")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Diagnosis and Screening of Patients with Fabry Disease
Authors Vardarli I, Rischpler C , Herrmann K, Weidemann F
Received 31 January 2020
Accepted for publication 9 May 2020
Published 22 June 2020 Volume 2020:16 Pages 551—558
DOI https://doi.org/10.2147/TCRM.S247814
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Irfan Vardarli,1,2 Christoph Rischpler,3 Ken Herrmann,3 Frank Weidemann1,2
1Department of Medicine I, Klinikum Vest, Knappschaftskrankenhaus Recklinghausen, Academic Teaching Hospital, Ruhr-University Bochum, Recklinghausen, Germany; 2Herz- Und Gefäßzentrum Klinikum Vest, Recklinghausen, Germany; 3Department of Nuclear Medicine, University Hospital Essen, Essen, Germany
Correspondence: Irfan Vardarli
Department of Medicine I, Klinikum Vest, Knappschaftskrankenhaus Recklinghausen, Academic Teaching Hospital, Ruhr-University Bochum, Dorstener Str. 151, Recklinghausen 45657, Germany
Tel +49 2361 563406
Fax +49 2361 563498
Email [email protected]
Abstract: Fabry disease (FD) is an X-linked lysosomal storage disorder caused by absence or deficient activity of α-galactosidase A (α-Gal A) due to mutations in the α-galactosidase A gene (GLA), leading to progressive accumulation of globotriaosylceramide (Gb3) in tissues and organs including heart, kidney, the eyes, vascular endothelium, the nervous system and the skin. Cardiac involvement is leading to fatal complications and reduced life expectancy. FD is treatable with disease-specific treatment (enzyme replacement therapy (ERT) or with chaperone therapy). Therefore, the early diagnosis of FD is crucial for reducing the morbidity and mortality. Screening of high-risk populations (eg, patients with unexplained left ventricular hypertrophy (LVH), young patients with unexplained stroke, and patients with unexplained renal failure proteinuria or microalbuminuria) yields good results. The diagnostic algorithm is gender-specific. Initially, the measurement of α-Gal A activity is recommended in males, and optionally in females. In males with non-diagnostic residual activity (5– 10%) activity, genetic testing is afterwards done for confirming the diagnosis. In fact, diagnosis of FD is not possible without genetic testing for both males and females. Globotriaosysphingosine (lyso-Gb3) for identification of atypical FD variants and high- sensitive troponin T (hsTNT) for identification of cardiac involvement are also important diagnostic biomarkers. The aim of this review was to provide an update on diagnosis and screening of patients with FD.
Keywords: metabolic disease, genetics, hypertrophic cardiomyopathy, proteinuria, algorithm, heart failure
Introduction
Fabry disease (FD) is an X-linked rare multisystemic lysosomal storage disorder resulting from α-galactosidase A (α-Gal A) deficiency and is disease-specific, but not curatively treatable with enzyme replacement therapy (ERT) since 2001 and chaperone therapy with Migalastat (Galafold®, Amicus Therapeutics, Cranbury, NJ, USA) since 2016.1–4 The recombinant enzyme is available as agalsidase-alpha (Replagal®, Shire Human Genetic Therapies AB, Stockholm, Sweden) and agalsidase-beta (Fabrazyme®, Sanofi Genzyme, Cambridge, MA, USA).5 However, early diagnosis of FD is crucial to reduce morbidity and mortality. Cardiovascular complications are the major cause of death in patients with FD.6,7
Epidemiology and Pathogenesis of Fabry Disease
The prevalence of FD has been estimated between 1:40,000 and 1:117,000 individuals.8 However, this prevalence is underestimated. In newborn screenings, higher prevalence values were described, for example in Spain, where the prevalence of FD in males was estimated at 1:7575 (0.013%).9 In genetic screening studies of high-risk populations, the overall prevalence of individuals with α-galactosidase A (GLA) gene variants, including genetic variants of unknown significance (GVUS), was 0.62%; prevalence of definitive FD diagnosis was 0.12%.10 In patients (male and female) receiving hemodialysis treatment in Brazil, the estimated prevalence for FD was 0.87%, close to that reported in other trials.11–14 In young patients with stroke a prevalence of 0–4% was found.15–19 In patients with unexplained hypertrophic cardiomyopathy (HCM) FD should be considered. In two cohorts of men with unexplained LVH (≥ 13mm) the prevalence of decreased α-gal A activity was 6.3% and 3.0%, respectively.20,21 In a cohort of females with unexplained HCM, up to 12% of these patients were affected by FD.22 In large cohorts of patients with unexplained LVH, α-gal A mutations were described in 0.5% and 1.0%, respectively.23,24 Doheny et al reanalyzed all published screening studies for FD from 1995 through 2017 and provided more valid prevalence estimates of previously unrecognized FD in high-risk cohorts, ranging from 0% in female renal transplant patients to about 0.9% among both male and female cardiac cohorts.25 Previous prevalence values from 1.6-fold in male renal transplant patients to 33-fold and 15-fold in male and female patients with stroke, respectively, may be due to benign variants.25 Capuano et al reanalyzed the prevalence of GLA mutations in dialysis patients in FD screening studies published between 1995 and 2019 after assigning their correct phenotype. They found an overall prevalence recalculated on the basis of only pathogenetic mutations of 0.14%.26
FD is caused by a deficiency or absence of α-Gal A activity due to mutations in the GLA gene (located on Xq22.1), resulting in intracellular accumulation of globotriaosylceramide (Gb3) and multiorgan damage.27,28 Heterozygous female may have clinical manifestations of FD, too, but usually on average 10 years later than male patients. Phenotypic expression and penetrance varies among families with the same variant and, also, within the same family. There are various phenotypes, with early severe classic presentations, and the later, milder presentation.28 Two phenotypes of FD are relevant: classic FD and variant FD. Males with the classic FD develop early signs and symptoms in childhood or adolescence (e.g., periodic crisis of severe pain in the extremities (acroparesthesias), neuropathic pain, telangiectasias and vascular lesions called angiokeratomas (in the groin, hip and periumbilical areas), sweating abnormalities (anhidrosis and hypohidrosis), gastrointestinal symptoms, corneal (cornea verticillate) and lenticular opacities).29–31 Vascular complications, renal failure with unknown etiology, including unexplained proteinuria or microalbuminuria, unexplained LVH mimicking sarcomeric HCM, or neurological manifestations (e.g., cryptogenic stroke, transient ischemic attack [TIA], sensoneuronal hearing loss, migraine) may be clinical features in adults.10,30,32–34 Patients with atypical variants may develop late-onset renal failure, LVH, cerebrovascular disease, or a combination thereof.30,35 Symptoms in female subjects vary. Presentation ranges from asymptomatic to severely affected, but subjects are typically older compared to male subjects.30,34–36 Gb3-deposition and its deacylated metabolite globotriaosysphingosine (lyso-Gb3) are associated with the cellular involvement.37 This may lead to alterations of tissue (interstitial fibrosis and replacement fibrosis, inflammation, apoptosis, hypertrophy).38 The cellular damage is multifactorial and partly caused by cellular signaling or microvascular ischemia via affected endothelial cells or both, leading to organ dysfunction, renal and heart failure, for example.39 Plasma lyso-Gb3 concentrations appear to correlate with disease stages, the mutation severity and decreasing during ERT.37 There is still debate about which patients or variants really do show correlation of LysoGb3 levels as treatment response (or both ERT and chaperone).5,40
Diagnosis of Fabry Disease
Diagnosis of classic FD in males may be straightforward, whereas in females and in individuals with genetic variants the diagnosis can be challenging.41
A diagnostic approach involving a detailed history, family history, physical examination, clinical and biochemical findings, genetic testing, various imaging procedures, and expert opinion is recommended.10,42,43 Some clinical findings and signs, e.g., acroparesthesia, angiokeratomas and cornea verticillate may have a high specificity.10
In males suspected to have FD, α-Gal A activity should be measured. Alpha Gal A activity < 1% is highly suggestive for the diagnosis of classic FD.44 Even in the presence of clinical signs, the α-Gal A activity is variable and can be within the normal range in females.45,46 Most of these females develop clinically significant disease.27,47
Genetic confirmatory testing is mandatory in males and females.10 Mutations in the GLA gene may be associated with the classic FD, a variant phenotype or a GVUS.10 We present an updated diagnostic algorithm in Figure 1.10,27,31,43,48,49 At the diagnosis of FD, determination of amenability for pharmacological chaperone therapy with migalastat is possible with good laboratory practice (GLP)-validated human embryogenic kidney cell-based in vitro assay.50,51 This in vitro assay is currently the only approved method for this evaluation.52
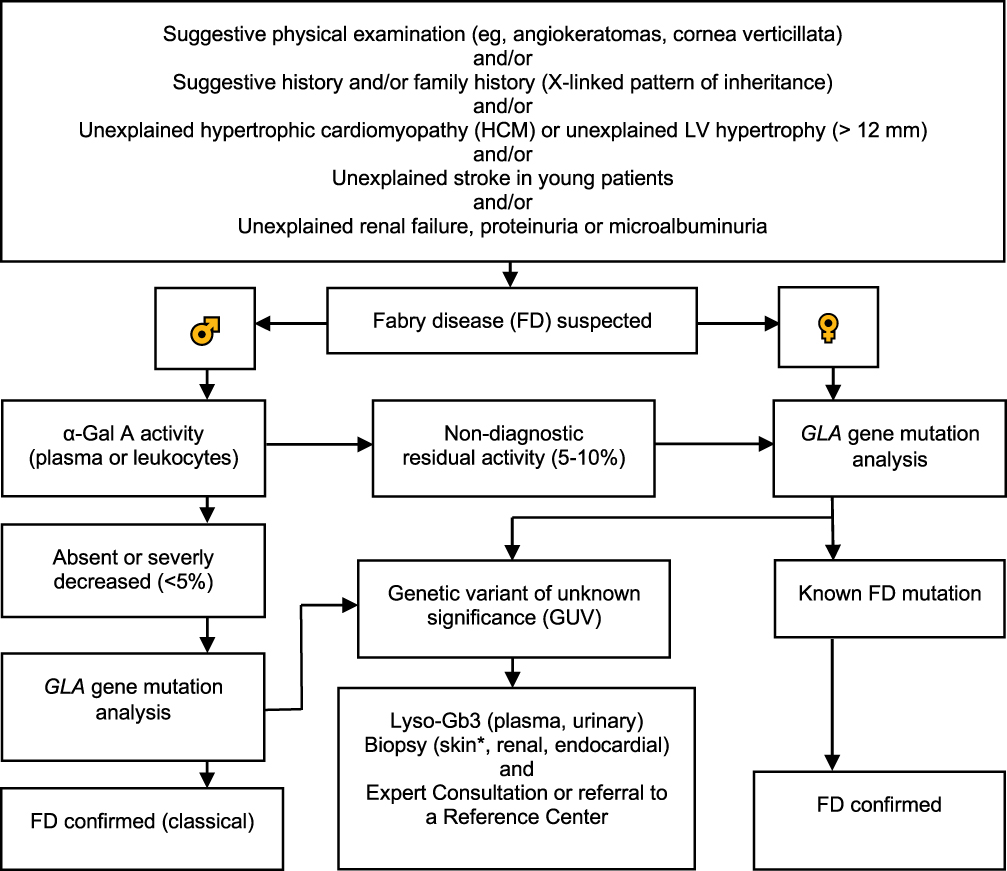 |
Figure 1 Updated diagnostic algorithm for FD. Adapted with permission from van der Tol L, Smid BE, Poorthuis BJ, et al. A systematic review on screening for Fabry disease: prevalence of individuals with genetic variants of unknown significance. J Med Genet. 2014;51:1–9. Copyright © 2014, BMJ Publishing Group Ltd.10 Adapted with permission from Yogasundaram H, Kim D, Oudit O, Thompson RB, Weidemann F, Oudit GY. Clinical Features, Diagnosis, and Management of Patients With Anderson-Fabry Cardiomyopathy. Can J Cardiol. 2017;33:883–897. © 2017 Canadian Cardiovascular Society.27 Adapted with permission from Putko BN, Wen K, Thompson RB, et al. Anderson-Fabry cardiomyopathy: prevalence, pathophysiology, diagnosis and treatment. Heart Fail Rev. 2015;20:179–191. Copyright © 2014, Springer Nature.49 Adapted with permission from Laney DA, Bennett RL, Clarke V, et al. Fabry disease practice guidelines: recommendations of the National Society of Genetic Counselors. J Genet Couns. 2013;22:555–564. © 2013 National Society of Genetic Counselors, Inc.43 Data from Niemann et al 48 and Putko et al.49 α-Galactosidase A (α-Gal A), α-galactosidase A gene (GLA gene), globotriaosysphingosine (Lyso-Gb3), left ventricular (LV). *Punch biopsy, taken from proximal (thigh: 15 cm above the patella) and distal (leg: 10 cm above the lateral malleolus) hairy skin sites or from other lesions to evaluate Gb3 deposits, and for other histopathological evaluations.31 |
Lyso-Gb3 has also clinical relevance and emerged a powerful biomarker for FD, particularly the variable α-Gal A activity and mutations.37,48 In patients with GVUS, to determine if the mutation is possibly clinically significant, and if the diagnosis of FD should be made, the lyso-Gb3 test can be used. Lyso-Gb3 can be used for stratifying patients as classic and variant phenotypes, even before clinical manifestation is present.48 Lyso-Gb3 is especially helpful as a phenotype discriminative marker because of phenotypic variability, most prominently in females.42
Diagnosis of Cardiac Involvement
The first step to assess the Fabry cardiomyopathy is transthoracic echocardiography.53,54 Typical signs for FD are concentric LVH and a prominent papillary muscle. In addition, most patients show signs of diastolic dysfunction. Global left ventricular function assessed by ejection fraction (EF) during echocardiography is only reduced in end-stage patients.53,54
Patients with FD suspected to have cardiac involvement should undergo cardiac magnetic resonance imaging (CMR) imaging, echocardiography, a 24-h Holter ECG to assess cardiac structure and function, as early as possible.55
The diagnostic gold standard for cardiac fibrosis in cardiomyopathy patients with FD is CMR imaging examination with late gadolinium enhancement (LGE-CMR).55 In cases of patients with contraindications, e.g., in patients with an implanted cardioverter or pacemaker, or with end-stage renal disease, echocardiographic modalities (e.g., speckle-tracking echocardiography [STE]), are available for indirect evaluation of cardiac involvement.56,57 Starting ERT is recommended as soon as clinical signs of organ involvement are seen, e.g., when detection of LGE first becomes apparent.42,55 T1-MRI mapping has been proposed to discriminate FD from other causes of LVH.58 In FD patients with LVH, abnormal native myocardial T1 values are highly prevalent.59,60 Cardiac MRI-derived myocardial mapping may have a high sensitivity and specificity to discriminate FD patients.58,59 Nappi et al showed the feasibility of PET-MR imaging for the early detection of cardiac involvement, even in patients with non-hypertrophic stage.61 PET-MR examinations can provide unique and simultaneous results to detect myocardial inflammation.58
In cardiology, high-sensitive Troponin T (hsTNT) is established to detect myocardial tissue damage. However, for Fabry cardiomyopathy only few data are available regarding biomarkers to detect cardiac involvement.
Seydelmann et al showed, that hsTNT is the most promising biomarker for cardiac involvement in FD, with Values >14ng/mL, suggesting pathological late enhancement in MRI.62 This biomarker was correlated with the progression of FD, and patients with elevation of hsTNT during observation showed the progress of myocardial fibrosis, and a decrease of EF.62 Elevation of hsTNT can predict an increment in the magnitude of fibrosis.63
In patients with FD and cardiac involvement, assessed by echocardiographic findings, NT-proBNP levels are elevated and correlate with electrocardiographic findings, but not with myocardial fibrosis.62,64 However, NT-proBNP levels are not correlated with disease progression.64,65 The evaluation of myocardial fibrosis defines the stage of the cardiomyopathy and is associated with a poor prognosis.53,66
The noninvasive biochemical markers, imaging strategies and genetic tests for the diagnosis of FD are sufficient, therefore the place value for endocardial biopsy has diminished.27
Diagnosis of Renal Involvement
General diagnostic tools for the baseline assessment of renal function, e.g., serum creatinine, calculated GFR and urine protein diagnostics (24 hour collection would be ideal, but spot urine total protein/creatinine and albumin/creatinine ratios has been established) can be used initially in FD patients.67 Additionally, the creatinine and cystatin C based GFR-calculation is recommended for the assessment of renal function.63 Serum cystatin C concentration is a more sensitive marker than creatinine to detect early renal dysfunction.65 Urine lyso-GB3 showed no correlation to renal involvement in FD.68,69 Proteinuria and albuminuria is the most important indicator for early renal involvement in FD; regular measurement is essential.70 Measurement of albumin-creatinine ratio in spot urine is highly recommended for early detection of microalbuminuria. Glomerular hyperfiltration (age-corrected eGFR > 130 mL/min/1.73/m2 corrected for age) may be an early marker for renal involvement.69 Furthermore, a kidney ultrasound should be carried out.71
In FD patients with persistent albuminuria and/or proteinuria a renal biopsy should be performed.72
Diagnosis of Neurologic Involvement
Unfortunately there are no specific blood biomarkers available for the assessment of neurological signs in FD patients. In cases of suggestive physical examination and/or history (and/or) family history (e.g., paresthesias/dysesthesias, hypohidrosis/anhidrosis, chronic burning pain, attacks of excruciating pain, sensory losses, tinnitus, hearing loss, nausea, dizziness, abdominal cramp, [post-prandial] diarrhea, bloating) further neurological diagnostics should be initiated.73–75 Neurological involvement is usually assessed by brain MRI, audiometry, etc. White matter hyperintensities, dolichoectasia, and infarcts are characteristic signs found on brain MR imaging in classic FD.76,77
Screening Strategies for Fabry Disease
In epidemiology, screening is defined
as the examination of asymptomatic people in order to classify them as likely or unlikely to have the disease that is the object of screening. People who appear likely to have the disease are investigated further to arrive at a final diagnosis. Those people who are then found to have the disease are treated.78
To be suitable for general use, a procedure for early detection and treatment should meet many criteria in addition to reducing morbidity or mortality.78 For cost-effectiveness, a high positive predictive value (PVP) is required in the population screened.78 Therefore, screening for FD is currently only recommended in high-risk populations and in families of index patients.11,79 Due to various reasons, systematic newborn screening for FD was not introduced.9 Several screening strategies are possible. For males, initially, biomarker-based screening methods (α-Gal A or lyso-Gb3) are used.49,80 Enzyme-based screening methods are not appropriate for females.19 Vascular lesions, especially the retinal vessel diameter can help during screening for FD.81
High-Risk Screening
Screening of the following high-risk populations could increase the diagnostic rate of FD:82 1) Patients with unexplained HCM or with unexplained LVH (>12 mm);20,23 2) Patients with end-stage renal disease (receiving hemodialysis treatment), patients after kidney transplantation or patients with unexplained proteinuria or microalbuminuria;80 3) Individuals (aged 15–55 years) with unexplained stroke.15,16,18,19
Many screening studies in high-risk populations revealed an unexpectedly large number of mutations or GVUS in the galactosidase alpha gene (GLA) and/or impaired α-gal A activity.10 In contrast to the absent or near absent α-gal A activity in classically affected males, most male patients with GVUS have residual activity of α-gal A.10
The diagnostic algorithm (Figure 1) may also be adapted for screening for FD.
Family Screening
Pedigree analysis and genetic counseling is crucial for patients with FD, relatives may need to be genetically tested.43
Conclusion
FD is a rare, progressive multi-system disease with a reduction in life expectancy. Therefore, early diagnosis of FD is essential, using diagnostic and screening procedures adapted to consensus guidelines, which we present in this review.
Disclosure
Dr. Vardarli and Dr. Rischpler report no conflicts of interest in this work. Dr. Herrmann reports personal fees from Bayer, stock options (less than 1%) from Sofie Biosciences, personal fees from SIRTEX, non-financial support from ABX, personal fees from Adacap, personal fees from Curium, personal fees from Endocyte, grants and personal fees from BTG, personal fees from IPSEN, personal fees from Siemens Healthineers, personal fees from GE Healthcare, personal fees from Amgen, personal fees from Novartis, personal fees from Y-mAbs, outside the submitted work. Dr. Weidemann has received research grants from Genzyme and Shire and speaker honoraria from Amicus, Genzyme, and Shire.
References
1. Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276(21):1163–1167. doi:10.1056/NEJM196705252762101
2. Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138:338–346. doi:10.7326/0003-4819-138-4-200302180-00014
3. Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16. doi:10.1056/NEJM200107053450102
4. Kramer J, Lenders M, Canaan-Kuhl S, et al. Fabry disease under enzyme replacement therapy-new insights in efficacy of different dosages. Nephrol Dial Transplant. 2018;33:1362–1372. doi:10.1093/ndt/gfx319
5. Muntze J, Gensler D, Maniuc O, et al. Oral chaperone therapy migalastat for treating fabry disease: enzymatic response and serum biomarker changes after 1 year. Clin Pharmacol Ther. 2019;105:1224–1233. doi:10.1002/cpt.1321
6. Mehta A, Clarke JT, Giugliani R, et al. Natural course of Fabry disease: changing pattern of causes of death in FOS - fabry outcome survey. J Med Genet. 2009;46:548–552. doi:10.1136/jmg.2008.065904
7. Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry registry. Genet Med. 2009;11:790–796. doi:10.1097/GIM.0b013e3181bb05bb
8. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi:10.1001/jama.281.3.249
9. Colon C, Ortolano S, Melcon-Crespo C, et al. Newborn screening for Fabry disease in the north-west of Spain. Eur J Pediatr. 2017;176:1075–1081. doi:10.1007/s00431-017-2950-8
10. van der Tol L, Smid BE, Poorthuis BJ, et al. A systematic review on screening for Fabry disease: prevalence of individuals with genetic variants of unknown significance. J Med Genet. 2014;51(1):1–9. doi:10.1136/jmedgenet-2013-101857
11. Sodre LSS, Huaira R, Bastos MG, Colugnati FAB, Coutinho MP, Fernandes N. Screening for fabry disease in kidney disease: a cross-sectional study in males and females. Kidney Blood Press Res. 2017;42(6):1258–1265. doi:10.1159/000485929
12. Saito O, Kusano E, Akimoto T, et al. Prevalence of Fabry disease in dialysis patients: japan Fabry disease screening study (J-FAST). Clin Exp Nephrol. 2016;20(2):284–293. doi:10.1007/s10157-015-1146-7
13. Herrera J, Miranda CS. Prevalence of Fabry’s disease within hemodialysis patients in Spain. Clin Nephrol. 2014;81:112–120. doi:10.5414/CN108053
14. Kleinert J, Kotanko P, Spada M, et al. Anderson-Fabry disease: a case-finding study among male kidney transplant recipients in Austria. Transpl Int. 2009;22:287–292. doi:10.1111/j.1432-2277.2008.00791.x
15. Rolfs A, Bottcher T, Zschiesche M, et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. 2005;366(9499):1794–1796. doi:10.1016/S0140-6736(05)67635-0
16. Brouns R, Sheorajpanday R, Braxel E, et al. Middelheim Fabry Study (MiFaS): a retrospective Belgian study on the prevalence of Fabry disease in young patients with cryptogenic stroke. Clin Neurol Neurosurg. 2007;109:479–484. doi:10.1016/j.clineuro.2007.03.008
17. Lanthier S, Saposnik G, Lebovic G, et al. Prevalence of fabry disease and outcomes in young canadian patients with cryptogenic ischemic cerebrovascular events. Stroke. 2017;48:1766–1772. doi:10.1161/STROKEAHA.116.016083
18. Kinoshita N, Hosomi N, Matsushima H, et al. Screening for fabry disease in japanese patients with young-onset stroke by measuring alpha-galactosidase a and globotriaosylsphingosine. J Stroke Cerebrovasc Dis. 2018;27:3563–3569. doi:10.1016/j.jstrokecerebrovasdis.2018.08.025
19. Lee TH, Yang JT, Lee JD, et al. Genomic screening of Fabry disease in young stroke patients: the Taiwan experience and a review of the literature. Eur J Neurol. 2019;26:553–555. doi:10.1111/ene.13775
20. Sachdev B, Takenaka T, Teraguchi H, et al. Prevalence of anderson-fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105:1407–1411. doi:10.1161/01.CIR.0000012626.81324.38
21. Nakao S, Takenaka T, Maeda M, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288–293. doi:10.1056/NEJM199508033330504
22. Chimenti C, Pieroni M, Morgante E, et al. Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation. 2004;110:1047–1053. doi:10.1161/01.CIR.0000139847.74101.03
23. Elliott P, Baker R, Pasquale F, et al. Prevalence of anderson-fabry disease in patients with hypertrophic cardiomyopathy: the European anderson-fabry disease survey. Heart. 2011;97:1957–1960. doi:10.1136/heartjnl-2011-300364
24. Monserrat L, Gimeno-Blanes JR, Marin F, et al. Prevalence of fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50:2399–2403. doi:10.1016/j.jacc.2007.06.062
25. Doheny D, Srinivasan R, Pagant S, Chen B, Yasuda M, Desnick RJ. Fabry disease: prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J Med Genet. 2018;55:261–268. doi:10.1136/jmedgenet-2017-105080
26. Capuano I, Garofalo C, Buonanno P, et al. Identifying Fabry patients in dialysis population: prevalence of GLA mutations by renal clinic screening, 1995–2019. J Nephrol. 2019. doi:10.1007/s40620-019-00663-6.
27. Yogasundaram H, Kim D, Oudit O, Thompson RB, Weidemann F, Oudit GY. Clinical features, diagnosis, and management of patients with anderson-fabry cardiomyopathy. Can J Cardiol. 2017;33:883–897. doi:10.1016/j.cjca.2017.04.015
28. Favalli V, Disabella E, Molinaro M, et al. Genetic screening of anderson-fabry disease in probands referred from multispecialty clinics. J Am Coll Cardiol. 2016;68:1037–1050. doi:10.1016/j.jacc.2016.05.090
29. Laney DA, Peck DS, Atherton AM, et al. Fabry disease in infancy and early childhood: a systematic literature review. Genet Med. 2015;17:323–330. doi:10.1038/gim.2014.120
30. Mehta A, Hughes DA. Fabry Disease. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. Genereviews(R) [Internet]. Seattle (WA). August 5, 2002;1993–2019. Available from: http:www.ncbi.nlm.nih.gov/books/NBK1292/.
31. Liguori R, Incensi A, de Pasqua S, et al. Skin globotriaosylceramide 3 deposits are specific to Fabry disease with classical mutations and associated with small fibre neuropathy. PLoS One. 2017;12:e0180581. doi:10.1371/journal.pone.0180581
32. Wozniak MA, Kittner SJ, Tuhrim S, et al. Frequency of unrecognized Fabry disease among young European-American and African-American men with first ischemic stroke. Stroke. 2010;41:78–81. doi:10.1161/STROKEAHA.109.558320
33. van der Tol L, Svarstad E, Ortiz A, et al. Chronic kidney disease and an uncertain diagnosis of Fabry disease: approach to a correct diagnosis. Mol Genet Metab. 2015;114:242–247. doi:10.1016/j.ymgme.2014.08.007
34. Nagueh SF. Anderson-Fabry disease and other lysosomal storage disorders. Circulation. 2014;130:1081–1090. doi:10.1161/CIRCULATIONAHA.114.009789
35. El-Abassi R, Singhal D, England JD. Fabry’s disease. J Neurol Sci. 2014;344:5–19. doi:10.1016/j.jns.2014.06.029
36. Gambarin FI, Disabella E, Narula J, et al. When should cardiologists suspect Anderson-Fabry disease? Am J Cardiol. 2010;106:1492–1499. doi:10.1016/j.amjcard.2010.07.016
37. Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008;105:2812–2817. doi:10.1073/pnas.0712309105
38. Moon JC, Sachdev B, Elkington AG, et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J. 2003;24:2151–2155. doi:10.1016/j.ehj.2003.09.017
39. Pastores GM, Hughes DA. To see a world in a grain of sand: elucidating the pathophysiology of Anderson-Fabry disease through investigations of a cellular model. Kidney Int. 2009;75:351–353. doi:10.1038/ki.2008.606
40. Germain DP, Elliott PM, Falissard B, et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: a systematic literature review by a European panel of experts. Mol Genet Metab Rep. 2019;19:100454. doi:10.1016/j.ymgmr.2019.100454
41. Schiffmann R, Fuller M, Clarke LA, Aerts JM. Is it Fabry disease? Genet Med. 2016;18:1181–1185. doi:10.1038/gim.2016.55
42. Smid BE, van der Tol L, Cecchi F, et al. Uncertain diagnosis of Fabry disease: consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int J Cardiol. 2014;177:400–408. doi:10.1016/j.ijcard.2014.09.001
43. Laney DA, Bennett RL, Clarke V, et al. Fabry disease practice guidelines: recommendations of the national society of genetic counselors. J Genet Couns. 2013;22:555–564. doi:10.1007/s10897-013-9613-3
44. Clarke JT. Narrative review: fabry disease. Ann Intern Med. 2007;146:425–433. doi:10.7326/0003-4819-146-6-200703200-00007
45. Havndrup O, Christiansen M, Stoevring B, et al. Fabry disease mimicking hypertrophic cardiomyopathy: genetic screening needed for establishing the diagnosis in women. Eur J Heart Fail. 2010;12:535–540. doi:10.1093/eurjhf/hfq073
46. Linthorst GE, Bouwman MG, Wijburg FA, Aerts JM, Poorthuis BJ, Hollak CE. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet. 2010;47:217–222. doi:10.1136/jmg.2009.072116
47. Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. 2007;9:34–45. doi:10.1097/GIM.0b013e31802d8321
48. Niemann M, Rolfs A, Stork S, et al. Gene mutations versus clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet. 2014;7:8–16. doi:10.1161/CIRCGENETICS.113.000249
49. Putko BN, Wen K, Thompson RB, et al. Anderson-Fabry cardiomyopathy: prevalence, pathophysiology, diagnosis and treatment. Heart Fail Rev. 2015;20:179–191. doi:10.1007/s10741-014-9452-9
50. Benjamin ER, Della Valle MC, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19:430–438. doi:10.1038/gim.2016.122
51. Schiffmann R, Bichet DG, Benjamin E, Wu X, Giugliani R. The migalastat GLP-HEK assay is the gold standard for determining amenability in patients with Fabry disease. Mol Genet Metab Rep. 2019;20:100494. doi:10.1016/j.ymgmr.2019.100494
52. Lukas J, Cimmaruta C, Liguori L, et al. Assessment of gene variant amenability for pharmacological chaperone therapy with 1-deoxygalactonojirimycin in fabry disease. Int J Mol Sci. 2020;21(3):956.
53. Weidemann F, Breunig F, Beer M, et al. The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease. Eur Heart J. 2005;26:1221–1227. doi:10.1093/eurheartj/ehi143
54. Linhart A, Kampmann C, Zamorano JL, et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28:1228–1235. doi:10.1093/eurheartj/ehm153
55. Weidemann F, Beer M, Kralewski M, Siwy J, Kampmann C. Early detection of organ involvement in Fabry disease by biomarker assessment in conjunction with LGE cardiac MRI: results from the SOPHIA study. Mol Genet Metab. 2019;126:169–182. doi:10.1016/j.ymgme.2018.11.005
56. Pieroni M, Chimenti C, Ricci R, Sale P, Russo MA, Frustaci A. Early detection of Fabry cardiomyopathy by tissue Doppler imaging. Circulation. 2003;107:1978–1984. doi:10.1161/01.CIR.0000061952.27445.A0
57. Kramer J, Niemann M, Liu D, et al. Two-dimensional speckle tracking as a non-invasive tool for identification of myocardial fibrosis in Fabry disease. Eur Heart J. 2013;34:1587–1596. doi:10.1093/eurheartj/eht098
58. Imbriaco M, Nappi C, Ponsiglione A, et al. Hybrid positron emission tomography-magnetic resonance imaging for assessing different stages of cardiac impairment in patients with Anderson-Fabry disease: AFFINITY study group. Eur Heart J Cardiovasc Imaging. 2019;20:1004–1011. doi:10.1093/ehjci/jez039
59. Pica S, Sado DM, Maestrini V, et al. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2014;16:99. doi:10.1186/s12968-014-0099-4
60. Sado DM, White SK, Piechnik SK, et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging. 2013;6:392–398. doi:10.1161/CIRCIMAGING.112.000070
61. Nappi C, Altiero M, Imbriaco M, et al. First experience of simultaneous PET/MRI for the early detection of cardiac involvement in patients with Anderson-Fabry disease. Eur J Nucl Med Mol Imaging. 2015;42:1025–1031. doi:10.1007/s00259-015-3036-3
62. Seydelmann N, Liu D, Kramer J, et al. High-sensitivity troponin: a clinical blood biomarker for staging cardiomyopathy in fabry disease. J Am Heart Assoc. 2016;5:e002839. doi:10.1161/JAHA.115.002839
63. Kramer J, Weidemann F. Biomarkers for diagnosing and staging of fabry disease. Curr Med Chem. 2018;25:1530–1537. doi:10.2174/0929867324666170616102112
64. Coats CJ, Parisi V, Ramos M, et al. Role of serum N-terminal pro-brain natriuretic peptide measurement in diagnosis of cardiac involvement in patients with anderson-fabry disease. Am J Cardiol. 2013;111:111–117. doi:10.1016/j.amjcard.2012.08.055
65. Torralba-Cabeza MA, Olivera S, Hughes DA, Pastores GM, Mateo RN, Perez-Calvo JI. Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease. Mol Genet Metab. 2011;104:301–307. doi:10.1016/j.ymgme.2011.06.021
66. Kramer J, Niemann M, Stork S, et al. Relation of burden of myocardial fibrosis to malignant ventricular arrhythmias and outcomes in Fabry disease. Am J Cardiol. 2014;114:895–900. doi:10.1016/j.amjcard.2014.06.019
67. Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8:539–548. doi:10.1097/01.gim.0000237866.70357.c6
68. Schiffmann R, Waldek S, Benigni A, Auray-Blais C. Biomarkers of Fabry disease nephropathy. Clin J Am Soc Nephrol. 2010;5:360–364. doi:10.2215/CJN.06090809
69. Riccio E, Sabbatini M, Capuano I, Pisani A. Early biomarkers of fabry nephropathy: a review of the literature. Nephron. 2019;143:274–281. doi:10.1159/000502907
70. Wanner C, Oliveira JP, Ortiz A, et al. Prognostic indicators of renal disease progression in adults with Fabry disease: natural history data from the Fabry Registry. Clin J Am Soc Nephrol. 2010;5:2220–2228. doi:10.2215/CJN.04340510
71. Glass RB, Astrin KH, Norton KI, et al. Fabry disease: renal sonographic and magnetic resonance imaging findings in affected males and carrier females with the classic and cardiac variant phenotypes. J Comput Assist Tomogr. 2004;28:158–168. doi:10.1097/00004728-200403000-00002
72. Tondel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008;51:767–776. doi:10.1053/j.ajkd.2007.12.032
73. Burlina AP, Sims KB, Politei JM, et al. Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: the report of an expert panel. BMC Neurol. 2011;11:61.
74. Moller AT, Jensen TS. Neurological manifestations in Fabry’s disease. Nat Clin Pract Neurol. 2007;3:95–106. doi:10.1038/ncpneuro0407
75. Low M, Nicholls K, Tubridy N, et al. Neurology of Fabry disease. Intern Med J. 2007;37:436–447. doi:10.1111/j.1445-5994.2007.01366.x
76. Reisin RC, Romero C, Marchesoni C, et al. Brain MRI findings in patients with Fabry disease. J Neurol Sci. 2011;305:41–44. doi:10.1016/j.jns.2011.03.020
77. Lee HJ, Hung SC, Hsu TR, et al. Brain MR imaging findings of cardiac-type fabry disease with an IVS4+919G>A mutation. AJNR Am J Neuroradiol. 2016;37:1044–1049. doi:10.3174/ajnr.A4677
78. Morrison AS. Screening. In: Rothman KJ, Greenland S, editors. Modern Epidemiology.
79. Hagege A, Reant P, Habib G, et al. Fabry disease in cardiology practice: literature review and expert point of view. Arch Cardiovasc Dis. 2019;112:278–287. doi:10.1016/j.acvd.2019.01.002
80. Auray-Blais C, Lavoie P, Abaoui M, et al. High-risk screening for Fabry disease in a Canadian cohort of chronic kidney disease patients. Clin Chim Acta. 2019;501:234–240.
81. Sodi A, Nicolosi C, Vicini G, Lenzetti C, Virgili G, Rizzo S. Computer-assisted retinal vessel diameter evaluation in Fabry disease. Eur J Ophthalmol. 2019;1120672119886985.
82. Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: improving Global Outcomes” (KDIGO) controversies conference. Kidney Int. 2017;91:284–293.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.