating polyradiculoneuropathy, lumbar spinal stenosis, diabetic neuropathies, Charcot-Marie-Tooth neuropathy or motor neuron disease. In patients with a peripheral neuropathy of otherwise undetermined aetiology, early search for associated clinical features, especially for cardiac involvement, can help reveal amyloidosis.
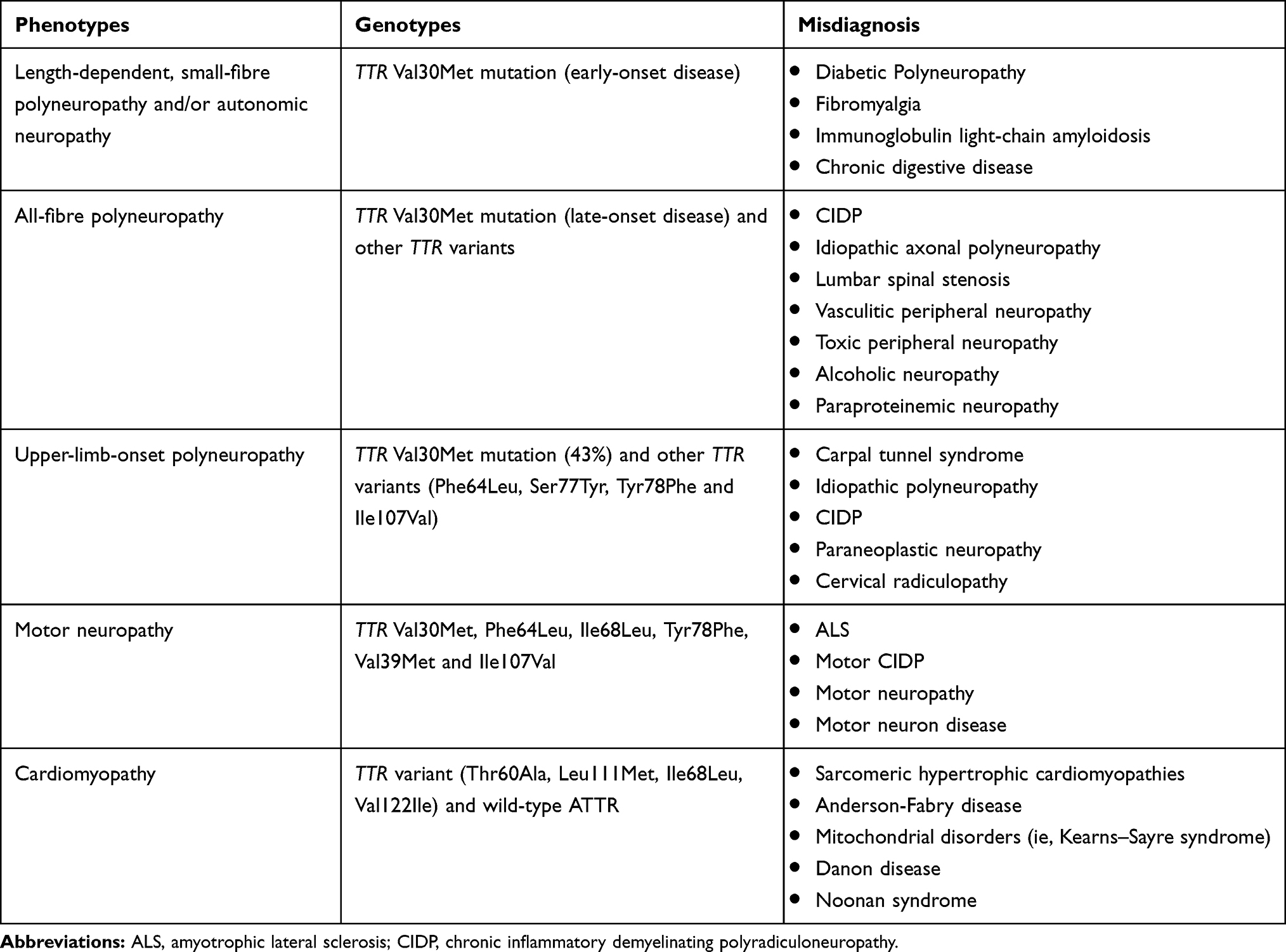 |
Table 2 Frequent Misdiagnosis in Patients with Transthyretin Amyloidosis |
The diagnosis can be confirmed by demonstration of amyloid in a biopsy sample and/or detection of an amyloidogenic mutation by TTR gene sequencing.31,32
Characteristic amyloid deposits can be detected in different tissues, such as skin, nerve, myocardium, kidney, fat pad, labial salivary gland or gastrointestinal mucosa.42,48 Therefore, following a clinical suspicion, tissue biopsy, ideally of an affected organ, may be performed. Staining with Congo red, which typically gives an apple-green birefringence under polarized light, can reveal amyloid deposition.1,42 Once amyloid is demonstrated, the type of precursor protein should be identified: confirmation of TTR as precursor protein can be made by either immunohistochemistry or mass spectroscopy.42 However, the patchy and uneven distribution of amyloid fibrils in tissues may yield false-negative or false-positive results.23
Hence, a negative biopsy does not rule out the diagnosis of hATTR amyloidosis: the sensitivity (besides relying on the skill of the pathologist) varies depending on the biopsied tissue, the TTR mutation, and the patient’s age; multiple biopsies might be required for the diagnosis in some cases.44
In some European countries, biopsy of the labial salivary gland can be used to look for evidence of amyloidosis in unexplained progressive axonal neuropathy.49 Nerve biopsy (Figures 2–4), despite being usually considered a second-line investigation for diagnosis, has a sensitivity of nearly 80%.42,50,51 Amyloid can also be visualised in other specimens, such as muscle or abdominal fat, showing a variable sensitivity of 14–83%.42,44,50
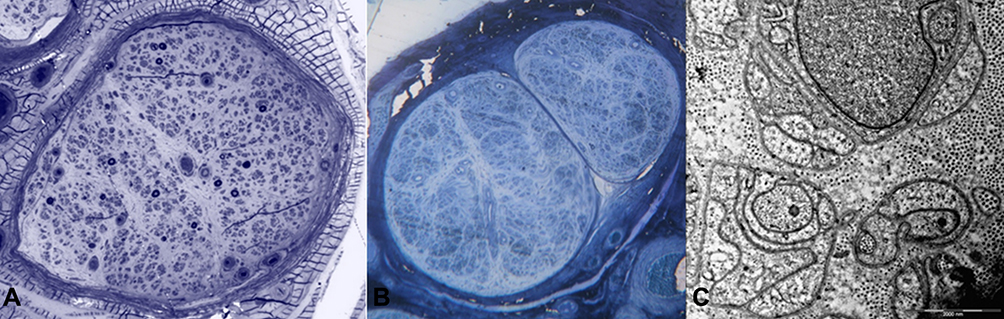 |
Figure 2 Sural nerve biopsies from a patient with late-onset FAP. Semithin sections stained with toluidine blue showed a severe reduction of myelinated fibres (A). A second biopsy performed four years later revealed complete loss of myelinated fibres (B), while unmyelinated fibres were relatively preserved (C). |
 |
Figure 3 Sural nerve biopsy from a patient with late-onset FAP. Congo red staining showed a small amyloid deposition (A). Ultrastructural analysis with electron microscope confirmed short and thin amyloid fibrils (B). |
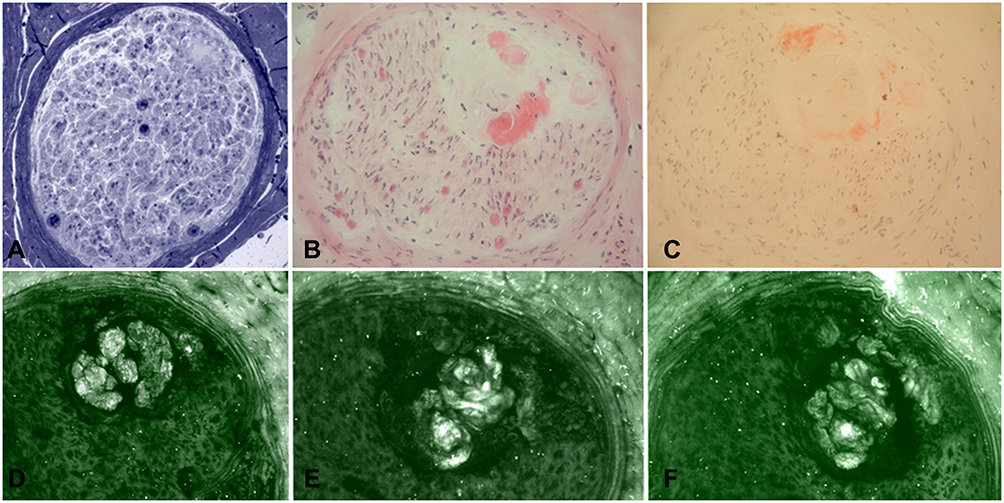 |
Figure 4 Sural nerve biopsy from a patient with late-onset FAP. Semithin section stained with Toluidine blue (A) showed amyloid deposition, also confirmed by H&E (B) and Congo red (C) staining. However, immunofluorescence with anti-TTR (D), anti-kappa light chain (E), and anti-lambda light chain (F) resulted not specific for TTR amyloidosis. |
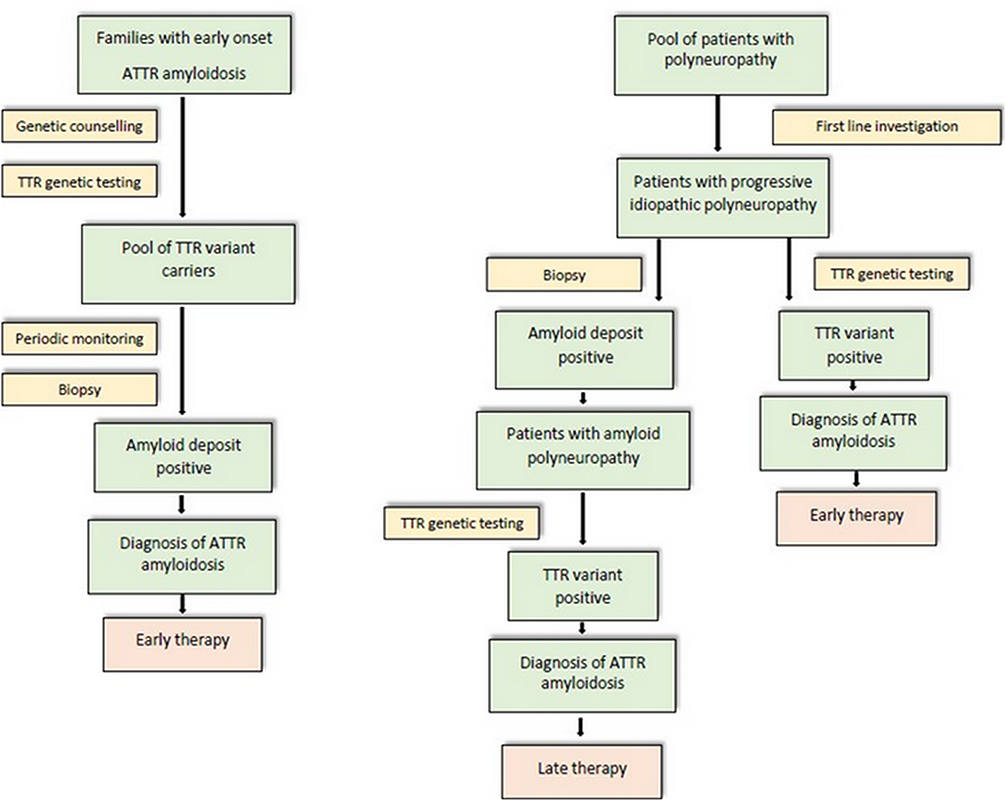 |
Figure 5 Flow-chart strategy for the diagnosis of FAP in endemic (left) and non-endemic (right) areas. |
However, if clinical suspicion is high, TTR gene sequencing should be performed in all patients, and Sanger sequencing represents now the gold standard (Figure 5).2
Once the diagnosis has been reached, additional extensive investigations are required to evaluate the extent and severity of organ involvement.
Traditional nerve conduction studies are performed to detect the presence and severity of a peripheral neuropathy.32,52 Even if skin biopsy still remains the gold standard for the diagnosis of a small fibre neuropathy, evaluation of sudomotor function via electrochemical skin conductance,53–55 as well as measurement of heart rate variability,56 and testing for orthostatic hypotension can all be useful to investigate an autonomic neuropathy.
Cardiac investigations in hATTR amyloidosis are mainly aimed at detecting a possible infiltrative (hypertrophic) cardiomyopathy and, above all, any potential serious conduction disorders that may require the implantation of a prophylactic pacemaker to decrease the risk of sudden death. For specific mutations causing a predominantly/exclusively cardiac phenotype, particularly common in USA (Val122Ile),57 UK (Thr60Ala),58 or Italy (Ile68Leu),24 a differential diagnosis with other cardiomyopathies may be necessary (Table 2). Useful investigations for cardiac assessment include echocardiogram, 24 hr Holter monitoring, and echocardiography with strain imaging; cardiac magnetic resonance imaging (MRI) and intracardiac electrophysiological studies, even if not always available, are useful when necessary.2
Scintigraphy to detect cardiac uptake of bone tracers such as 99mTc-2,3-dicarboxypropane-1,1-diphosphonate (DPD), 99mTc-hydroxymethylene diphosphonate (HMDP) or 99mTc-pyrophosphate (PYP)2,3,59 is a non-invasive investigation that may facilitate early diagnosis, helping to recognize an amyloid origin of an hypertrophic cardiomyopathy. It can be used, although taking into account radioactivity exposure, as an equivalent for cardiac biopsy in this context, owing to its usually high sensitivity and specificity.59 On the other hand, the possibility of a low sensitivity for specific mutations, including Phe64Leu, should be considered.60
Cardiac serum biomarkers, specifically brain natriuretic peptide (BNP) or its N-terminal prohormone (NT-proBNP) and cardiac troponins (T or I), are useful and have a prognostic value in amyloid cardiomyopathy.3,61 NT-proBNP is a natriuretic peptide without biological activity whose plasma levels are increased in association with high intracardiac pressure, as observed in heart failure with preserved ejection fraction. NT-proBNP plasma levels are abnormal even in the early stages of cardiac amyloid infiltration, and correlate with left ventricular mass (evaluated on cardiac MRI) and with late gadolinium enhancement, suggesting their utility as a measure of cardiac amyloidosis severity.61 In addition, high levels of troponin are observed in the most severe forms or in advanced stages of the disease.
Once the genetic diagnosis is confirmed, an ophthalmological assessment should be carried out as well. When ocular manifestations are present, the frequency of ophthalmological evaluations varies depending on severity of eye involvement, and should include measurement of visual acuity and of intraocular pressure, Schirmer test, ocular fundus and slit-lamp examination.38
Renal evaluation is also crucial, and usually based on measurement of serum creatinine, proteinuria and microalbuminuria, and on the estimated glomerular filtration rate (eGFR).2,36 Determination of serum cystatin C may represent another valid marker for non-invasive estimation of GFR: since serum cystatin C concentration seems to be independent of sex, age, and muscle mass, it has been suggested to be a more sensitive marker of renal dysfunction compared with serum creatinine level.62
Finally, a modified version of the body mass index (mBMI), that corrects for the effect of hypalbuminaemia, provides a marker of nutritional status, which is, to some extent, influenced by the duration and severity of gastrointestinal symptoms and malabsorption in hATTR patients.63
Outcome Measures
To evaluate the extent of disability in hATTR-PN patients, useful tools in clinical practice include the FAP staging system64 and the Polyneuropathy Disability (PND) score.63 However, these clinical scales provide only a generic indicator of overall disease status and are not sensitive to track disease progression in the short-term period.
Hence, to better evaluate all the aspects of the polyneuropathy, clinical trials have tested different Neuropathy Impairment Score (NIS)-based measures, which provide a better chance to detect a treatment effect. Nevertheless, the NIS score,65 a clinical compound score based on examination of muscle weakness, sensory loss and stretch reflexes in the limbs, and its subset, the NIS-lower limbs (NIS-LL) score,6 have shown some limitations, requiring gradual modifications.
In a trial published in 2013, the NIS+7,66 which combines clinical assessment with seven electrophysiological tests, was used to better characterize and quantify neuropathic impairment.
In recent trials, run by Alnylam and by Ionis,8,9 two different variants of the modified NIS+7 (mNIS+7) were used. The mNIS+7Alnylam and the mNIS+7Ionis, specifically designed to assess disease impairment and progression in hATTR amyloidosis clinical trials, better quantify sensory abnormalities over the whole body, nerve conduction abnormalities, and autonomic function.67
Other useful clinical scales include: the Composite Autonomic Symptom Scale-31 (COMPASS-31) questionnaire68 and the Compound Autonomic Dysfunction Test (CADT) questionnaire69 for assessment of autonomic symptoms; the Rasch-built Overall Disability Scale (R-ODS) survey70 for evaluation of activities of daily living; the Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) questionnaire71 to estimate quality of life; the 6 min walking test, the 10-meter walking test, and handgrip strength test (dynamometer) to assess specific motor function.72
Instrumental examination, as Sudoscan, also proved to be a good tool for monitoring disease progression in late-onset hATTR.53–55
Considering the amount of available scales and/or questionnaires, it is difficult to imagine the actual application of all of them in clinical practice. In the Authors’ opinion, some of these, including FAP staging system, PND and NIS-LL scores, could be currently used in clinical daily activity, while others, including NIS+7 and its variants, should be limited to protocols. Regarding autonomic involvement, which is quite hard to quantify, CADT questionnaire could be the best questionnaire, and Sudoscan the best instrumental technique.
Therapy
Being a systemic disease, management of hATTR amyloidosis patients requires a multidisciplinary approach.42 Care should include anti-amyloid therapy to inhibit further production and/or deposition of amyloid aggregates, symptomatic therapy, and treatment of cardiac, renal and ocular involvement, considering also the eventuality of heart or kidney transplantation in advanced disease.42
After the diagnosis of hATTR, it is advisable to timely start treatment to delay disease progression.73
Systematic and regular monitoring of TTR asymptomatic carriers is essential to detect early signs of disease and to promptly initiate anti-amyloid therapy.74
In the last few years, many therapies have been developed for hATTR amyloidosis (Figure 6), especially for the early stages of the disease (stage 1 and 2), while currently patients with an advanced disease (stage 3 hATTR-PN) do not have any available treatment.
 |
Figure 6 Overview of therapeutic strategies in hATTR with polyneuropathy. |
Liver Transplantation
Since TTR is mainly produced by the liver, the first therapeutic approach for these patients was liver transplantation (LT), introduced for treatment of hATTR amyloidosis in 1990 to prevent amyloid formation.10 The outcome of liver transplantation is influenced by various patients’ characteristics, including age, severity of disease at the time of surgery, and TTR mutation.75 In a retrospective analysis performed on 1940 patients from the FAP World Transplant Registry, the 20-year survival after LT was 55.3%, with cardiovascular complications representing the leading causes of death (22%).76
This procedure, that replaces the mutant TTR protein with the wild-type protein, can indeed fail to stop subsequent progressive cardiac deposition.77,78 Clinical observations post-liver transplantation suggest that preformed fibrils present in the organs of hATTR patients at the time of surgery could template and accelerate polymerization of soluble circulating TTR: it is hypothesized that this “seeding” may contribute to cardiomyopathy by cardiac deposition of wild-type TTR secreted by the new liver.77
TTR Stabilizers
TTR tetramer stabilizers are agents designed to stabilize the normal circulating form of TTR, and hence are hypothesized to prevent the protein from dissociating and undergoing the conformational changes that lead to its aggregation as amyloid.79 However, TTR stabilizers do not inhibit “seeding” of ATTR: this data may help to explain results of clinical trials.78
Two drugs that act as TTR stabilizers have been assessed in controlled trials in hATTR amyloidosis:
Tafamidis
Tafamidis is an oral, kinetic TTR stabilizer, approved in Europe and selected countries of Asia and South America for the treatment of hATTR in adults with stage 1 symptomatic polyneuropathy.80
Tafamidis treatment in hATTR-PN amyloidosis was assessed in a multicentre, placebo-controlled, Phase III clinical trial involving 128 patients, most of whom of Portuguese origin, with an early-onset ATTR-Val30Met amyloidosis and a mild neuropathy at baseline.6 Tafamidis treatment demonstrated TTR stabilization in 98% of the patients and lack of neuropathy progression in 60% of the patients (versus 38% in the placebo group), with maintained quality of life (QoL).6
The short-term and long-term efficacy and safety of tafamidis in late-onset hATTR (Val30Met and non-Val30Met mutations) have been evaluated in three open-label studies.81–83 Tafamidis was well tolerated in all the studies. However, it did not prevent progression of disability at 1 year after initiation of treatment.81,82 Neuropathy progression correlated with an older age and a poorer clinical status at baseline.83
More recently, based on the results of the ATTRACT trial, tafamidis has been approved by the Food and Drug Administration (FDA) for the treatment of patients with hATTR-CM, and it has been presented to the European Medicines Agency (EMA) for the same indication. In the ATTRACT trial, two dosages of tafamidis were compared with placebo in symptomatic patients with hATTR-CM (25%) and ATTRwt amyloid cardiomyopathy (75%).84 The primary analysis disclosed that at 18 months of therapy all-cause mortality and rates of cardiovascular-related hospitalizations were lower in the tafamidis group than in the placebo group, as well as a reduced deterioration in functional capacity and in QoL in patients with either hATTR-CM or ATTRwt cardiomyopathy.84
Diflunisal
Diflunisal, a nonsteroidal anti–inflammatory drug (NSAID), acts as well as a TTR stabilizer.23
A randomized, placebo-controlled, double-blind, multicentre, international phase III study, including patients with late-onset hATTR amyloidosis associated with different TTR mutations and high NIS at baseline, demonstrated that treatment with diflunisal for 2 years significantly reduces the rate of neuropathy progression and preserves QoL, compared to placebo.66 However, half of the study population discontinued the treatment because of disease progression and liver transplantation. Furthermore, long-term use of NSAID typically induces an increased risk of gastrointestinal, renal, and cardiac adverse events.79
Gene-Silencing Therapies
Lately, two new first-in-class drugs, patisiran (Onpattro, Alnylam Pharmaceuticals, Inc.), and inotersen (Tegsedi, Akcea Therapeutics, Inc.), have been approved in the United States for the treatment of hATTR amyloidosis and in Europe for the treatment of stage 1 or 2 polyneuropathy in adults with hATTR amyloidosis. These novel therapies aim to decrease both variant and wild-type TTR hepatic production by targeting its mRNA.2 However, long-term studies will be necessary to evaluate the effect of gene-silencing therapies on the functional role of TTR, especially regarding transport of vitamin A.
RNA Interference
RNA interference (RNAi) is a biological process in which non-coding, small interfering RNAs (siRNAs) inhibit the expression of specific genes by mediating the degradation of targeted mRNAs.85 siRNAs are encapsulated in lipid nanoparticles that, following intravenous administration, are opsonized by apolipoprotein and predominantly delivered to the liver.85,86
Use of RNAi therapy in hATTR amyloidosis has been tested in both preclinical models86 and clinical trials.72,84,87
In preclinical models, significant reduction of TTR deposits in peripheral tissues was observed following RNAi-mediated TTR suppression, with regression correlating to serum TTR concentration.86
A marked TTR knockdown was also seen in a Phase I study conducted on both healthy volunteers and hATTR patients, where patisiran leaded to a rapid, dose-dependent and persistent decrease of both mutant and non-mutant TTR.87
Patisiran was then assessed in a Phase II, open-label, multidose, dose-escalation study including 29 patients with stage 1 or 2 hATTR-PN amyloidosis. In patients treated with patisiran at a dose of 0.3 mg/kg body weight every 3 weeks, the maximum mean reduction in TTR level was 87%.84 Patients in the Phase II trial were eligible to continue treatment in an open-label extension study that showed stability of the measures of neuropathy impairment over 24 months of treatment.
A large randomized, double-blind, placebo-controlled, phase III study (APOLLO) showed the efficacy and safety of patisiran over 18 months of treatment.72 The primary end point was the change from baseline to 18 months in the mNIS+7Alnylam score. Secondary end points included the effect of patisiran on QoL (assessed by the Norfolk QoL-DN), motor strength (NIS-weakness subset), disability (score on the R-ODS survey), gait speed (10-meter walk test), nutritional status (mBMI), and patient-reported autonomic symptoms (COMPASS-31 questionnaire).72 Two hundred and twenty-five patients with different severity of disease (NIS score of 5 to 130) underwent randomization in a 2:1 ratio (148 to the patisiran arm vs 77 to the placebo group). Randomization was stratified according to NIS, Val30Met variant associated with early-onset disease (age <50 years) versus late-onset Val30Met cases and all other pathogenic mutations, and previous use of a TTR stabilizer versus no known previous treatment. Compared to placebo, patisiran showed a statistically significant benefit both in the primary end point and in all the secondary end points in all subgroups, regardless of the disease stage, TTR variant and age at onset. Improvement from baseline in mNIS+7 scores occurred in 56% of participants who received patisiran versus only 4% in the placebo group.8 Adverse events more frequently observed in the patisiran group than in the placebo one included peripheral edema (30% versus 22%) and infusion-related reactions (19% versus 9%), but these events were mild or moderate. The frequency of severe adverse events (28% in the patisiran group and 36% in the placebo group) and serious adverse events (36% and 40%, respectively) was instead comparable in the two groups.
Revusiran is another RNAi drug designed for the treatment of hATTR amyloidosis, administered subcutaneously, consisting of a TTR-targeting siRNA conjugated to a N-acetylgalactosamine (GalNAc) ligand.86,88 A randomized, double-blind, placebo-controlled, phase III trial (ENDEAVOUR) was designed to test its efficacy and safety in patients with hATTR-CM. However, this trial was stopped in 2016 owing to observation of a greater mortality in the treatment group than in the placebo arm.
Antisense Oligonucleotides
Inotersen (IONIS-TTRRx) is a second-generation 2′-O-methoxyethyl–modified antisense oligonucleotide (ASO) designed to inhibit the hepatic production of both mutant and wild-type TTR.9 Inotersen targets TTR mRNA, causing its degradation through a RNase H1 mechanism of action, thus preventing the translation of TTR mRNA into TTR protein.89
A robust (>80%), dose-dependent reduction in plasma TTR levels were observed both in animal models and in healthy human volunteers treated with IONIS-TTRRx.89
The safety and efficacy of inotersen in hATTR amyloidosis have been tested in an international, randomized, double-blind, placebo-controlled, 15-months, pivotal phase III trial (NEURO-TTR).9 A total of 172 patients with a diagnosis of stage 1 or 2 hATTR-PN (NIS score of 10 to 130) was enrolled and randomized in a 2:1 ratio to receive inotersen 300 mg (112 patients) or placebo (60 patients). The drug was administered subcutaneously three times during the first week to obtain near steady-state drug levels, followed by a once-weekly subcutaneous injection for the next 64 weeks. Patients were stratified according to TTR mutations (Val30Met versus non-Val30Met), disease stage (stage 1 versus stage 2), and previous treatment with tafamidis or diflunisal. Primary end points were the change from baseline to week 66 in the mNIS+7Ionis score and in the total score on the Norfolk QoL-DN questionnaire. Both primary efficacy end points favoured inotersen, independently of TTR mutation, disease stage, or presence of a cardiomyopathy at baseline.9 36.6% of patients treated with inotersen remained stable or improved on mNIS+7 score at 15 months versus 19.2% in the placebo arm. Five deaths were observed in the inotersen group versus none in the placebo group. The most common serious adverse events in the treatment group were glomerulonephritis (3%) and thrombocytopenia (3%), with one death associated with grade 4 thrombocytopenia. Subsequently, all patients received enhanced monitoring for thrombocytopenia and renal dysfunction.
The FDA and EMA approved inotersen for the treatment of patients with stage 1 or stage 2 hATTR-PN.
Emerging Therapeutic Approaches
Several agents that stabilize TTR, such as epigallocatechin-3-gallate (EGCG), have been studied. EGCG is a catechin in green tea that is likely to increase TTR tetramer stabilization and to reduce TTR deposition.90
Monoclonal antibodies directed against non-native forms of TTR protein represent another possible therapeutic option for hATTR amyloidosis. These antibodies selectively bound the target epitope on monomeric and non-native, misfolded forms of TTR, strongly inhibiting TTR fibril formation in vitro.91
A Phase I–II study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of AKCEA-TTR-LRx (ION-682884), an antisense inhibitor of transthyretin production, administered subcutaneously via a single subcutaneous injection once every 4 weeks to healthy volunteers and patients with hATTR amyloidosis, is ongoing (ClinicalTrials.gov Identifier: NCT03728634).
A phase III study has been initiated and is currently recruiting patients to assess the efficacy and safety of vutrisiran (ALN-TTRSC02), a RNAi drug administered subcutaneously once every 3 months, in patients with hATTR amyloidosis. Participants will receive vutrisiran or the reference comparator patisiran during the treatment period. This study will use the placebo arm of the APOLLO study (NCT01960348) as an external comparator for the primary and most other efficacy end points (ClinicalTrials.gov Identifier: NCT03759379).
Conclusion
hATTR amyloidosis is a severe, clinically and genetically heterogeneous, multisystem disease, distributed worldwide. Recent results of clinical studies have widened the therapeutic arsenal to include TTR stabilizers and TTR gene silencers, with a global decline in the utilization of liver transplantation. Nowadays, an increasing interest is especially addressed to gene-silencing therapies that are likely to change the natural history of the condition, regardless of the stage, although long-term benefits or side effects are still to be proved, as a further progression of the disease could be explained by the persistent production of transthyretin in the brain and in the eye. The future challenges will be to find an effective therapy in patients with an advanced disease, and to assess the effects of amyloid deposit clearance with monoclonal antibodies, the potential of curative gene replacement therapy, and the potential benefit of combined therapies with different mechanism of action, as well as the effects of disease-modifying therapies in pre-symptomatic individuals.
Abbreviations
9mTc-DPD, technetium-99m radiolabeled 2,3-dicarboxypropane-1,1-diphosphonate; 99mTc-HMDP, technetium-99m radiolabeled hydroxymethylene diphosphonate; 99mTc-PYP, technetium-99m radiolabeled pyrophosphate; ASO, antisense oligonucleotide; ATTRwt, wild-type transthyretin amyloidosis; BNP, brain natriuretic peptide; CADT, Compound Autonomic Dysfunction Test; CNS, central nervous system; COMPASS-31, Composite Autonomic Symptom Scale-31; CSF, cerebrospinal fluid; EGCG, epigallocatechin-3-gallate; eGFR, estimated glomerular filtration rate; EMA, European Medicines Agency; FAC, familial amyloid cardiomyopathy; FAP, familial amyloid polyneuropathy; FDA, Food and Drug Administration; GalNAc, N-acetylgalactosamine; hATTR, hereditary transthyretin amyloidosis; hATTR-CM, hereditary transthyretin amyloidosis with cardiomyopathy; hATTR-PN, hereditary transthyretin amyloidosis with polyneuropathy; LT, liver transplantation; mBMI, modified body mass index; mNIS+7, modified Neuropathy Impairment Score +7; MRI, magnetic resonance imaging; NIS, Neuropathy Impairment Score; NIS+7, Neuropathy Impairment Score +7; NIS-LL, Neuropathy Impairment Score-Lower Limbs; Norfolk QoL-DN, Norfolk Quality of Life-Diabetic Neuropathy; NSAID, nonsteroidal anti-inflammatory drug; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; PND, Polyneuropathy Disability score; QoL, quality of life; RNAi, RNA interference; RNase H1, ribonuclease H1; R-ODS, Rasch-built Overall Disability Scale; siRNAs, small interfering RNAs; T4, thyroxine; TTR, transthyretin.
Disclosure
Dr Luigetti received financial grants (honoraria and speaking) from Akcea, Alnylam and Pfizer, and travel grants from Pfizer, Kedrion and Grifols; Dr Bisogni received financial grants (honoraria and speaking) from Alnylam, and travel grants from Pfizer, and Grifols; Dr Romano received travel grants from Akcea and Pfizer; Dr Di Paolantonio received travel grants from Akcea and Pfizer; Dr Sabatelli received financial grants (honoraria and speaking) from Akcea. The authors report no other conflicts of interest in this work.
References
1. Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10(12):1086–1097. doi:10.1016/S1474-4422(11)70246-0
2. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387–404. doi:10.1038/s41582-019-0210-4
3. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–1300. doi:10.1161/CIRCULATIONAHA.111.078915
4. Pinney JH, Whelan CJ, Petrie A, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2(2):e000098. doi:10.1161/JAHA.113.000098
5. Saraiva MJ, Birken S, Costa PP, Goodman DS. Family studies of the genetic abnormality in transthyretin (prealbumin) in Portuguese patients with familial amyloidotic polyneuropathy. Ann N Y Acad Sci. 1984;435:86–100. doi:10.1111/j.1749-6632.1984.tb13742.x
6. Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792. doi:10.1212/WNL.0b013e3182661eb1
7. Adams D, Cauquil C, Labeyrie C, Beaudonnet G, Algalarrondo V, Théaudin M. TTR kinetic stabilizers and TTR gene silencing: a new era in therapy for familial amyloidotic polyneuropathies. Expert Opin Pharmacother. 2016;17(6):791–802. doi:10.1517/14656566.2016.1145664
8. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi:10.1056/NEJMoa1716153
9. Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi:10.1056/NEJMoa1716793
10. Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40(3):242–246. doi:10.1111/j.1399-0004.1991.tb03085.x
11. Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain J Neurol. 1952;75(3):408–427. doi:10.1093/brain/75.3.408
12. Dardiotis E, Koutsou P, Papanicolaou EZ, et al. Epidemiological, clinical and genetic study of familial amyloidotic polyneuropathy in Cyprus. Amyloid. 2009;16(1):32–37. doi:10.1080/13506120802676948
13. Reinés JB, Vera TR, Martín MU, et al. Epidemiology of transthyretin-associated familial amyloid polyneuropathy in the Majorcan area: Son Llàtzer Hospital descriptive study. Orphanet J Rare Dis. 2014;9:29. doi:10.1186/1750-1172-9-29
14. Tsuzuki T, Mita S, Maeda S, Araki S, Shimada K. Structure of the human prealbumin gene. J Biol Chem. 1985;260(22):12224–12227.
15. Koike H, Misu K, Ikeda S, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002;59(11):1771–1776. doi:10.1001/archneur.59.11.1771
16. Parman Y, Adams D, Obici L, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016;29(Suppl 1):S3–S13. doi:10.1097/WCO.0000000000000288
17. Saporta MAC, Zaros C, MW C, et al. Penetrance estimation of TTR familial amyloid polyneuropathy (type I) in Brazilian families. Eur J Neurol. 2009;16(3):337–341. doi:10.1111/j.1468-1331.2008.02429.x
18. Planté-Bordeneuve V, Carayol J, Ferreira A, et al. Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. 2003;40(11):e120. doi:10.1136/jmg.40.11.e120
19. Hellman U, Alarcon F, Lundgren H-E, Suhr OB, Bonaiti-Pellié C, Planté-Bordeneuve V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008;15(3):181–186. doi:10.1080/13506120802193720
20. Soares ML, Coelho T, Sousa A, et al. Haplotypes and DNA sequence variation within and surrounding the transthyretin gene: genotype-phenotype correlations in familial amyloid polyneuropathy (V30M) in Portugal and Sweden. Eur J Hum Genet. 2004;12(3):225–237. doi:10.1038/sj.ejhg.5201095
21. Bonaïti B, Alarcon F, Bonaïti-Pellié C, Planté-Bordeneuve V. Parent-of-origin effect in transthyretin related amyloid polyneuropathy. Amyloid. 2009;16(3):149–150. doi:10.1080/13506120903093944
22. Richardson SJ. Cell and molecular biology of transthyretin and thyroid hormones. Int Rev Cytol. 2007;258:137–193. doi:10.1016/S0074-7696(07)58003-4
23. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. doi:10.1186/1750-1172-8-31
24. Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520–528. doi:10.1093/eurheartj/ehs123
25. Maurer MS, Hanna M, Grogan M, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (transthyretin amyloid outcome survey). J Am Coll Cardiol. 2016;68(2):161–172. doi:10.1016/j.jacc.2016.03.596
26. Yang N-C-C, Lee M-J, Chao -C-C, et al. Clinical presentations and skin denervation in amyloid neuropathy due to transthyretin Ala97Ser. Neurology. 2010;75(6):532–538. doi:10.1212/WNL.0b013e3181ec7fda
27. González-Duarte A, Soto KC, Martínez-Baños D, et al. Familial amyloidosis with polyneuropathy associated with TTR Ser50Arg mutation. Amyloid. 2012;19(4):171–176. doi:10.3109/13506129.2012.712925
28. Sousa A, Andersson R, Drugge U, Holmgren G, Sandgren O. Familial amyloidotic polyneuropathy in Sweden: geographical distribution, age of onset, and prevalence. Hum Hered. 1993;43(5):288–294. doi:10.1159/000154146
29. Coelho T, Sousa A, Lourenço E, Ramalheira J. A study of 159 Portuguese patients with familial amyloidotic polyneuropathy (FAP) whose parents were both unaffected. J Med Genet. 1994;31(4):293–299. doi:10.1136/jmg.31.4.293
30. Suhr OB, Lundgren E, Westermark P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017;281(4):337–347. doi:10.1111/joim.12585
31. Koike H, Tanaka F, Hashimoto R, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry. 2012;83(2):152–158. doi:10.1136/jnnp-2011-301299
32. Mariani -L-L, Lozeron P, Théaudin M, et al. Genotype-phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol. 2015;78(6):901–916. doi:10.1002/ana.24519
33. Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(2):222–228.e1. doi:10.1016/j.ahj.2012.04.015
34. Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799–2806. doi:10.1093/eurheartj/ehx589
35. Théaudin M, Lozeron P, Algalarrondo V, et al. Upper limb onset of hereditary transthyretin amyloidosis is common in non-endemic areas. Eur J Neurol. 2019;26(3):497–e36. doi:10.1111/ene.13845
36. Lobato L, Rocha A. Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol. 2012;7(8):1337–1346. doi:10.2215/CJN.08720811
37. Beirão JM, Malheiro J, Lemos C, Beirão I, Costa P, Torres P. Ophthalmological manifestations in hereditary transthyretin (ATTR V30M) carriers: a review of 513 cases. Amyloid. 2015;22(2):117–122. doi:10.3109/13506129.2015.1015678
38. Martins AC, Rosa AM, Costa E, Tavares C, Quadrado MJ, Murta JN. Ocular manifestations and therapeutic options in patients with familial amyloid polyneuropathy: a systematic review. Biomed Res Int. 2015;2015:282405. doi:10.1155/2015/282405
39. Hagiwara K, Ochi H, Suzuki S, et al. Highly selective leptomeningeal amyloidosis with transthyretin variant Ala25Thr. Neurology. 2009;72(15):1358–1360. doi:10.1212/WNL.0b013e3181a0fe74
40. JL Z, Greicius MD, Zhu W, AN O, CM A, Plowey ED. Neuropathologic analysis of Tyr69His TTR variant meningovascular amyloidosis with dementia. Acta Neuropathol Commun. 2015;3:43. doi:10.1186/s40478-015-0216-0
41. Mitsuhashi S, Yazaki M, Tokuda T, et al. Biochemical characteristics of variant transthyretins causing hereditary leptomeningeal amyloidosis. Amyloid. 2005;12(4):216–225. doi:10.1080/13506120500352404
42. Adams D, Suhr OB, Hund E, et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016;29(Suppl 1):S14–S26. doi:10.1097/WCO.0000000000000289
43. Dohrn MF, Röcken C, De Bleecker JL, et al. Diagnostic hallmarks and pitfalls in late-onset progressive transthyretin-related amyloid-neuropathy. J Neurol. 2013;260(12):3093–3108. doi:10.1007/s00415-013-7124-7
44. Luigetti M, Conte A, Del Grande A, et al. TTR-related amyloid neuropathy: clinical, electrophysiological and pathological findings in 15 unrelated patients. Neurol Sci. 2013;34(7):1057–1063. doi:10.1007/s10072-012-1105-y
45. Lozeron P, Lacroix C, Theaudin M, et al. An amyotrophic lateral sclerosis-like syndrome revealing an amyloid polyneuropathy associated with a novel transthyretin mutation. Amyloid. 2013;20(3):188–192. doi:10.3109/13506129.2013.818535
46. Lozeron P, Mariani -L-L, Dodet P, et al. Transthyretin amyloid polyneuropathies mimicking a demyelinating polyneuropathy. Neurology. 2018;91(2):e143–e152. doi:10.1212/WNL.0000000000005777
47. Cortese A, Vegezzi E, Lozza A, et al. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. 2017;88(5):457–458. doi:10.1136/jnnp-2016-315262
48. Westermark P. Diagnosing amyloidosis. Scand J Rheumatol. 1995;24(6):327–329. doi:10.3109/03009749509095175
49. Do Amaral B, Coelho T, Sousa A, Guimarães A. Usefulness of labial salivary gland biopsy in familial amyloid polyneuropathy Portuguese type. Amyloid. 2009;16(4):232–238. doi:10.3109/13506120903421850
50. Cappellari M, Cavallaro T, Ferrarini M, et al. Variable presentations of TTR-related familial amyloid polyneuropathy in seventeen patients. J Peripher Nerv Syst. 2011;16(2):119–129. doi:10.1111/j.1529-8027.2011.00331.x
51. Koike H, Hashimoto R, Tomita M, et al. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: a practical analysis. Amyloid. 2011;18(2):53–62. doi:10.3109/13506129.2011.565524
52. Koike H, Kawagashira Y, Iijima M, et al. Electrophysiological features of late-onset transthyretin Met30 familial amyloid polyneuropathy unrelated to endemic foci. J Neurol. 2008;255(10):1526–1533. doi:10.1007/s00415-008-0962-z
53. Castro J, Miranda B, Castro I, de Carvalho M, Conceição I. The diagnostic accuracy of Sudoscan in transthyretin familial amyloid polyneuropathy. Clin Neurophysiol. 2016;127(5):2222–2227. doi:10.1016/j.clinph.2016.02.013
54. Lefaucheur J-P, Zouari HG, Gorram F, Nordine T, Damy T, Planté-Bordeneuve V. The value of electrochemical skin conductance measurement using Sudoscan® in the assessment of patients with familial amyloid polyneuropathy. Clin Neurophysiol. 2018;129(8):1565–1569. doi:10.1016/j.clinph.2018.05.005
55. Luigetti M, Bisogni G, Romano A, et al. Sudoscan in the evaluation and follow-up of patients and carriers with TTR mutations: experience from an Italian Centre. Amyloid. 2018;25(4):242–246. doi:10.1080/13506129.2018.1545640
56. Niklasson U, Olofsson BO, Bjerle P. Autonomic neuropathy in familial amyloidotic polyneuropathy. A clinical study based on heart rate variability. Acta Neurol Scand. 1989;79(3):182–187. doi:10.1111/j.1600-0404.1989.tb03736.x
57. Buxbaum JN, Ruberg FL. Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med off J Am Coll Med Genet. 2017;19(7):733–742. doi:10.1038/gim.2016.200
58. Sattianayagam PT, Hahn AF, Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33(9):1120–1127. doi:10.1093/eurheartj/ehr383
59. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–2412. doi:10.1161/CIRCULATIONAHA.116.021612
60. Musumeci MB, Cappelli F, Russo D, et al. Low sensitivity of bone scintigraphy in detecting Phe64Leu mutation-related transthyretin cardiac amyloidosis. JACC Cardiovasc Imaging. 2019. doi:10.1016/j.jcmg.2019.10.015
61. Lehrke S, Steen H, Kristen AV, et al. Serum levels of NT-proBNP as surrogate for cardiac amyloid burden: new evidence from gadolinium-enhanced cardiac magnetic resonance imaging in patients with amyloidosis. Amyloid. 2009;16(4):187–195. doi:10.3109/13506120903421538
62. Coll E, Botey A, Alvarez L, et al. Serum cystatin C as a new marker for noninvasive estimation of glomerular filtration rate and as a marker for early renal impairment. Am J Kidney Dis. 2000;36(1):29–34. doi:10.1053/ajkd.2000.8237
63. Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med. 1994;235(5):479–485. doi:10.1111/j.1365-2796.1994.tb01106.x
64. Coutinho PM, Lázaro da Silva A, Lopes J, et al. Forty years of experience with type I amyloid neuropathy. Review of 483 cases. In: editors, Glenner GG, Pinho e Costa P, Falcao de Freitas A. Amyloid and Amyloidosis. Amsterdam: Excerpta Medica; 1980:88–98. Available from: https://www.scienceopen.com/document?vid=3a81c019-a30c-4c5a-9d7a-2db26a111dd2.
65. Dyck PJ, Davies JL, Litchy WJ, O’Brien PC. Longitudinal assessment of diabetic polyneuropathy using a composite score in the Rochester Diabetic Neuropathy Study cohort. Neurology. 1997;49(1):229–239. doi:10.1212/wnl.49.1.229
66. Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–2667. doi:10.1001/jama.2013.283815
67. Dyck PJB, González-Duarte A, Obici L, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS + 7. J Neurol Sci. 2019;405:116424. doi:10.1016/j.jns.2019.116424
68. Suarez GA, Opfer-Gehrking TL, Offord KP, Atkinson EJ, O’Brien PC, Low PA. The autonomic symptom profile: a new instrument to assess autonomic symptoms. Neurology. 1999;52(3):523–528. doi:10.1212/wnl.52.3.523
69. Denier C, Ducot B, Husson H, et al. A brief compound test for assessment of autonomic and sensory-motor dysfunction in familial amyloid polyneuropathy. J Neurol. 2007;254(12):1684–1688. doi:10.1007/s00415-007-0617-5
70. van Nes SI, Vanhoutte EK, van Doorn PA, et al. Rasch-built overall disability scale (R-ODS) for immune-mediated peripheral neuropathies. Neurology. 2011;76(4):337–345. doi:10.1212/WNL.0b013e318208824b
71. Vinik EJ, Hayes RP, Oglesby A, et al. The development and validation of the Norfolk QOL-DN, a new measure of patients’ perception of the effects of diabetes and diabetic neuropathy. Diabetes Technol Ther. 2005;7(3):497–508. doi:10.1089/dia.2005.7.497
72. Adams D, Suhr OB, Dyck PJ, et al. Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 2017;17(1):181. doi:10.1186/s12883-017-0948-5
73. Conceição I, Damy T, Romero M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019;26(1):3–9. doi:10.1080/13506129.2018.1556156
74. Obici L, Kuks JB, Buades J, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29(Suppl 1):S27–S35. doi:10.1097/WCO.0000000000000290
75. Wilczek HE, Larsson M, Ericzon B-G, FAPWTR. Long-term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR). Amyloid. 2011;18(Suppl 1):193–195. doi:10.3109/13506129.2011.574354072
76. Ericzon B-G, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–1854. doi:10.1097/TP.0000000000000574
77. Saelices L, Chung K, Lee JH, et al. Amyloid seeding of transthyretin by ex vivo cardiac fibrils and its inhibition. Proc Natl Acad Sci U S A. 2018;115(29):E6741–E6750. doi:10.1073/pnas.1805131115
78. Saelices L, Nguyen BA, Chung K, et al. A pair of peptides inhibits seeding of the hormone transporter transthyretin into amyloid fibrils. J Biol Chem. 2019;294(15):6130–6141. doi:10.1074/jbc.RA118.005257
79. Hawkins PN, Ando Y, Dispenzeri A, Gonzalez-Duarte A, Adams D, Suhr OB. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–638. doi:10.3109/07853890.2015.1068949
80. Said G, Grippon S, Kirkpatrick P. Tafamidis. Nat Rev Drug Discov. 2012;11(3):185–186. doi:10.1038/nrd3675
81. Lozeron P, Théaudin M, Mincheva Z, et al. Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur J Neurol. 2013;20(12):1539–1545. doi:10.1111/ene.12225
82. Cortese A, Vita G, Luigetti M, et al. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J Neurol. 2016;263(5):916–924. doi:10.1007/s00415-016-8064-9
83. Planté-Bordeneuve V, Gorram F, Salhi H, et al. Long-term treatment of transthyretin familial amyloid polyneuropathy with tafamidis: a clinical and neurophysiological study. J Neurol. 2017;264(2):268–276. doi:10.1007/s00415-016-8337-3
84. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
85. Akinc A, Querbes W, De S, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther J Am Soc Gene Ther. 2010;18(7):1357–1364. doi:10.1038/mt.2010.85
86. Butler JS, Chan A, Costelha S, et al. Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid. 2016;23(2):109–118. doi:10.3109/13506129.2016.1160882
87. Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. doi:10.1056/NEJMoa1208760
88. Nair JK, Willoughby JLS, Chan A, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136(49):16958–16961. doi:10.1021/ja505986a
89. Ackermann EJ, Guo S, Benson MD, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid. 2016;23(3):148–157. doi:10.1080/13506129.2016.1191458
90. Ferreira N, Saraiva MJ, Almeida MR. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: “in vivo” evidence from FAP mice models. PLoS One. 2012;7(1):e29933. doi:10.1371/journal.pone.0029933
91. Higaki JN, Chakrabartty A, Galant NJ, et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid. 2016;23(2):86–97. doi:10.3109/13506129.2016.1148025
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.