")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Diagnosis and management of glucokinase monogenic diabetes in pregnancy: current perspectives
Authors Rudland VL
Received 2 April 2019
Accepted for publication 22 June 2019
Published 10 July 2019 Volume 2019:12 Pages 1081—1089
DOI https://doi.org/10.2147/DMSO.S186610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Steven F. Abcouwer
Victoria L Rudland1,2
1Department of Diabetes and Endocrinology, Westmead Hospital, Sydney, NSW, Australia; 2Faculty of Medicine and Health, The University of Sydney, Sydney, NSW, Australia
Abstract: Glucokinase–maturity-onset diabetes of the young (GCK-MODY) is an autosomal dominant disorder caused by heterozygous inactivating GCK gene mutations. GCK-MODY is one the most common MODY subtypes, affecting 0.1% of the population and 0.4–1% of women with gestational diabetes mellitus. Glucokinase is predominantly expressed in pancreatic beta cells and catalyzes the phosphorylation of glucose to glucose-6-phosphate. The unique kinetics of glucokinase enable it to change the rate of glucose phosphorylation according to the glucose concentration, thereby regulating insulin secretion. Individuals with GCK-MODY have mildly elevated fasting blood glucose levels (5.5–8.0 mmol/L) and regulate glucose perturbations to a higher set-point, resulting in a relatively flat glucose profile on a 75 g oral glucose tolerance test. The hyperglycemia is usually subclinical and may only be detected on incidental glucose testing. It is important to correctly identify GCK-MODY as the clinical course and management differs substantially from other types of diabetes. Diabetes-related complications are relatively uncommon, so glucose-lowering treatment is not usually required. The exception is pregnancy, where fetal growth and therefore glucose-lowering treatment are predominantly determined by whether or not the fetus inherits the GCK mutation. The fetal genotype is not usually known but can be inferred from serial fetal ultrasound measurements. If there is evidence of accelerating fetal abdominal circumference on serial ultrasounds, the fetus is assumed to not have the GCK mutation and treatment of maternal hyperglycemia is indicated to reduce the risk of macrosomia, Caesarean section and neonatal hypoglycemia. If there is no evidence of accelerating fetal growth, the fetus is assumed to have inherited the GCK mutation and will have a similarly elevated glucose set-point as their mother, so maternal hyperglycemia is not treated. With recent advances in genetic technology, such as next-generation sequencing and noninvasive fetal genotyping, the detection and management of GCK-MODY in pregnancy should continue to improve.
Keywords: GCK, MODY, genetics, mutation, gestational diabetes, fetal
Introduction
Monogenic diabetes encompasses several types of diabetes that are caused by single gene mutations. Overall, monogenic diabetes accounts for <5% of all cases of diabetes. One subtype of monogenic diabetes that has particular relevance in pregnancy is glucokinase–maturity-onset of the young (GCK-MODY). The population prevalence of GCK-MODY is 1.1 in 1000 (0.1%).1 GCK-MODY is the most common MODY subtype in Japan2 and the second most common MODY subtype in Chinese3 and Anglo-Celtic4 populations. However, due to its subtle clinical presentation and limited availability of diagnostic molecular genetic testing, GCK-MODY is often undiagnosed.5 It is important to diagnose GCK-MODY, particularly in relation to pregnancy, because the clinical course and management differs substantially from other types of diabetes, including gestational diabetes mellitus (GDM).
This review focusses on the genetics and physiology, clinical features, and diagnosis and management of GCK-MODY, with particular reference to pregnancy.
Genetics of GCK-MODY
GCK-MODY is caused by heterozygous inactivating mutations in the glucokinase (GCK) gene, which is located on chromosome 7p. The GCK gene consists of 12 exons that span 45,168bp and encode for a 465 amino acid protein.6 There are three GCK isoforms, based on different-sized exon 1. Exon 1a is expressed in the pancreatic beta cell. Exons 1b and 1c are expressed in the liver. GCK mutations have been identified in the promoter7 and exons 1a, 2–10 of the pancreatic beta cell GCK isoform.6 The majority of GCK mutations are missense or nonsense mutations, followed by small deletions and splice site mutations. Partial or whole gene deletions, which may not be detected by sequencing, account for 3.5% of GCK-MODY cases in the UK.8 There are no common mutations.
Despite substantial variation in the type and severity of the GCK mutation, the clinical phenotype of GCK-MODY is relatively constant.9 The stable phenotype may be explained by overexpression of the normal allele, potentially via post-translational mechanisms,10 which may partly compensate for the mutant allele.11
GCK-MODY has an autosomal dominant mode of inheritance. Therefore, for women with GCK-MODY, there is theoretically a 50% chance with each pregnancy that the child will inherit the GCK mutation. In addition to inherited GCK mutations, de novo GCK mutations have been reported.12
Physiology of GCK-MODY
Glucokinase is a hexokinase isoenzyme that catalyzes the phosphorylation of glucose to glucose-6-phosphate, which is the first rate-limiting step in glucose metabolism.13 Glucokinase is predominantly expressed in pancreatic beta cells, where it regulates insulin secretion by altering the rate of glucose phosphorylation according to the glucose concentration. Glucokinase is, therefore, the glucose sensor for the pancreas. Glucokinase has unique kinetics that distinguish it from other hexokinases and enable it to function as an effective glucose sensor, including its lower affinity for glucose (Km ~10 mmol/L), moderate cooperative binding with glucose and lack of inhibitory feedback by glucose-6-phosphate.14
Functional studies have demonstrated that heterozygous inactivating mutations in the GCK gene decrease the ability of the glucokinase enzyme to phosphorylate glucose to glucose-6-phosphate, which causes hyperglycaemia.6 Individuals with GCK-MODY have mild fasting hyperglycemia, typically in the range of 5.5–8.0 mmol/L.13 Studies investigating insulin secretion rates following intravenous glucose infusions have demonstrated that the first-phase insulin response is preserved in individuals with GCK-MODY.15 However, there is a right shift in the glucose to insulin secretion rate dose-response curve, such that, for any given glucose, individuals with GCK-MODY have a reduced insulin secretion rate.13 In normal subjects, the pancreatic beta cell is most sensitive to an increase in glucose at concentrations of 5.5–6.0 mmol/L. In individuals with GCK-MODY, the peak beta cell sensitivity occurs at higher glucose concentrations of 6.5–7.5 mmol/L.13 Therefore, the glucose threshold for insulin release is 1.0–2.5 mmol/L higher in individuals with GCK-MODY compared with normal subjects.
When individuals with GCK-MODY undergo a 75 g oral glucose tolerance test (OGTT), the shape of the glucose response curve is similar to that of normal subjects, but shifted upwards. The pattern is one of mild fasting hyperglycemia, followed by an early glucose rise with early (30 min) insulin secretion, such that glucose peaks at 30–60 mins before declining towards the elevated baseline glucose level.9 Unlike other types of diabetes, the shape of the glucose response curve is relatively “flat,” which results in a small increment between the 2 hr glucose and fasting glucose. UK data suggests that the increment (2 hr glucose minus fasting glucose) is <3.0 mmol/L in 70% of individuals with GCK-MODY9 and <4.6 mmol/L in 90% of individuals with GCK-MODY.16
Similarly, in individuals with GCK-MODY, counterregulatory responses to hypoglycemia are activated at a higher glucose level. Following induction of hypoglycemia by a hyperinsulinemic clamp, individuals with GCK-MODY had a higher glucose threshold for increasing glucose production (5.0 vs 3.9 mmol/L) and stimulating glucagon (4.5 vs 3.7 mmol/L) compared with normal subjects.17 These counterregulatory responses restore blood glucose levels to the elevated baseline level.
In addition to pancreatic expression, glucokinase is expressed in the liver and contributes to the storage of glucose as glycogen. In GCK-MODY, there is decreased hepatic glycogen production and increased hepatic gluconeogenesis after meals, which may contribute to the pathogenesis of hyperglycaemia.18 Interestingly, the liver does not seem to have a threshold response to glucose, so is not thought to have a major role in the determination of an individual’s glucose set-point.19
Clinical features of GCK-MODY
Individuals with GCK-MODY have mild fasting hyperglycaemia, typically in the range of 5.5–8.0 mmol/L. Following a 75g OGTT, 2 hr glucose levels are relatively preserved, so the increment between the 2 hr glucose and fasting glucose is usually <4.6 mmol/L.16 The HbA1c is typically in the range of 38–60 mmol/mol (5.6–7.6%).20
The hyperglycemia associated with GCK-MODY is present at birth and persists lifelong, but is usually subclinical so may only be detected on routine glucose testing. Unlike other types of diabetes, the degree of hyperglycemia only slightly worsens with increasing age and does not appear to be related to an individual’s body mass index (BMI).9
Given the mild nature of the hyperglycemia, many individuals with GCK-MODY do not meet the diagnostic thresholds for diabetes. In one study, only 38% of individuals with GCK-MODY met the fasting glucose threshold for diabetes (≥7.0 mmol/L) and only 19% met the 2 hr glucose threshold for diabetes (≥11.1 mmol/L).9 A further 46% met the World Health Organization (WHO) threshold for impaired fasting glucose (≥6.1 mmo/L) and 53% met the threshold for impaired glucose tolerance (≥7.8 mmol/L).9 Therefore, many individuals with GCK-MODY are considered as having “normal” glucose levels, so may never be tested for GCK-MODY.
GCK-MODY has an autosomal pattern of inheritance. Therefore, a family history of “type 2 diabetes,” fasting hyperglycemia (5.5–8.0 mmol/L) or GDM is often present in a first degree relative. However, the absence of family history does not exclude the diagnosis, as de novo GCK mutations can occur.
Diabetes-related complications are relatively uncommon in GCK-MODY, with several studies reporting a similar rate of microvascular and macrovascular complications as that of the general population.12,21 The exception is the rate of retinopathy, which was detected in 30% of individuals who had GCK-MODY for a median duration of 49 years, compared with 14% of normal subjects (p=0.007).21 In that study, most cases of retinopathy in individuals with GCK-MODY were classified as mild background retinopathy, with less than five microaneurysms.21 None of the GCK-MODY individuals had pre-proliferative or proliferative retinopathy, maculopathy or required laser therapy.21
Diagnosis of GCK-MODY
Molecular genetic testing of the GCK gene is expensive and not readily available. However, it is extremely important that individuals with suspected GCK-MODY undergo genetic testing in order to avoid misdiagnosis as another type of diabetes, usually type 2 diabetes. A diagnosis of GCK-MODY prevents unnecessary glucose-lowering treatment and medical review, and has been demonstrated to have a positive impact on the health and wellbeing of many individuals with GCK-MODY.22 In the UK, genetic diabetes nurses are trained as regional experts in monogenic diabetes and help identify individuals with possible monogenic diabetes who warrant genetic testing.23 This approach has led to considerable cost savings.23
Best practice guidelines for the molecular genetic diagnosis of GCK-MODY were developed in 2008 and remain applicable today. Screening criteria for GCK genetic testing include: (1) fasting glucose 5.5–8.0 mmol/L; (2) 2 hr increment <4.6 mmol/L on a 75 g OGTT; (3) family history of type 2 diabetes or GDM (although the absence of family history does not exclude the diagnosis).16,24
The detection rate of monogenic diabetes is further improved by excluding individuals with probable type 1 diabetes, identified by low C-peptide and/or the presence of positive glutamic acid decarboxylase (GAD) autoantibodies, insulinoma-associated antigen-2 (IA-2) autoantibodies or zinc transporter 8 (ZnT8) autoantibodies.25,26
Genetic testing of the GCK gene involves sequencing the promoter, exons 1a, 2–10 and splice sites of the GCK gene.6,27,28 In addition, gene dosage analysis using multiplex ligation-dependent probe amplification (MLPA) is recommended to detect large gene deletions, insertions or duplications that may be missed with sequencing alone.29 Alternatively, targeted next-generation sequencing enables the simultaneous analysis of multiple monogenic diabetes genes.30
Diagnosis of GCK-MODY in pregnancy
Hyperglycemia that is first detected during pregnancy is invariably diagnosed as GDM. However, some of these women will have undiagnosed GCK-MODY. The prevalence of GCK-MODY in the GDM population is 0.4–1%.12,31,32 Differentiating GCK-MODY from GDM is important because the management during and after pregnancy differs.
Clinical features that suggest GCK-MODY in pregnancy include: (1) persistent fasting hyperglycemia (5.5–8.0 mmol/L) before, during and after pregnancy; (2) 2 hr increment <4.6 mmol/L on a 75 g OGTT before, during or after pregnancy; (3) history of “type 2 diabetes,” fasting hyperglycemia (≥5.5 mmol/L) or GDM in a first degree relative (although the absence of family history does not exclude the diagnosis).16,33,34
Several studies have validated the use of the fasting glucose (≥5.5 mmol/L) criterion for identifying pregnant women with GCK-MODY.1,24,31 However, neither the 2 hr increment nor family history appear to adequately differentiate GCK-MODY from GDM.12 A small study that screened women with GDM for GCK-MODY demonstrated that all women with GCK-MODY had a 2 hr increment <3.0 mmol/L on the 75g antepartum OGTT,34 which suggests the standard increment of <4.6 mmol/L may be too generous in pregnancy.
More recently, combined criteria of fasting glucose ≥5.5 mmol/L and normal pre-pregnancy BMI <25 kg/m2 have been proposed. These combined criteria had a 68% sensitivity, 96% specificity and number needed to test of 2.7 women with GDM to identify one case of GCK-MODY.1 This study was conducted in a predominantly Anglo-Celtic population and the use of these combined criteria in Anglo-Celtic populations is supported by another study.31 However, the normal BMI criterion may exclude some cases of GCK-MODY in other ethnic populations and ethnic-specific screening criteria need to be developed.31
Management of GCK-MODY
The current recommendation for individuals with GCK-MODY is that they do not require treatment of their hyperglycemia, except during pregnancy. Glucose-lowering therapy, including insulin, is reported to have minimal impact on glucose levels in individuals with GCK-MODY, which is thought to be related to the rapid onset of counterregulatory responses following a downward perturbation in glucose.12,17,35,36 There is also evidence of reduced endogenous insulin secretion in individuals with GCK-MODY who are receiving exogenous insulin treatment.12 In one study, there was no difference in HbA1c between individuals with GCK-MODY who were receiving glucose-lowering treatment (oral glucose-lowering agents or insulin) and those who were not receiving any treatment.37 Further, when the glucose-lowering treatment was ceased, the HbA1c did not deteriorate,37 a result that was replicated in a recent prospective study.38 Therefore, the recommendation for individuals with newly diagnosed GCK-MODY, who may have been receiving glucose-lowering treatment prior to the diagnosis, is to cease this treatment.12
Individuals with GCK-MODY have the same background risk of type 1 and type 2 diabetes as the general population.12 Therefore, individuals with GCK-MODY who develop symptoms of hyperglycemia or blood glucose levels that are above the range typically seen in GCK-MODY should be investigated for coexistent type 1 or type 2 diabetes. HbA1c may have a role in monitoring individuals with GCK-MODY for the development of other types of diabetes. An HbA1c of 60 mmol/mol (7.6%) represents the 95% confidence limit for GCK-MODY,12 so an HbA1c above this level may be useful in considering coexistent type 1 or type 2 diabetes.
Family members of an individual with GCK-MODY should be counseled regarding cascade testing. Cascade testing involves screening for fasting hyperglycemia (5.5–8.0 mmol/L) unless the family member already has a diagnosis of diabetes or prediabetes, then performing limited sequencing of the exon with the known GCK mutation, which is less expensive than standard GCK genetic testing.23 A correct diagnosis of GCK-MODY may prevent unnecessary investigations and treatment for family members, which has broader health and economic implications.23 However, for each family member, the potential benefits of cascade testing should be balanced with the possible negative consequences of testing, such as psychosocial and insurance-related issues.39
Management of GCK-MODY in pregnancy
This section on management of GCK-MODY in pregnancy applies to women with a confirmed diagnosis of GCK-MODY. If a woman with GDM is suspected of having GCK-MODY, the standard treatment approach for GDM should be continued until a genetic diagnosis of GCK-MODY is confirmed.
Preconception
The rate of congenital anomalies in pregnancies of women with GCK-MODY is unknown and difficult to ascertain given the limited availability of GCK genetic testing and small numbers of women with diagnosed GCK-MODY.12 For women with type 1 or type 2 diabetes, the rate of congenital anomalies has been reported to linearly increase with increasing hyperglycemia above an HbA1c of 45 mmol/mol (6.3%).40 The HbA1c range for individuals with GCK-MODY is typically 38–60 mmol/mol (5.6–7.6%),20 so it is biologically plausible that maternal hyperglycemia secondary to GCK-MODY may confer an increased risk of congenital anomaly.39 Recently, there was a case report of a congenital anomaly in a fetus of a woman with GCK-MODY. In that case, the fetus had sacral agenesis, which is a subtype of caudal regression syndrome.39 However, the fetal genotype was unknown, which raises the possibility that the sacral agenesis may have been a “chance” event.39 Whilst further research and reporting is required, the possibility of an increased congenital anomaly risk in women with GCK-MODY raises the question of whether preconception high dose folic acid should be considered in these women.
Antepartum
It is well established that GCK-MODY can affect fetal growth. If either parent has GCK-MODY, there is a 50% chance that the fetus will inherit the GCK mutation. Fetal inheritance of the GCK mutation largely determines fetal growth.41,42
For maternal GCK-MODY, if the fetus inherits the GCK mutation, the fetus will have the same elevated glucose set-point as their mother and will regulate their blood glucose levels to that higher set-point.41,42 As a result, neonates who inherit a maternal GCK mutation typically have a normal birth weight if maternal hyperglycemia is not treated.41,42 Treatment of maternal hyperglycemia in this situation may adversely reduce the birth weight, so is not recommended.43
For maternal GCK-MODY, if the fetus does not inherit the GCK mutation, the fetus will be exposed to maternal hyperglycemia, which increases the risk of macrosomia, Caesarean section and neonatal hypoglycaemia.35,41,42 The increased fetal growth can be explained by the Pedersen hypothesis which asserts that maternal hyperglycemia stimulates fetal hyperinsulinism and insulin-mediated fetal growth.44 Studies have demonstrated a 550–700 g increase in birth weight in neonates who do not inherit the maternal GCK mutation, compared to neonates who inherit the GCK mutation, with associated increases in birth weight centile and rate of macrosomia (Table 1).41,42,45 In this situation, insulin treatment is recommended, aimed at lowering maternal blood glucose levels to normal pregnancy levels in order to reduce the risk of complications.12,43 Supraphysiological insulin doses (>1 unit/kg/day) are usually required to overcome maternal counterregulatory mechanisms.35,43 Management is further challenged by maternal symptoms of hypoglycemia at “normal” glucose levels.36 Delivery at 38 weeks’ gestation should be considered.12
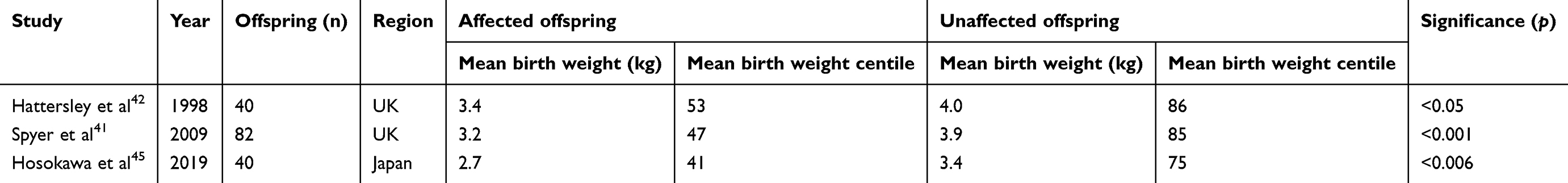 |
Table 1 Studies investigating the birth weight of offspring born to women with GCK-maturity-onset diabetes of the young (MODY) |
For paternal GCK-MODY, fetal inheritance of the GCK mutation also affects fetal growth. If the fetus does not inherit the paternal GCK mutation, the fetus will have the same glucose set-point as their mother and will have normal fetal growth (48th vs 47th centile; p=0.99).41 However, if the fetus inherits the paternal GCK mutation, the fetus will have a higher glucose set-point than their mother, which will result in reduced fetal insulin secretion and reduced insulin-mediated fetal growth. In these neonates, birth weight is typically reduced by ~400 g, with associated reduction in birth weight centile (30th vs 47th centile; p<0.005).41
The clinical dilemma in the management of pregnant women with GCK-MODY is that fetal genotype is not usually known during pregnancy. The current recommendation is to use serial fetal ultrasound measurements to assess fetal growth and guide management.12 Fetal ultrasounds should be performed every 2 weeks from 26 weeks’ gestation.12 If there is ultrasound evidence that the fetal abdominal circumference is increasing disproportionately above the 75th centile, the fetus is assumed to not have the GCK mutation and insulin treatment of maternal hyperglycemia should be commenced. The use of fetal ultrasound measurements to guide management decisions may become less reliable in the future if the trend of increasing pre-pregnancy overweight and obesity continues.46–48 In addition, excessive gestational weight gain is independently associated with accelerating fetal abdominal circumference in late pregnancy, which may further complicate management decisions.49
The current management approach for pregnant women with GCK-MODY is supported by two case reports where the fetus was diagnosed with a GCK mutation in utero. Both fetuses were diagnosed with a GCK mutation by opportunistic genetic testing of DNA from chorionic villus sampling, which had been performed following high-risk aneuploidy screening.50 Knowledge of the fetal GCK mutation dictated that the maternal hyperglycemia was not treated. Both fetuses had a normal birth weight and there were no peripartum complications, despite the maternal hyperglycemia.50
Given that knowledge of the fetal genotype guides management, if a pregnant woman with GCK-MODY has chorionic villus sampling or amniocentesis for another reason, the fetal DNA should be tested for the GCK mutation.50 However, these invasive procedures are not indicated for fetal GCK genotyping alone because the benefit of knowing the fetal genotype does not outweigh the miscarriage risk associated with these procedures.51 Recent advances in non-invasive fetal genotyping using cell-free fetal DNA from maternal plasma sampling are promising.52–54 Non-invasive fetal GCK genotyping would more accurately determine whether maternal hyperglycemia requires treatment and whether delivery at 38 weeks’ gestation should be considered (Table 2).
 |
Table 2 Management of pregnant women with GCK-maturity-onset diabetes of the young (MODY) according to fetal genotype |
Post-partum
Post-partum, any maternal antepartum insulin treatment should be ceased immediately. Testing for neonatal hypoglycemia should be performed. Women should be advised to seek preconception counseling prior to future pregnancies. A yearly HbA1c is recommended to monitor for the development of concurrent type 1 or type 2 diabetes.
For offspring of individuals with GCK-MODY, there is currently no evidence to suggest adverse long-term outcomes. In two large studies, adult offspring had a similar BMI, irrespective of maternal or fetal GCK genotype.55,56 In addition, exposure to the relatively mild maternal hyperglycemia of GCK-MODY in utero did not appear to impact glucose tolerance, insulin secretion, insulin sensitivity, blood pressure or lipid profiles in adulthood.55,56
Conclusion
GCK-MODY is a unique subtype of monogenic diabetes which, despite a mild clinical phenotype, has important implications for pregnancy. Understanding the maternal-fetal GCK genotype interactions is essential in order to optimize maternal and fetal outcomes.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Chakera AJ, Spyer G, Vincent N, et al. The 0.1% of the population with glucokinase monogenic diabetes can be recognized by clinical characteristics in pregnancy: the Atlantic diabetes in pregnancy cohort. Diabetes Care. 2014;37:1230–1236. doi:10.2337/dc13-2248
2. Kawakita R, Hosokawa Y, Fujimaru R, et al. Molecular and clinical characterization of glucokinase maturity-onset diabetes of the young (GCK-MODY) in Japanese patients. Diabetic Med. 2014;31:1357–1362. doi:10.1111/dme.12487
3. Xu JY, Dan QH, Chan V, et al. Genetic and clinical characteristics of maturity-onset diabetes of the young in Chinese patients. Eur J Hum Genet. 2005;13:422–427. doi:10.1038/sj.ejhg.5201347
4. McCarthy MI, Hattersley AT. Learning from molecular genetics: novel insights arising from the definition of genes for monogenic and type 2 diabetes. Diabetes. 2008;57:2889–2898. doi:10.2337/db08-0343
5. Shields BM, Hicks S, Shepherd MH, et al. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53:2504–2508. doi:10.1007/s00125-010-1799-4
6. Osbak KK, Colclough K, Saint-Martin C, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–1526. doi:10.1002/humu.21110
7. Gasperikova D, Tribble ND, Stanik J, et al. Identification of a novel beta-cell glucokinase (GCK) promoter mutation (−71G>C) that modulates GCK gene expression through loss of allele-specific Sp1 binding causing mild fasting hyperglycemia in humans. Diabetes. 2009;58:1929–1935. doi:10.2337/db09-0070
8. Ellard S, Thomas K, Edghill EL, et al. Partial and whole gene deletion mutations of the GCK and HNF1A genes in maturity-onset diabetes of the young. Diabetologia. 2007;50:2313–2317. doi:10.1007/s00125-007-0798-6
9. Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45:427–435. doi:10.1007/s00125-001-0770-9
10. Burke CV, Buettger CW, Davis EA, et al. Cell-biological assessment of human glucokinase mutants causing maturity-onset diabetes of the young type 2 (MODY-2) or glucokinase-linked hyperinsulinaemia (GK-HI). Biochem J. 1999;342:345–352.
11. Sturis J, Kurland IJ, Byrne MM, et al. Compensation in pancreatic beta-cell function in subjects with glucokinase mutations. Diabetes. 1994;43:718–723. doi:10.2337/diab.43.5.718
12. Chakera AJ, Steele AM, Gloyn AL, et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care. 2015;38:1383–1392. doi:10.2337/dc14-2769
13. Byrne MM, Sturis J, Clement K, et al. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest. 1994;93:1120–1130. doi:10.1172/JCI117064
14. Kelly A, Li C, Gao Z, et al. Glutaminolysis and insulin secretion: from bedside to bench and back. Diabetes. 2002;51:S421–S426. doi:10.2337/diabetes.51.2007.s421
15. Velho G, Froguel P, Clement K, et al. Primary pancreatic beta-cell secretory defect caused by mutations in glucokinase gene in kindreds of maturity onset diabetes of the young. Lancet. 1992;340:444–448. doi:10.1016/0140-6736(92)91768-4
16. Ellard S, Bellanne-Chantelot C, Hattersley AT. European Molecular Genetics Quality Network MODY Group. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia. 2008;51:546–553. doi:10.1007/s00125-008-0942-y
17. Guenat E, Seematter G, Philippe J, et al. Counterregulatory responses to hypoglycemia in patients with glucokinase gene mutations. Diabetes Metab. 2000;26:377–384.
18. Velho G, Petersen KF, Perseghin G, et al. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J Clin Invest. 1996;98:1755–1761. doi:10.1172/JCI118974
19. Gloyn AL, Noordam K, Willemsen MA, et al. Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations. Diabetes. 2003;52:2433–2440. doi:10.2337/diabetes.52.9.2433
20. Steele AM, Wensley KJ, Ellard S, et al. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PLoS One. 2013;8:e65326. doi:10.1371/journal.pone.0065326
21. Steele AM, Shields BM, Wensley KJ, et al. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA. 2014;311:279–286. doi:10.1001/jama.2013.283980
22. Shepherd M, Hattersley AT. ‘I don’t feel like a diabetic any more’: the impact of stopping insulin in patients with maturity onset diabetes of the young following genetic testing. Clin Med (Northfield Il). 2004;4:144–147. doi:10.7861/clinmedicine.4-2-144
23. Shepherd M, Colclough K, Ellard S, et al. Ten years of the national genetic diabetes nurse network: a model for the translation of genetic information into clinical care. Clin Med (Northfield Il). 2014;14:117–121. doi:10.7861/clinmedicine.14-2-117
24. Diabetes Research Department and the Centre for Molecular Genetics at the University of Exeter Medical School and Royal Devon and Exeter Hospital, UK. Guidelines for genetic testing in MODY. Available from: https://www.diabetesgenes.org/tests-for-diabetes-subtypes/guidelines-for-genetic-testing-in-mody/.
25. Shields BM, Shepherd M, Hudson M, et al. Population-based assessment of a biomarker-based screening pathway to aid diagnosis of monogenic diabetes in young-onset patients. Diabetes Care. 2017;40:1017–1025. doi:10.2337/dc17-0224
26. Patel KA, Weedon MN, Shields BM, et al. Zinc transporter 8 autoantibodies (ZnT8A) and a type 1 diabetes genetic risk score can exclude individuals with type 1 diabetes from inappropriate genetic testing for monogenic diabetes. Diabetes Care. 2019;42:e16–e17. doi:10.2337/dc18-0373
27. Thomson KL, Gloyn AL, Colclough K, et al. Identification of 21 novel glucokinase (GCK) mutations in UK and European Caucasians with maturity-onset diabetes of the young (MODY). Hum Mutat. 2003;22:417. doi:10.1002/humu.9173
28. Diabetes Research Department and the Centre for Molecular Genetics at the University of Exeter Medical School and Royal Devon and Exeter Hospital, UK. GCK gene analysis in maturity-onset diabetes of the young (GCK MODY). Available from: https://www.diabetesgenes.org/what-is-mody/gck-gene-analysis-in-maturity-onset-diabetes-of-the-young-gck-mody/.
29. Sellner LN, Taylor GR. MLPA and MAPH: new techniques for detection of gene deletions. Hum Mutat. 2004;23:413–419. doi:10.1002/humu.20035
30. Diabetes Research Department and the Centre for Molecular Genetics at the University of Exeter Medical School and Royal Devon and Exeter Hospital, UK. Targeted next generation sequencing. Available from: https://www.diabetesgenes.org/tests-for-diabetes-subtypes/targeted-next-generation-sequencing-analysis-of-45-monogenic-diabetes-genes/#.
31. Rudland VL, Hinchcliffe M, Pinner J, et al. Identifying glucokinase monogenic diabetes in a multiethnic gestational diabetes mellitus cohort: new pregnancy screening criteria and utility of HbA1c. Diabetes Care. 2016;39:50–52. doi:10.2337/dc15-1001
32. Wang Z, Ping F, Zhang Q, et al. Preliminary screening of mutations in the glucokinase gene of Chinese patients with gestational diabetes. J Diabetes Investig. 2018;9:199–203. doi:10.1111/jdi.12664
33. Diabetes Research Department and the Centre for Molecular Genetics at the University of Exeter Medical School and Royal Devon and Exeter Hospital, UK. The management of pregnancy in patients with hyperglycaemia due to mutations of the glucokinase (GCK) gene: GCK and pregnancy guidelines 18.01.2018. Available from: https://www.diabetesgenes.org/what-is-mody/what-is-glucokinase-gck/.
34. Ellard S, Beards F, Allen LI, et al. A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia. 2000;43:250–253. doi:10.1007/s001250050038
35. Hattersley AT, Pearson ER. Minireview: pharmacogenetics and beyond: the interaction of therapeutic response, beta-cell physiology, and genetics in diabetes. Endocrinology. 2006;147:2657–2663. doi:10.1210/en.2006-0152
36. Chakera AJ, Hurst PS, Spyer G, et al. Molecular reductions in glucokinase activity increase counter-regulatory responses to hypoglycemia in mice and humans with diabetes. Mol Metab. 2018;17:17–27. doi:10.1016/j.molmet.2018.08.001
37. Stride A, Shields B, Gill-Carey O, et al. Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia. 2014;57:54–56. doi:10.1007/s00125-013-3075-x
38. Shepherd MH, Shields BM, Hudson M, et al. A UK nationwide prospective study of treatment change in MODY: genetic subtype and clinical characteristics predict optimal glycaemic control after discontinuing insulin and metformin. Diabetologia. 2018;61:2520–2527. doi:10.1007/s00125-018-4728-6
39. Shepherd M. Genetic testing in maturity onset diabetes of the young (MODY) - practical guidelines for professionals. Pract Diab Int. 2003;20:103–110. doi:10.1002/pdi.458
40. Bell R, Glinianaia SV, Tennant PW, et al. Peri-conception hyperglycaemia and nephropathy are associated with risk of congenital anomaly in women with pre-existing diabetes: a population-based cohort study. Diabetologia. 2012;55:936–947. doi:10.1007/s00125-012-2455-y
41. Spyer G, Macleod KM, Shepherd M, et al. Pregnancy outcome in patients with raised blood glucose due to a heterozygous glucokinase gene mutation. Diabetic Med. 2009;26:14–18. doi:10.1111/j.1464-5491.2008.02622.x
42. Hattersley AT, Beards F, Ballantyne E, et al. Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet. 1998;19:268–270. doi:10.1038/953
43. Spyer G, Hattersley AT, Sykes JE, et al. Influence of maternal and fetal glucokinase mutations in gestational diabetes. Am J Obstet Gynecol. 2001;185:240–241. doi:10.1067/mob.2001.113127
44. Pedersen J. Diabetes and pregnancy; blood sugar of newborn infants during fasting and glucose administration. Nord Med. 1952;47:1049.
45. Hosokawa Y, Higuchi S, Kawakita R, et al. Pregnancy outcome of Japanese patients with glucokinase-maturity-onset diabetes of the young. J Diabetes Investig. 2019. doi:10.1111/jdi.13046
46. Chen C, Xu X, Yan Y. Estimated global overweight and obesity burden in pregnant women based on panel data model. PLoS One. 2018;13:e0202183. doi:10.1371/journal.pone.0202183
47. Deputy NP, Dub B, Sharma AJ. Prevalence and trends in prepregnancy normal weight - 48 States, New York City, and District of Columbia, 2011–2015. MMWR. 2018;66:1402–1407. doi:10.15585/mmwr.mm665152a3
48. Maxwell C, Glanc P. Imaging and obesity: a perspective during pregnancy. AJR. 2011;196:311–319. doi:10.2214/AJR.10.5849
49. Hellebust H, Johnsen SL, Rasmussen S, et al. Maternal weight gain: a determinant for fetal abdominal circumference in the second trimester. Acta Obstet Gynecol Scand. 2011;90:666–670. doi:10.1111/j.1600-0412.2011.01129.x
50. Chakera AJ, Carleton VL, Ellard S, et al. Antenatal diagnosis of fetal genotype determines if maternal hyperglycemia due to a glucokinase mutation requires treatment. Diabetes Care. 2012;35:1832–1834. doi:10.2337/dc12-0151
51. Chakera AJ, Carleton VL, Shields B, et al. Response to comment on: Chakera et al. Antenatal diagnosis of fetal genotype determines if maternal hyperglycemia due to a glucokinase mutation requires treatment. Diabetes Care. Diabetes care. 2013;36:e15, 2012;35:1832–1834. doi:10.2337/dc12-0151
52. Lun FM, Tsui NB, Chan KC, et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci U S A. 2008;105:19920–19925. doi:10.1073/pnas.0810373105
53. Colclough K, Saint-Martin C, Timsit J, et al. Clinical utility gene card for: maturity-onset diabetes of the young. Eur J Hum Genet. 2014;22:e1–e6. doi:10.1038/ejhg.2014.14
54. De Franco E, Caswell R, Houghton JA, et al. Analysis of cell-free fetal DNA for non-invasive prenatal diagnosis in a family with neonatal diabetes. Diabetic Med. 2017;34:582–585. doi:10.1111/dme.13180
55. Velho G, Hattersley AT, Froguel P. Maternal diabetes alters birth weight in glucokinase-deficient (MODY2) kindred but has no influence on adult weight, height, insulin secretion or insulin sensitivity. Diabetologia. 2000;43:1060–1063. doi:10.1007/s001250051490
56. Singh R, Pearson ER, Clark PM, et al. The long-term impact on offspring of exposure to hyperglycaemia in utero due to maternal glucokinase gene mutations. Diabetologia. 2007;50:620–624. doi:10.1007/s00125-006-0541-8
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.