")
Back to Archived Journals » Research and Reports in Endocrine Disorders » Volume 5
Diabetes reversal via gene transfer: building on successes in animal models
Authors Gerace D, Martiniello-Wilks R, Simpson A
Received 18 September 2014
Accepted for publication 3 November 2014
Published 29 January 2015 Volume 2015:5 Pages 15—29
DOI https://doi.org/10.2147/RRED.S51644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Mingzhao Xing
Video abstract presented by Dario Gerace
Views: 1167
Dario Gerace,1,* Rosetta Martiniello-Wilks,1,2,* Ann M Simpson1
1School of Medical and Molecular Biosciences, Centre for Health Technologies, 2Translational Cancer Research Group, University of Technology Sydney, Sydney, NSW, Australia
*These authors contributed equally to this work
Abstract: Type 1 diabetes (T1D) is caused by the autoimmune destruction of the insulin-producing pancreatic β-cells. People with T1D manage their hyperglycemia using daily insulin injections; however, this does not prevent the development of long-term diabetic complications such as retinopathy, nephropathy, neuropathy, and various macrovascular disorders. Currently, the only "cure" for T1D is pancreas transplantation or islet-cell transplantation; however, this is hampered by the limited number of donors and the requirement for life-long immunosuppression. As a result, the need for alternative therapies is vital. One of the strategies employed to correct T1D is the use of gene transfer to generate the production of an “artificial” β-cell that is capable of secreting insulin in response to fluctuating glucose concentrations that normally occurs in people without T1D. The treatment of many diseases using cell and gene therapy is generating significant attention in the T1D research community; however, for a cell therapy to enter clinical trials, success and safety must first be shown in an appropriate animal model. Animal models have been used in diabetes research for over a century, have improved our understanding of the pathophysiology of diabetes, and have led to the discovery of useful drugs for the treatment of the disease. Currently, the nonobese diabetic mouse is the animal model of choice for the study of T1D as it most closely reflects disease development in humans. The aim of this review is to evaluate the success of cell and gene therapy to reverse T1D in animal models for future clinical application.
Keywords: β-cell transcription factors, animal models, gene therapy, insulin
Introduction
Type 1 diabetes (T1D) results from the autoimmune destruction of the insulin-producing β-cells of the pancreas.1,2 As a consequence, insulin-responsive tissues cannot take up glucose effectively, resulting in hyperglycemia. Currently, people with T1D manage their hyperglycemia using daily insulin injections,3 but insulin therapy does not replicate the physiological regulation of glycemia and patients tend to develop the long-term complications associated with extended periods of hyperglycemia.4,5 In addition, the stressful condition of hypoglycemic unawareness, where a person with long-term diabetes eventually does not recognize the classic symptoms of their hypoglycemia, can become life-threatening.4,6
Currently, pancreas or islet transplantation is the only cure; however, this is hampered by the limited number of pancreas donors and the requirement for life-long immunosuppression.7 As a result, alternative therapeutic approaches that overcome both the requirement for immunosuppression and recurrent autoimmunity are required. Currently, a number of therapeutic approaches are under investigation including restoration of immune tolerance,8,9 endogenous β-cell regeneration,9,10 transplantation of encapsulated artificial β-cells,11,12 and the “artificial pancreas”.13,14 In addition, gene transfer of pancreatic transcription factors and insulin for the production of “artificial” β-cells that are capable of synthesizing and secreting insulin in response to metabolic signals is a promising alternative. Two common methods of generating surrogate β-cells are the dedifferentiation and directed transdifferentiation of autologous or allogeneic cells ex vivo15–17 followed by transplantation, and the in vivo18–20 transdifferentiation of target tissue via gene transfer of transcription factors or insulin within viral vectors.
Animal models have been used in T1D research since the discovery of insulin by Banting and Best in 192221 for studying the pathogenesis of the disease and its complications and for the discovery of new treatments. For any diabetes research to have clinical applications, potential treatments must be performed in animal models to provide proof of the principle. The most common animal model for T1D research is the nonobese diabetic (NOD) mouse. This review will outline the common types of animal models of T1D, the generation of artificial β-cells as a promising alternative therapy for the treatment of T1D, and how animal model research is applied to demonstrate the safety and success in reversing diabetes, which are important for clinical development.
Animal models of T1D
The use of animal models in diabetes research (Table 1) has been extensively pursued. Most studies are performed in small animal models such as rodents and mice to provide proof of concept for the development of Phase I clinical trials. However, large animal models of diabetes in pigs, dogs, and monkeys are becoming increasingly popular due to the criticism that rodents do not adequately represent the human condition of diabetes.
 | Table 1 Animal models of T1D |
In the past, one of the most popular methods for inducing diabetes in animals was by complete pancreatectomy.22 However, this does not correctly represent the clinical manifestation of diabetes due to the loss of potential feedback mechanisms as a consequence of the absence of pancreatic hormone-producing cells, which are still present in people with diabetes. As a result, nonsurgical methods of inducing a diabetic state in animal models via the administration of toxins such as streptozotocin (STZ) and alloxan are now routinely used. STZ is a nitrosourea derivative isolated from Streptomyces achromogenes,23 with either a single high-dose administration or multiple low-dose administrations. A single high-dose in mice ranges from 100 mg/kg to 200 mg/kg, with complete destruction of β-cells and the development of hyperglycemia.24 Alternatively, multiple low-dose administrations, ranging from 20 mg/kg/day to 40 mg/kg/day over a 5-day period, lead to insulitis in mice.25 Alloxan, which is commonly administered in mice at a dose of 50–200 mg/kg, generates superoxide free-radicals that destroy β-cells as they do not possess any defense mechanisms against the chemical.26,27 These models are particularly useful for studying the success of transplantation of pancreatic tissue or putative artificial β-cells, and cytokine-targeted therapies. However, some of the limitations of chemically inducing diabetes are the possible toxicity to other organs and the potential for β-cell regeneration following high-dose STZ administration.28
Currently, the most popular choices of the T1D animal model are the spontaneous NOD mouse and the biobreeding (BB) rat. These models manifest with autoimmune diabetes similar to that observed in humans and currently dominate the literature. The NOD mouse was developed at the Shionogi Research Laboratories, Osaka, Japan, in 1974.29 NOD mice typically develop insulitis at around 3–4 weeks of age, during which the pancreatic islets are infiltrated by CD4+ and CD8+ lymphocytes and to a lesser degree by B cells and natural killer (NK) cells.30 They develop typical diabetes between 12 weeks and 30 weeks of age. Due to the parallel in T1D clinical manifestation between NOD mice and humans, the former have been useful in identifying pathways that lead to T1D and are also suitable for testing therapies that modulate the autoimmune response.31 More recently, NOD mice are being used to study the success of cell therapies to either modulate the autoimmune response32–34 or directly control hyperglycemia by artificial β-cells.35–37 Nonetheless, some major differences do exist, such as the inbred nature of this model, allowing the genetic susceptibility to diabetes development in NOD mice to be easily traced. This is evidenced by the fact that female mice are susceptible whereas males are not.38 In humans, susceptibility to the development of T1D is governed by a much more complex interaction of genetic and environmental stimuli. In addition, there is evidence that NOD mice develop lymphopenia, a characteristic not seen in the human situation.39 An isolated genetic contamination of the NOD mouse outcrossed with a C57BL mouse produced an insulitis-resistant and diabetes-free strain named the nonobese resistant (NOR) mouse, which is now routinely used as a major histocompatibility complex-matched control mouse for studies using NOD mice.40 The BB rat, which spontaneously develops autoimmune diabetes, was derived from outbred Wistar rats in 1974.41 They develop diabetes between 8 weeks and 16 weeks of age, and similar to NOD mice display insulitis with the infiltration of T cells, B cells, macrophages, and NK cells. However, like NOD mice, these animals develop lymphopenia and therefore are not as routinely used as NOD mice.39,42 Other animal models of spontaneous autoimmune diabetes include the LEW.1AR1/Ztm-iddm rats,43 the Keeshond dog,44 and the Celebes black ape (Macaca nigra).45
In addition to these main animal models of T1D, genetically and virally induced models are available for research purposes. The AKITA mouse was derived in Akita, Japan, from a C57BL/6NSlc mouse with a spontaneous mutation in the insulin 2 gene, which results in a severe insulin-dependent diabetes beginning at 3–4 weeks of age, and is used to study the success of islet transplantation.46 Since the implication of viruses in the pathogenesis of T1D, viruses have been used to induce diabetes in animal models. These include the coxsackie B virus,47 the encephalomyocarditis virus,48 and the Kilham rat virus.49 The remainder of this review will focus on the use of viral gene transfer to a variety of cell types for the production of artificial β-cells and assessment of the success of these cell therapies in animal models of diabetes via autologous or allogeneic transplantation.
Selecting a suitable vector for gene transfer
Viruses possess the natural capacity to infect and deliver genes to cells and, as a consequence, have been engineered to not replicate yet efficiently transduce infected cells with genes of interest for a number of purposes. The ability to integrate the genes of interest into the genome of a target cell allows for long-term expression of the transgene, resulting in a sustained therapeutic effect. The engineering of β-cells for the treatment of T1D would preferably employ integrating viral vectors to provide a sustained therapeutic benefit over the life of the patient without the need for multiple administration of the treatment.
The most commonly used gene delivery vector is the retroviral vector, which is derived from disabled murine virus.50 Although these vectors are capable of integrating into the host genome, a limitation of their use is their risk of insertional mutagenesis.51 A study on the use of retroviral vectors for the treatment of severe combined immunodeficiency (Scid) resulted in the development of leukemia in four of the nine patients, and demonstrated the site-specific preferences of integration for retroviruses to be in close proximity to the protooncogenes.52,53 The use of retroviral vectors is also limited by their ability to only transduce dividing cells, posing a challenge for the transduction of nondividing cells such as the liver. When transducing bone marrow-derived mesenchymal stem cells (BMSCs) with a retroviral vector containing the insulin gene under the control of the cytomegalovirus (CMV) promoter, it was found that these cells were able to secrete insulin, maintain normal blood glucose levels, and evade autoimmune destruction upon transplantation in STZ-diabetic mice for 42 days.54
Adenoviral vectors transduce nondividing cells episomally and therefore only provide transient gene expression.50,55 In some cases, immune responses against the viral proteins have been reported,56,57 and to overcome the immunogenicity of the viral capsid proteins, a “gutless” adenovirus was developed.58 Despite displaying a reduction in immunogenicity, immunosuppressants are still required to manage immune responses activated following treatment.59 In addition, preexisting immunity to adenovirus in humans limits the use of multiple administrations of the vector that would be required for sustained therapeutic effects. The ability of adeno-associated viral (AAV) vectors to transduce both dividing and nondividing cells makes them a suitable vector of choice for gene transfer. However, they have a limited gene cargo capacity of <5 kb.60 Due to their site-specific nature of gene integration in target cells, AAV insertion sites can be predicted and potentially oncogenic consequences avoided. A study using AAV vectors to directly deliver the preproinsulin gene to livers of chemically induced STZ mice61 transiently reduced blood glucose levels, and supports the utility of AAV for insulin gene transfer.
An attractive candidate for gene therapy is lentiviral vectors (LVs) because they are capable of transducing both nondividing and dividing cells.62 LVs are derived from the human immunodeficiency virus (HIV), so biosafety was a concern for their application as therapeutics. By introducing deletions in the long terminal repeat (LTR) promoter, the likelihood of generating a replication-competent virus was reduced and greater safety for clinical application was achieved.63 Within our laboratory, LV is currently the gene transfer vector of choice for corrective gene therapy. We have successfully used a LV, HMD, which has a murine stem cell virus promoter to deliver furin-cleavable insulin (INS-FUR) to the livers of STZ-diabetic rats,18 NOD mice,19 and pancreatectomized Westran pigs.20 In these animal models, we observed spontaneous expression of β-cell transcription factors, formation of storage granules, and permanent reversal of diabetes.
Target cells for T1D gene therapy
Monkey kidney cells and fibroblasts were the first targets of somatic cell gene therapy for T1D.64,65 Unfortunately, these cells do not possess characteristics similar to those of β-cells, and as a result were not able to produce biologically active insulin. Despite employing extensive genetic manipulation to such cell types, the generation of functional artificial β-cells was not achieved. Similarly, targeting of muscle cells has been examined sparingly due to their lack of β-cell characteristics. However, a study using vascular smooth muscle cells transduced with INS-FUR under the control of a glucose-regulatable promoter was able to reduce blood glucose levels in spontaneously diabetic congenic BB rats for a period of 6 weeks, after which exogenous insulin therapy was required.66 A more sustained reversal of diabetes was achieved in STZ-diabetic mice for >4 months following the dual expression of insulin and glucokinase (GK) in muscle.67 However, it was quickly determined that the ideal target cells for the successful generation of functional artificial β-cells would be derived from an endodermal origin and possess characteristics similar to those of β-cells such as a glucose-sensing system, proinsulin-processing enzymes, and exocytosis system.68
Pituitary cells possess both proinsulin-processing enzymes and secretory granules, and have been modified to produce insulin via the transfer of a recombinant plasmid containing human preproinsulin cDNA.15 Although these cells produced biologically active insulin, they lacked glucose responsiveness and therefore required further genetic modification via the transduction of GLUT2 and GK. Following the additional modifications, the modified cells became glucose-responsive, albeit at subphysiological levels. In addition, following transplantation, the insulin function was inhibited by the in vivo secretion of adrenocorticotropic-hormone-stimulated glucocorticoid synthesis, limiting their therapeutic efficacy.69
A more promising target for gene transfer are liver cells as they are derived from the same endodermal origin as the pancreas and possess the key glucose-responsive elements (G1REs) GLUT2 and GK.70 Despite lacking proinsulin-processing enzymes and secretory granules, the induced expression of mature insulin is possible via the targeted expression of INS-FUR.16,18 Our laboratory has shown that the expression of insulin in a liver cell line that had endogenous expression of β-cell transcription factors led to pancreatic transdifferentiation, formation of secretory granules, and a regulated response to glucose with reversal of diabetes.16 Many studies that have attempted to reproduce this phenomenon by simply expressing either insulin or insulin analogs in liver without the expression of β-cell transcription factors have only reported constitutive release of insulin that is not stored or secreted in a regulated fashion.71–73
More recently, mesenchymal stem cells (MSCs) have been targeted for gene transfer due to their extensive immunomodulatory capacity and reported ability to evade immune rejection.74–77 Most use of MSCs in diabetes reversal in animal models has been in the form of native MSC transplantations aimed at modulating immune responses.78,79 However, currently most MSC research is being driven toward the generation of artificial β-cells via chemically induced differentiation protocols or gene transfer.80 Artificial β-cells can be obtained from BMSCs via the use of a high-glucose culture medium81 or a nicotinamide-enriched medium to induce differentiation.82 Similarly, a three-step differentiation protocol has been shown to produce glucose-responsive artificial β-cells from BMSCs with high expression levels of Pdx-1, insulin, and glucagon.83 BMSCs retrovirally transduced with Pdx-1 generated artificial β-cells that reduced blood glucose concentrations beginning 12 days post transplantation in STZ-diabetic/Scid mice, and displayed a normal glucose tolerance until 6–8 weeks post transplantation.35
Glucose-responsive production of insulin
Ultimately, T1D gene therapy aims to create artificial β-cells that reproduce the mechanism of insulin secretion that occurs normally within β-cells. Normally, insulin is initially translated as a proinsulin precursor in the endoplasmic reticulum (ER); it is then transported from the ER to the Golgi apparatus and crosses the Golgi network before being sorted into clathrin-coated immature insulin secretory granules (ISGs).84,85 Various biochemical modifications trigger the maturation of ISGs, and following glucose stimulation the mature ISGs secrete their insulin content into the extracellular space. Although the pancreatic β-cell contains ~10,000 ISGs, only 100–200 ISGs are capable of quickly releasing their insulin content in response to increases in cytosolic [Ca2+].84 This means that there is a “reserve pool” of ISGs which can be induced to release insulin, resulting in the minute-to-minute regulation of insulin secretion in response to changes in blood glucose levels. One of the major hurdles for T1D gene therapy is the inability to reproduce the regulated mechanism of insulin secretion in pancreatic β-cells. Consequently, the use of glucose-responsive promoters attached to pancreatic transcription factors or the insulin gene have been investigated in an attempt to engineer the physiologically regulated production of insulin.
Table 2 summarizes a number of studies in which the insulin gene has been expressed in various cell types with the aim of reversing T1D. Three promoters have been identified (L-type pyruvate kinase [LPK], Spot 14, and glucose-6-phosphate) that regulate the expression of a variety of genes in the liver in response to extracellular glucose concentrations.62 These promoters have been used in association with the insulin gene for gene transfer in attempts to reproduce the glucose-responsive production of insulin that occurs in normal β-cells. The G1RE found in the LPK promoter has been found to regulate the transcription of proinsulin in response to glucose.86,87 In particular, the work by Thule et al showed that, by injecting diabetic rats with a construct containing three copies of the G1RE from the LPK promoter, an inhibitory element from the insulin-like growth factor binding protein (IGFBP)-1 promoter and a modified proinsulin gene, normoglycemia was nearly achieved.87,88 STZ-induced diabetic mice expressing a human proinsulin gene under the control of the LPK promoter expressed proinsulin mRNA in the liver, with the transcription of proinsulin mRNA regulated by diet. However, STZ-induced diabetic mice did not increase the expression of insulin mRNA due to inhibition of GK by glucagon.89 This was overcome by inducing basal levels of expression of transgenic insulin with a simian virus 40 (SV40) enhancer that stimulated GK expression, which in turn activated expression of transgenic insulin via the LPK promoter.86 Promising work using a glucose-6-phosphate promoter showed that glucose-responsive production of insulin could be induced in rat hepatoma cells. However, this was limited by low levels of insulin production as a consequence of the negative feedback by the insulin produced.90
 | Table 2 Insulin used for the reversal of T1D in animal models |
More recently, transcriptional analysis of BMSCs grown under varying concentrations of glucose identified a number of glucose-responsive promoters, and of particular interest, the early growth response-1 (EGR1) promoter which was capable of expressing insulin in transduced BMSC in a glucose-responsive manner.91 Upon implantation of the cells in STZ-diabetic-induced NOD/Scid mice, the transduced cells were capable of restoring normoglycemia, glucose tolerance, and body weight in this model. Interestingly, the EGR1 promoter was not sensitive to transgenic expression of insulin, because it is known that the EGR1 promoter can be activated by both glucose and insulin.92 However, as NOD/Scid mice are not a model of autoimmune diabetes, such work should ideally be performed in immune-competent models of autoimmune diabetes, such as NOD mice, that intrinsically develop insulitis around 3–4 weeks of age and progress to full-fledged diabetes between 12 weeks and 30 weeks.93
Investigating tissue-specific regulation of pancreatic hormones or proteins would hopefully reveal the underlying mechanisms governing the expression of those factors. As a result, the insulin promoter that is activated by glucose has been intensively studied to identify these G1REs in the hopes of better recreating the physiological regulation of insulin production.94,95 A study by Sander et al discovered a strong G1RE (Z element) in the distal region of the human insulin promoter,94 elucidating one of the mechanisms that provides glucose-sensitive regulation of insulin in primary cultured islet cells. More recently, the pancreatic-derived factor (PANDER), a newly discovered cytokine-like protein that is strictly expressed in the pancreatic islets, was analyzed for its glucose-responsive nature of expression.96 It revealed that the 5′-untranslated region of the PANDER promoter contained the G1REs that drive PANDER expression that mimic insulin expression, and that the PANDER promoter could potentially be used to drive transgenic insulin expression in alternative cell targets.
One of the limitations of transcriptionally regulated insulin production in transplanted alternative cell targets is their delay in responding with immediate insulin secretion when challenged with glucose. As insulin secretion is linked to insulin transcription, the minute-to-minute glycemic control, which occurs in normal β-cells, is not present in these engineered target cells. This is due to the absence of secretory granules that store and immediately secrete insulin in response to fluctuations in blood glucose concentrations. To overcome this limitation, engineering a promoter expressing high levels of insulin in response to fluctuating glucose concentrations, yet possessing insulin-sensitivity as a feedback mechanism, could more closely mimic the normal physiology of insulin secretion. However, the successful adaptation of this technology in vivo would require the development of granules similar to ISGs to regulate glycemia.
Gene transfer of insulin
Our laboratory has shown that the ability of liver cells to store and secrete insulin, and undergo pancreatic differentiation, is linked to the induced expression of certain β-cell transcription factors. We were the first to show a number of cutting-edge developments in this field and, very importantly, have never seen the development of exocrine differentiation and tissue destruction often seen in studies of liver-directed gene therapy where Pdx-1 was used.17,97 This is, in part, due to the alternative choice of genes used for the viral delivery to hepatocytes.
In several animal models, we have delivered INS-FUR, within the LV human immunodeficiency virus, murine stem cell lentiviral vector, to the liver by using a surgical technique that isolated the liver from the circulation, allowing the LV to settle in the liver without the problem of excessive inactivation from the blood. We permanently reversed diabetes in STZ-diabetic rats18 and spontaneously diabetic NOD mice.19 In both studies, we reported spontaneous expression of key β-cell transcription factors (Pdx-1, Neurog3, and NeuroD1), which are important in the development of insulin storage and regulated insulin expression in pancreatic β-cells.98–101 Some later stage transcription factors (Pax4 and Nkx2.2) were also expressed. Our lentiviral transduction procedure may have represented a cellular insult, making progenitor cells permissive to a pancreatic developmental shift. Consistent with this, expression of Pdx-1, but only at the mRNA level, was also observed after treatment with the empty vector alone. Expression of insulin was necessary for protein expression of transcription factors. There was also expression of pancreatic hormones and development of ISGs, and normal intravenous glucose tolerance tests were observed in the STZ-diabetic rat and NOD mouse (Figure 1). Furthermore, insulin expression was restricted to the liver. In the NOD mouse study, there was no evidence of intrahepatic inflammation or autoimmune destruction of the insulin-secreting liver tissue. By contrast, in our NOD mouse study and a similar study by Elsner et al, a simple injection of insulin into the portal circulation resulted in unregulated constitutive release of insulin, no pancreatic transdifferentiation, and an abnormal glucose response.19,71 Pancreatic transdifferentiation of the liver has been seen in other situations: following a dose of the hepatotoxin carbon tetrachloride102 and when oval cells were cultured in high glucose.103,104 We are yet to define the mechanism that has resulted in pancreatic differentiation in our studies, which is being analyzed at the molecular level in our laboratory.
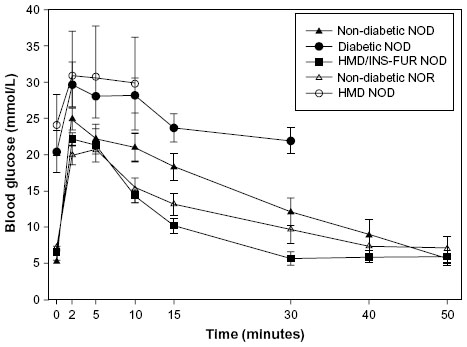 | Figure 1 Plasma glucose levels following an IVGTT in NOD mice treated with INS-FUR in a lentiviral vector (HMD). |
We have also reversed diabetes in a diabetic pig model, which was characterized by normal glucose tolerance, together with expression of β-cell transcription factors.20 However, reproducible results were problematic in the large animal due to the complexity of the surgical approach. Subsequently, viral delivery of INS-FUR has become a popular choice for gene therapy, with a number of studies showing amelioration of hyperglycemia in rodent models. However, abnormal glucose tolerance was observed as a result of a lack of pancreatic transdifferentiation and expression of β-cell transcription factors.105–108
Studies in our laboratory have also shown that these insulin-secreting liver cells are resistant to the detrimental effects of β-cell cytotoxins and proinflammatory cytokines that play a principle role in the pathogenesis of T1D.109,110 In other experiments, no infiltrates of immune cells were observed in NOD mice engineered to express insulin in their livers.19,111 These studies established that liver cells are appropriate candidates for the creation of artificial β-cells, and also highlighted that dual expression of insulin and β-cell transcription factors gave a better outcome than expression of either alone. Xu et al studied the retroviral transduction of BMSCs with an insulin gene under the control of the CMV promoter and their ability to restore normoglycemia in STZ-diabetic mice.54 It was found that these BMSCs successfully expressed insulin and were able to maintain normoglycemia for at least 42 days. In addition, the transduced BMSCs were able to evade the autoimmune destruction that ordinarily targets pancreatic islets.
Gene transfer of β-cell transcription factors
During embryonic development, the pancreas is derived from the early gut endoderm. Intrinsic and extrinsic factors direct the formation of the dorsal and ventral pancreatic buds, with the buds later rotating and fusing to form the early pancreas.112,113 Islet cell differentiation is regulated by the expression of β-cell transcription factors (Figure 2) during embryogenesis, and during adult life the transcription factors regulate the expression of pancreatic hormones. Endodermal formation is linked to expression of the Forkhead Box factors FoxA1 and FoxA2, where deletions of FoxA2 have been shown to interfere with the formation of endoderm in mouse models.114 Pdx-1 is considered the “master regulator” of pancreatic development, as it is involved in both the early development of the pancreas and in the functioning of mature β-cells during adulthood.113,115 Differentiation of the exocrine and endocrine pancreatic partitions occurs rapidly following the fusion of the dorsal and ventral buds, with expression of Hes-1 and Neurog3 in the precursor cells directing the corresponding compartmental fates via notch signaling.116 Persistent expression of NeuroD1 maintains endocrine cell fate.117 A study using homozygous NeuroD1-/- mice showed a decrease in the growth of insulin-producing cells.118
 | Figure 2 β-cell transcription factor hierarchy. |
Once endocrine cell fate has been established, the transcription factors Pax4 and Pax6 direct the differentiation of individual hormone-producing cells.113 Differentiation of β-cells is eventually driven by the expression of Nkx2.2 and Nkx6.1. Interestingly, expression of Nkx2.2 has been observed in α and PP-cells; however, its knockout results in the disruption of the development of β-cells.119 Taken together, these results suggest that Nkx2.2 and Nkx6.1 are imperative for β-cell differentiation.
Due to the limitations of pancreas and islet transplantation, the requirement for the generation of an alternative β-cell that produces insulin in a regulated manner while evading degradation by the immune system is of utmost importance. Table 3 summarizes a number of studies which have expressed beta cell transcription factors for the reversal of T1D in animal models. The generation of artificial β-cells for the reversal of diabetes through the transfer of pancreatic transcription factors has been extensively studied in liver tissue due to its common developmental origin with the pancreas,68 which makes it readily able to transdifferentiate. Ferber et al directly delivered the β-cell transcription factor Pdx-1 to liver tissue via a recombinant adenovirus in an endeavor to correct hyperglycemia by inducing the expression of insulin in the liver in vivo.17 The study showed that expression of Pdx-1 in the livers of diabetic mice resulted in insulin expression and secretion, and as a consequence, the maintenance of normal blood glucose levels. However, the restoration of normoglycemia was only for 8 days, which is considerably short. Furthermore, the development of hepatitis in the liver due to exocrine differentiation led to an increased likelihood of autoimmune destruction.17,97
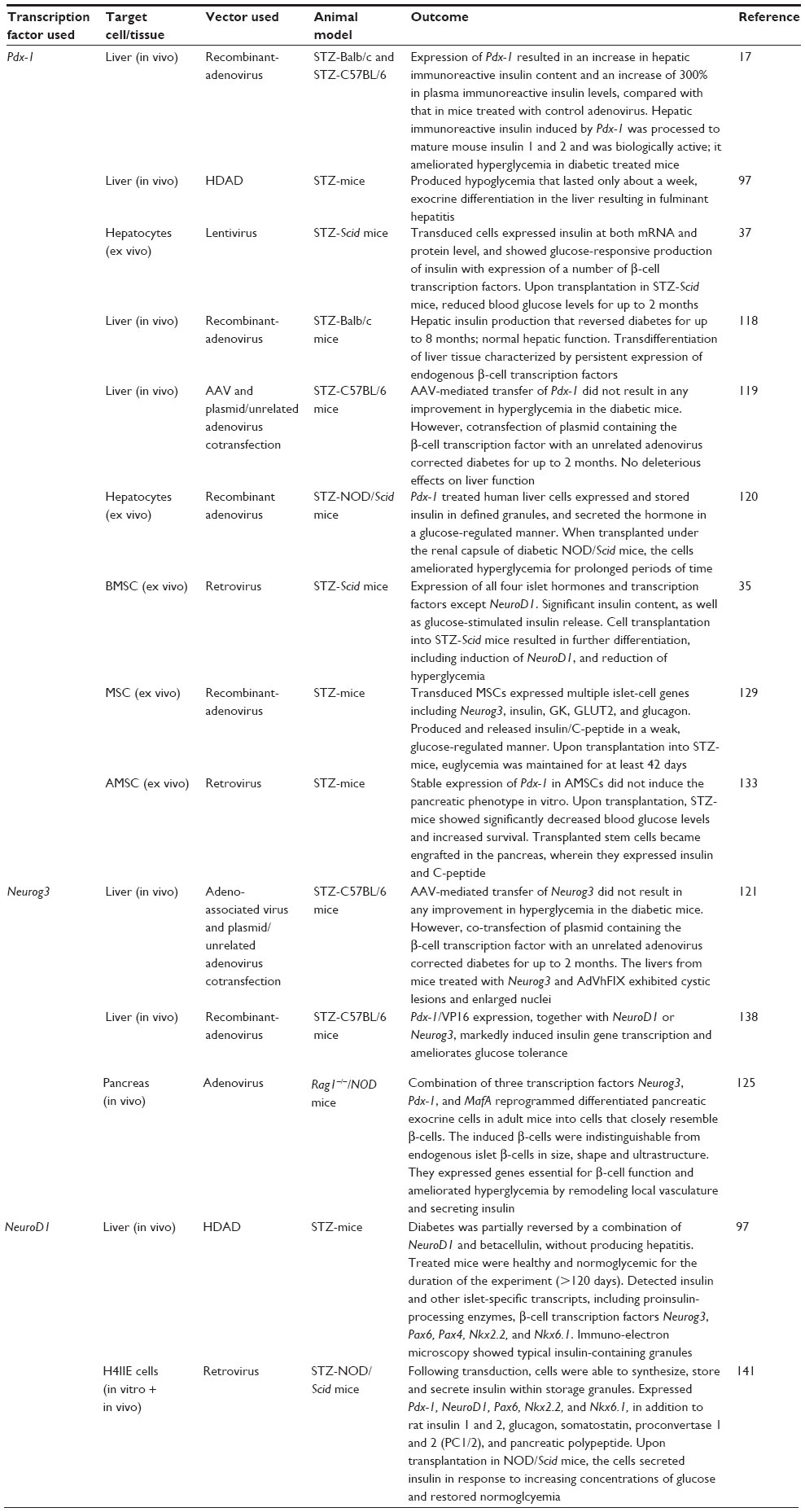 | Table 3 β-cell transcription factors used for the reversal of T1D in animal models |
Similarly, Kojima et al reported the development of exocrine differentiation in the livers of STZ-diabetic mice after delivering Pdx-1 with the use of a helper-dependent adenovirus.97 This was most likely due to the continuous expression of high levels of Pdx-1 as a consequence of the use of the ubiquitously expressed elongation factor-1α promoter. To date, the process of transdifferentiation from hepatocytes to pancreatic tissue via the direct delivery of Pdx-1 has been performed on multiple occasions.37,120–123 However, direct delivery of Pdx-1 has been pursued in a variety of other cell types, including mouse pancreas via the bile duct,124 rat intestinal epithelium-derived cells (IEC-6),125 and primary duct cells.126 Delivery of a combination of pancreatic transcription factors (Pdx-1, Neurog3, and MafA) was successful in converting pancreatic exocrine cells in vivo to closely resemble β-cells,127 and served as evidence for the use of transcription factor combinations. Despite the transduced cells showing all the characteristics of normal β-cells, they were limited by the low number of successfully converted exocrine cells and the fact they did not organize themselves into islet structures.
Due to the success of Pdx-1 to induce pancreatic transdifferentiation and generate artificial β-cells in a number of differentiated cell types, there was a logical transition to stem cells as targets to exploit their regenerative capabilities and plasticity. Considering that MSCs possess unique immune-evading capabilities, their use as targets for gene transfer has been pursued with great interest. Pdx-1 has been delivered to MSCs from a variety of sources, including BMSC,35,128–132 umbilical cord MSC,133 and adipose-derived MSC36,134,135 with varying success in the generation of glucose-responsive artificial β-cells. Embryonic stem cells (ESCs) have also been targeted for transfer of Pdx-1, with a study by Miyazaki et al showing that a murine ESC line (EB3) could be induced to differentiate into artificial β-cells. However, due to a lack of expression of pancreatic genes in vivo, there was no therapeutic potential.136 As a result, this was followed with a number of studies in other ESC lines137–139 attempting to improve the generation of artificial β-cells that would be suitable candidates for therapeutic adaptation.
Considering that Neurog3 has a pivotal role in defining the endocrine cell fate and is located lower in the β-cell transcription factor hierarchy, it could potentially overcome the problem of exocrine differentiation and may be used to produce artificial β-cells. However, most studies have reported low levels of insulin production after delivery of Neurog3.121,126,140–142 A study using adenoviral transfer of Neurog3 and betacellulin to oval cells resulted in the production of insulin and transdifferentiation;143 however the most effective use of Neurog3 delivery was in combination with other transcription factors.127 To successfully overcome the exocrine differentiation induced by the transfer of Pdx-1, Kojima et al expressed NeuroD1 and betacellulin in the livers of STZ-treated diabetic mice.97 They demonstrated the restoration of normoglycemia in these mice for more than 120 days, along with the expression of pancreatic transcription factors Neurog3, Pax6, Pax4, Nkx2.2, and Nkx6.1. Most importantly, they did not observe any exocrine differentiation or significant hepatotoxicity. The ability of NeuroD1 to strongly induce insulin expression also makes it an ideal alternative for the generation of artificial β-cells.126,144
Our laboratory has reported promising results using viral delivery of NeuroD1 to a genetically modified rat liver cell line (H4IIE) which does not express β-cell transcription factors. The H4IIE cells were engineered to express both insulin and NeuroD1,145 and following transduction were able to synthesize, store, and secrete insulin within storage granules. Upon transplantation in NOD/Scid mice, the cells secreted insulin in response to increasing concentrations of glucose and restored normoglycemia. They also induced the expression of Pdx-1, NeuroD1, Pax6, Nkx2.2, and Nkx6.1, in addition to rat insulin 1 and 2, glucagon, somatostatin, proconvertase 1 and 2 (PC1/2), and pancreatic polypeptide. This study provides evidence for the potential use of NeuroD1 in gene therapy protocols to induce safe differentiation.
Pax4 is necessary for defining β-cell fate and could be used for the generation of artificial β-cells. A study by Liew et al showed that overexpression of Pax4 in human ESCs enhances their ability to form putative β-cells.146 This was supported by a study that showed that insulin-producing cells generated via the overexpression of Pax4 in mouse ESCs and selected for nestin expression were capable of maintaining normal blood glucose levels for 14 days.147 The use of ESCs as targets for gene therapy is, however, limited by their propensity for teratoma formation, which limits their potential for clinical application.148,149
Since knockouts of Nkx6.1 in mice have shown a disruption in the development of β-cells, the use of the β-cell-specific transcription factor makes it a good candidate for gene transfer applications. It has been demonstrated that ectopic expression of Nkx6.1 alone is not a strong inducer of upper-hierarchy β-cell transcription factor expression, and that only upon coexpression with Pdx-1 was it capable of substantial insulin expression and glucose-responsive secretion of insulin.150 The lack of expression of the full hierarchy of β-cell transcription factors makes Nkx6.1 a mediocre choice for the generation of artificial β-cells for analysis in animal models of diabetes.
Conclusion
Reversal of T1D via gene transfer in animal models has had varying success to date. The choice of the model to assess the success of any potential therapy is of significant importance, as the future clinical application of T1D therapies in humans should be primarily assessed in a model of diabetes which closely resembles the human situation. Currently, the NOD mouse model is the most widely studied and shows similar pathophysiology to human diabetes. With the development of improved molecular techniques such as generalized knock-outs, tissue-specific knockouts, and knock-ins, it should be possible to generate a large number of new animal models for specific diabetes research. Considering the autoimmune nature of diabetes, overcoming recurrent autoimmunity toward engineered cell therapies is one of the major hurdles facing this area of research. In addition, mimicking the tightly regulated control of glucose concentrations which occurs within normal β-cells is a phenomenon that has yet to be exquisitely adapted in current cell therapies. In this review, we have suggested that gene transfer of β-cell transcription factors and insulin show considerable promise in overcoming these challenges. Looking to the future, if a cell therapy is to be brought to the clinic, we believe that the targeting of an allogeneic tissue source capable of circumventing the autoimmune response for the generation of artificial β-cells shows most promise in overcoming the current challenges limiting cell and gene therapies for the treatment of T1D.
Acknowledgments
Dario Gerace is supported by an Australian Postgraduate Award and the Arrow Bone Marrow Transplant Foundation/Hawkesbury Canoe Classic Scholarship. Research conducted by Ann M Simpson and Dario Gerace was supported by the National Health and Medical Research Council of Australia Project Grants (352909, 513100). Ann M Simpson and Rosetta Martiniello-Wilks also received grants from Diabetes Australia Research Trust and Rebecca L Cooper Medical Research Foundation. The authors would like to thank Richard Limburg for IT support.
Disclosure
Ann M Simpson is an inventor in the patent “Cells genetically modified to comprise pancreatic islet glucokinase and uses thereof”; WO 2009021276 A1, Ann M Simpson and Chang Tao, European patent: EP20080782908, Australian patent: AU 2008/001160, United States of America patent: US12/672,832. The authors declare no other conflicts of interest.
References
Atkinson MA, Maclaren NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med. 1994;19(331):1428–1436. | |
van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91(1):79–118. | |
Margulies D, Ergun-longmire B, Ten S, Maclaren N. Pediatric Endocrinology, Chapter 5: Diabetes Mellitus. South Dartmouth, MA: MDText.com, Inc.; 2010. Available from: http://www.endotext.org. | |
Melendez-Ramirez LY, Richards RJ, Cefalu WT. Complications of type 1 diabetes. Endocrinol Metab Clin North Am. 2010;39(3):625–640. | |
Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care. 2003;26(6):1902–1912. | |
Smith CB, Choudhary P, Pernet A, Hopkins D, Amiel SA. Hypoglycemia unawareness is associated with reduced adherence to therapeutic decisions in patients with type 1 diabetes: evidence from a clinical audit. Diabetes Care. 2009;32(7):1196–1198. | |
Marzorati S, Melzi R, Citro A, et al. Engraftment versus immunosuppression: cost-benefit analysis of immunosuppression after intrahepatic murine islet transplantation. Transplantation. 2014;97(10):1019–1026. | |
Haller MJ, Viener H-L, Wasserfall C, Brusko T, Atkinson MA, Schatz DA. Autologous umbilical cord blood infusion for type 1 diabetes. Exp Hematol. 2008;36(6):710–715. | |
Luo X, Yang H, Kim IS, et al. Systemic transforming growth factor-β1 gene therapy induces Foxp3+ regulatory cells, restores self-tolerance, and facilitates regeneration of beta cell function in overtly diabetic nonobese diabetic mice. Transplantation. 2005;79(9):1091–1096. | |
Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48(12):2270–2276. | |
Tuch BE, Hughes TC, Evans MDM. Encapsulated pancreatic progenitors derived from human embryonic stem cells as a therapy for insulin-dependent diabetes. Diabetes Metab Res Rev. 2011;27(8):928–932. | |
Jacobs-Tulleneers-Thevissen D, Chintinne M, Ling Z, et al; Beta Cell Therapy Consortium EU-FP7. Sustained function of alginate-encapsulated human islet cell implants in the peritoneal cavity of mice leading to a pilot study in a type 1 diabetic patient. Diabetologia. 2013;56(7):1605–1614. | |
Breton M, Farret A, Bruttomesso D, Anderson S, et al; International Artificial Pancreas Study Group. Fully integrated artificial pancreas in type 1 diabetes: modular closed-loop glucose control maintains near normoglycemia. Diabetes. 2012;61(9):2230–2237. | |
Hovorka R, Kumareswaran K, Harris J, et al. Overnight closed loop insulin delivery (artificial pancreas) in adults with type 1 diabetes: crossover randomised controlled studies. BMJ. 2011;342:d1855. | |
Stewart C, Taylor NA, Green IC, Docherty K, Bailey CJ. Insulin-releasing pituitary cells as a model for somatic cell gene therapy in diabetes mellitus. J Endocrinol. 1994;142(2):339–343. | |
Tuch BE, Szymanska B, Yao M, et al. Function of a genetically modified human liver cell line that stores, processes and secretes insulin. Gene Ther. 2003;10(6):490–503. | |
Ferber S, Halkin A, Cohen H, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6(5):568–572. | |
Ren B, O’Brien BA, Swan MA, et al. Long-term correction of diabetes in rats after lentiviral hepatic insulin gene therapy. Diabetologia. 2007;50(9):1910–1920. | |
Ren B, O’Brien BA, Byrne MR, et al. Long-term reversal of diabetes in non-obese diabetic mice by liver-directed gene therapy. J Gene Med. 2013;15(1):28–41. | |
Gerace D, Ren B, Hawthorne WJ, et al. Pancreatic transdifferentiation in porcine liver following lentiviral delivery of human furin–cleavable insulin. Transplant Proc. 2013;45(5):1869–1874. | |
Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J. 1922;12(3):141–146. | |
Barnes AJ, Bloom SR. Pancreatectomised man: a mode for diabetes without glucagon. Lancet. 1976;307(7953):219–221. | |
Bono VH Jr. Review of mechanism of action studies of the nitrosoureas. Cancer Treat Rep. 1976;60(6):699–702. | |
Dekel Y, Glucksam Y, Elron-Gross I, Margalit R. Insights into modeling streptozotocin-induced diabetes in ICR mice. Lab Anim. 2009;38(2):55–60. | |
Leiter EH. Multiple low-dose streptozotocin-induced hyperglycemia and insulitis in C57BL mice: influence of inbred background, sex, and thymus. Proc Natl Acad Sci U S A. 1982;79(2):630–634. | |
Nerup J, Mandrap-Poulsen T, Helqvist S, et al. On the pathogenesis of IDDM. Diabetologia. 1994;37(2):S82–S89. | |
Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50(6):537–546. | |
Grossman EJ, Lee DD, Tao J, et al. Glycemic control promotes pancreatic beta-cell regeneration in streptozotocin-induced diabetic mice. PLoS One. 2010;5(1):e8749. | |
Hanafusa T, Miyagawa J, Nakajima H, et al. The NOD mouse. Diabetes Res Clin Pract. 1994;24(Suppl):S307–S311. | |
Yoon JW, Jun HS. Cellular and molecular pathogenic mechanisms of insulin-dependent diabetes mellitus. Ann N Y Acad Sci. 2001;928:200–211. | |
Yang Y, Santamaria P. Lessons on autoimmune diabetes from animal models. Clin Sci. 2006;110(6):627–639. | |
Zhao Y, Jiang Z, Zhao T, et al. Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood-derived multipotent stem cells. BMC Med. 2012;10(1):3. | |
Zhao Y, Lin B, Darflinger R, Zhang Y, Holterman MJ, Skidgel RA. Human cord blood stem cell-modulated regulatory T lymphocytes reverse the autoimmune-caused type 1 diabetes in non-obese diabetic (NOD) mice. PLoS One. 2009;4(1):e4226. | |
Roep BO, Tree TIM. Immune modulation in humans: implications for type 1 diabetes mellitus. Nat Rev Endocrinol. 2014;10(4):229–242. | |
Karnieli O, Izhar-Prato Y, Bulvik S, Efrat S. Generation of insulin-producing cells from human bone marrow mesenchymal stem cells by genetic manipulation. Stem Cells. 2007;25(11):2837–2844. | |
Kajiyama H, Hamazaki TS, Tokuhara M, et al. Pdx1-transfected adipose tissue-derived stem cells differentiate into insulin-producing cells in vivo and reduce hyperglycemia in diabetic mice. Int J Dev Biol. 2010;54(4):699–705. | |
Fodor A, Harel C, Fodor L, et al. Adult rat liver cells transdifferentiated with lentiviral IPF1 vectors reverse diabetes in mice: an ex vivo gene therapy approach. Diabetologia. 2007;50(1):121–130. | |
Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol. 2011;33(1):67–87. | |
Rossini AA, Handler ES, Mordes JP, Greiner DL. Human autoimmune diabetes mellitus: lessons from BB rats and NOD mice – caveat emptor. Clin Immunol Immunopathol. 1995;74(1):2–9. | |
Prochazka M, Serreze DV, Frankel WN, Leiter EH. NOR/Lt mice: MHC-matched diabetes-resistant control strain for NOD mice. Diabetes. 1992;41(1):98–106. | |
Nakhooda AF, Like AA, Chappel CI, Murray FT, Marliss EB. The spontaneously diabetic Wistar rat. Metabolic and morphologic studies. Diabetes. 1977;26(2):100–112. | |
Mordes JP, Bortell R, Blankenhorn EP, Rossini AA, Greiner DL. Rat models of type 1 diabetes: genetics, environment, and autoimmunity. ILAR J. 2004;45(3):278–291. | |
Lenzen S, Tiedge M, Elsner M, et al. The LEW.1AR1/Ztm-iddm rat: a new model of spontaneous insulin-dependent diabetes mellitus. Diabetologia. 2001;44(9):1189–1196. | |
Kramer JW, Nottingham S, Robinette J, Lenz G, Sylvester S, Dessouky MI. Inherited, early onset, insulin-requiring diabetes mellitus of Keeshond dogs. Diabetes. 1980;29(7):558–565. | |
Howard CF. Spontaneous diabetes in Macaca nigra. Diabetes. 1972; 21(11):1077–1090. | |
Mathews CE, Langley SH, Leiter EH. New mouse model to study islet transplantation in insulin-dependent diabetes mellitus. Transplantation. 2002;73(8):1333–1336. | |
Yoon J-W, London WT, Curfman BL, Brown RL, Notkins AL. Coxsackie virus B4 produces transient diabetes in nonhuman primates. Diabetes. 1986;35(6):712–716. | |
Craighead JE, McLane MF. Diabetes mellitus: induction in mice by encephalomyocarditis virus. Science. 1968;162(3856):913–914. | |
Guberski D, Thomas V, Shek W, et al. Induction of type I diabetes by Kilham’s rat virus in diabetes-resistant BB/Wor rats. Science. 1991;254(5034):1010–1013. | |
Morgan RA, Anderson WF. Human gene therapy. Annu Rev Biochem. 1993;62:191–217. | |
Bushman FD. Retroviral integration and human gene therapy. J Clin Invest. 2007;117(8):2083–2086. | |
Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288(5466):669–672. | |
Laufs S, Nagy KZ, Giordano FA, Hotz-Wagenblatt A, Zeller WJ, Fruehauf S. Insertion of retroviral vectors in NOD/SCID repopulating human peripheral blood progenitor cells occurs preferentially in the vicinity of transcription start regions and in introns. Mol Ther. 2004;10(5):874–881. | |
Xu J, Lu Y, Ding F, Zhan X, Zhu M, Wang Z. Reversal of diabetes in mice by intrahepatic injection of bone-derived GFP-murine mesenchymal stem cells infected with the recombinant retrovirus-carrying human insulin gene. World J Surg. 2007;31(9):1872–1882. | |
Volpers C, Kochanek S. Adenoviral vectors for gene transfer and therapy. J Gene Med. 2004;6(S1):S164–S171. | |
Wold WS, Doronin K, Toth K, Kuppuswamy M, Lichtenstein DL, Tollefson AE. Immune responses to adenoviruses: viral evasion mechanisms and their implications for the clinic. Curr Opin Immunol. 1999;11(4):380–386. | |
McCaffrey AP, Fawcett P, Nakai H, et al. The host response to adenovirus, helper-dependent adenovirus, and adeno-associated virus in mouse liver. Mol Ther. 2008;16(5):931–941. | |
Alba R, Bosch A, Chillon M. Gutless adenovirus: last-generation adenovirus for gene therapy. Gene Ther. 2005;12:18–27. | |
Zhou HS, Liu DP, Liang CC. Challenges and strategies: the immune responses in gene therapy. Med Res Rev. 2004;24(6):748–761. | |
Muzyczka N. Use of adeno-associated virus as a general transduction vector for mammalian cells. In: Muzyczka N, editor. Viral Expression Vectors. Vol 158. Berlin, Heidelberg: Springer; 1992:97–129. | |
Sugiyama A, Hattori S, Tanaka S, et al. Defective adenoassociated viral-mediated transfection of insulin gene by direct injection into liver parenchyma decreases blood glucose of diabetic mice. Horm Metab Res. 1997;29(12):599–603. | |
Yoon JW, Jun HS. Recent advances in insulin gene therapy for type 1 diabetes. Trends Mol Med. 2002;8(2):62–68. | |
Zufferey R, Dull T, Mandel RJ, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72(12):9873–9880. | |
Laub O, Rutter WJ. Expression of the human insulin gene and cDNA in a heterologous mammalian system. J Biol Chem. 1983;258(10):6043–6050. | |
Iwata H, Ogawa N, Takagi T, Mizoguchi J. Preparation of insulin-releasing Chinese hamster ovary cell by transfection of human insulin gene. Polymers of Biological and Biomedical Significance. Vol 540. Washington, DC: American Chemical Society; 1993:306–313. | |
Barry SC, Ramesh N, Lejnieks D, et al. Glucose-regulated insulin expression in diabetic rats. Hum Gene Ther. 2001;12(2):131–139. | |
Mas A, Montané J, Anguela XM, et al. Reversal of type 1 diabetes by engineering a glucose sensor in skeletal muscle. Diabetes. 2006;55(6):1546–1553. | |
Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322(5907):1490–1494. | |
Hughes SD, Quaade C, Johnson JH, Ferber S, Newgard CB. Transfection of AtT-20ins cells with GLUT-2 but not GLUT-1 confers glucose-stimulated insulin secretion. Relationship to glucose metabolism. J Biol Chem. 1993;268(20):15205–15212. | |
Zaret KS. Genetic programming of liver and pancreas progenitors: lessons for stem-cell differentiation. Nat Rev Genet. 2008;9(5):329–340. | |
Elsner M, Terbish T, Jorns A, et al. Reversal of diabetes through gene therapy of diabetic rats by hepatic insulin expression via lentiviral transduction. Mol Ther. 2012;20(5):918–926. | |
Short DK, Okada S, Yamauchi K, Pessin JE. Adenovirus-mediated transfer of a modified human proinsulin gene reverses hyperglycemia in diabetic mice. Am J Physiol. 1998;275(5 pt 1):E748–E756. | |
Vollenweider F, Irminger JC, Gross DJ, Villa-Komaroff L, Halban PA. Processing of proinsulin by transfected hepatoma (FAO) cells. J Biol Chem. 1992;267(21):14629–14636. | |
Gebler A, Zabel O, Seliger B. The immunomodulatory capacity of mesenchymal stem cells. Trends Mol Med. 2012;18(2):128–134. | |
Vija L, Farge D, Gautier JF, et al. Mesenchymal stem cells: stem cell therapy perspectives for type 1 diabetes. Diabetes Metab. 2009; 35(2):85–93. | |
Abdi R, Fiorina P, Adra CN, Atkinson M, Sayegh MH. Immunomodulation by mesenchymal stem cells: a potential therapeutic strategy for type 1 diabetes. Diabetes. 2008;57(7):1759–1767. | |
da Silva Meirelles L, Fontes AM, Covas DT, Caplan AI. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev. 2009;20(5):419–427. | |
Lee RH, Seo MJ, Reger RL, et al. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci U S A. 2006;103(46):17438–17443. | |
Ezquer FE, Ezquer ME, Parrau DB, Carpio D, Yanez AJ, Conget PA. Systemic administration of multipotent mesenchymal stromal cells reverts hyperglycemia and prevents nephropathy in type 1 diabetic mice. Biol Blood Marrow Transplant. 2008;14(6):631–640. | |
Tang DQ, Cao LZ, Burkhardt BR, et al. In vivo and in vitro characterization of insulin-producing cells obtained from murine bone marrow. Diabetes. 2004;53(7):1721–1732. | |
Oh S-H, Muzzonigro TM, Bae S-H, LaPlante JM, Hatch HM, Petersen BE. Adult bone marrow-derived cells trans-differentiating into insulin-producing cells for the treatment of type I diabetes. Lab Invest. 2004;84(5):607–617. | |
Wu XH, Liu CP, Xu KF, et al. Reversal of hyperglycemia in diabetic rats by portal vein transplantation of islet-like cells generated from bone marrow mesenchymal stem cells. World J Gastroenterol. 2007;13(24):3342–3349. | |
Sun Y, Chen L, Hou XG, et al. Differentiation of bone marrow-derived mesenchymal stem cells from diabetic patients into insulin-producing cells in vitro. Chin Med J. 2007;120(9):771–776. | |
Brunner Y, Coute Y, Iezzi M, et al. Proteomics analysis of insulin secretory granules. Mol Cell Proteomics. 2007;6:1007–1017. | |
Howell SL. The mechanism of insulin secretion. Diabetologia. 1984;26(5):319–327. | |
Lee HC, Kim SJ, Kim KS, Shin HC, Yoon JW. Remission in models of type 1 diabetes by gene therapy using a single-chain insulin analogue. Nature. 2000;408(6811):483–488. | |
Thule PM, Liu J, Phillips LS. Glucose regulated production of human insulin in rat hepatocytes. Gene Ther. 2000;7:205–214. | |
Thule PM, Liu J. Regulated hepatic insulin gene therapy of STZ-diabetic rats. Gene Ther. 2000;7:1744–1752. | |
Mitanchez D, Doiron B, Chen R, Kahn A. Glucose-stimulated genes and prospects of gene therapy for type I diabetes. Endocr Rev. 1997;18(4):520–540. | |
Chen R, Meseck M, McEvoy RC, Woo SL. Glucose-stimulated and self-limiting insulin production by glucose 6-phosphatase promoter driven insulin expression in hepatoma cells. Gene Ther. 2000;7(21):1802–1809. | |
Chen NK, Tan SY, Udolph G, Kon OL. Insulin expressed from endogenously active glucose-responsive EGR1 promoter in bone marrow mesenchymal stromal cells as diabetes therapy. Gene Ther. 2010;17(5):592–605. | |
Harada S, Smith RM, Smith JA, Shah N, Hu DQ, Jarett L. Insulin-induced egr-1 expression in Chinese hamster ovary cells is insulin receptor and insulin receptor substrate-1 phosphorylation-independent. Evidence of an alternative signal transduction pathway. J Biol Chem. 1995;270(44):26632–26638. | |
Rees DA, Alcolado JC. Animal models of diabetes mellitus. Diabet Med. 2005;22(4):359–370. | |
Sander M, Griffen SC, Huang J, German MS. A novel glucose-responsive element in the human insulin gene functions uniquely in primary cultured islets. Proc Natl Acad Sci U S A. 1998;95(20):11572–11577. | |
Burkhardt BR, Loiler SA, Anderson JA, et al. Glucose-responsive expression of the human insulin promoter in HepG2 human hepatoma cells. Ann N Y Acad Sci. 2003;1005:237–241. | |
Burkhardt BR, Yang MC, Robert CE, et al. Tissue-specific and glucose-responsive expression of the pancreatic derived factor (PANDER) promoter. Biochim Biophys Acta. 2005;1730(3):215–225. | |
Kojima H, Fujimiya M, Matsumura K, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9(5):596–603. | |
Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–2365. | |
Lisowski L, Dane AP, Chu K, et al. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014; 506(7488):382–386. | |
Apelqvist A, Li H, Sommer L, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877–881. | |
Sommer L, Ma Q, Anderson DJ. Neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Mol Cell Neurosci. 1996;8(4):221–241. | |
Shanmukhappa K, Mourya R, Sabla GE, Degen JL, Bezerra JA. Hepatic to pancreatic switch defines a role for hemostatic factors in cellular plasticity in mice. Proc Natl Acad Sci U S A. 2005; 102(29):10182–10187. | |
Kim S, Shin J-S, Kim H-J, Fisher RC, Lee M-J, Kim C-W. Streptozotocin-induced diabetes can be reversed by hepatic oval cell activation through hepatic transdifferentiation and pancreatic islet regeneration. Lab Invest. 2007;87(7):702–712. | |
Yang L, Li S, Hatch H, et al. In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci U S A. 2002;99(12):8078–8083. | |
Han J, McLane B, Kim E-H, Yoon J-W, Jun H-S. Remission of diabetes by insulin gene therapy using a hepatocyte-specific and glucose-responsive synthetic promoter. Mol Ther. 2011;19(3):470–478. | |
Tatake RJ, O’Neill MM, Kennedy CA, et al. Glucose-regulated insulin production from genetically engineered human non-beta cells. Life Sci. 2007;81(17–18):1346–1354. | |
Tudurí E, Bruin JE, Kieffer TJ. Restoring insulin production for type 1 diabetes. J Diabetes. 2012;4(4):319–331. | |
Hsu P-J, Kotin R, Yang Y-W. Glucose- and metabolically regulated hepatic insulin gene therapy for diabetes. Pharm Res. 2008;25(6):1460–1468. | |
Tabiin MT, Tuch BE, Bai L, Han XG, Simpson AM. Susceptibility of insulin-secreting hepatocytes to the toxicity of pro-inflammatory cytokines. J Autoimmun. 2001;17(3):229–242. | |
Tuch BE, Beynon S, Tabiin MT, Sassoon R, Goodman RJ, Simpson AM. Effect of beta-cell toxins on genetically engineered insulin-secreting cells. J Autoimmun. 1997;10(3):239–244. | |
Tabiin MT, White CP, Morahan G, Tuch BE. Insulin expressing hepatocytes not destroyed in transgenic NOD mice. J Autoimmune Dis. 2004;1(1):3. | |
Sander M, German MS. The beta cell transcription factors and development of the pancreas. J Mol Med. 1997;75(5):327–340. | |
Chakrabarti SK, Mirmira RG. Transcription factors direct the development and function of pancreatic beta cells. Trends Endocrinol Metab. 2003;14(2):78–84. | |
Ang SL, Wierda A, Wong D, et al. The formation and maintenance of the definitive endoderm lineage in the mouse: involvement of HNF3/forkhead proteins. Development. 1993;119(4):1301–1315. | |
Korolija M, Hadzija MP, Hadzija M. Molecular mechanism in beta-cell development: the role of Pdx1, Ngn3 and Pax4 proteins. Period Biol. 2009;111(1):59–63. | |
Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. | |
Gasa R, Mrejen C, Lynn FC, et al. Induction of pancreatic islet cell differentiation by the neurogenin-neuroD cascade. Differentiation. 2008;76(4):381–391. | |
Naya FJ, Huang HP, Qiu Y, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11(18):2323–2334. | |
Sander N, Sussel L, Conners J, et al. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development. 2000;127(24):5533–5540. | |
Ber I, Shternhall K, Perl S, et al. Functional, persistent, and extended liver to pancreas transdifferentiation. J Biol Chem. 2003;278(34):31950–31957. | |
Wang AY, Ehrhardt A, Xu H, Kay MA. Adenovirus transduction is required for the correction of diabetes using Pdx-1 or Neurogenin-3 in the liver. Mol Ther. 2007;15(2):255–263. | |
Sapir T, Shternhall K, Meivar-Levy I, et al. Cell-replacement therapy for diabetes: generating functional insulin-producing tissue from adult human liver cells. Proc Natl Acad Sci U S A. 2005;102(22):7964–7969. | |
Nagaya M, Katsuta H, Kaneto H, Bonner-Weir S, Weir GC. Adult mouse intrahepatic biliary epithelial cells induced in vitro to become insulin-producing cells. J Endocrinol. 2009;201(1):37–47. | |
Taniguchi H, Yamato E, Tashiro F, Ikegami H, Ogihara T, Miyazaki J. Beta-cell neogenesis induced by adenovirus-mediated gene delivery of transcription factor pdx-1 into mouse pancreas. Gene Ther. 2003;10(1):15–23. | |
Yoshida S, Kajimoto Y, Yasuda T, et al. PDX-1 induces differentiation of intestinal epithelioid IEC-6 into insulin-producing cells. Diabetes. 2002;51(8):2505–2513. | |
Noguchi H, Xu G, Matsumoto S, et al. Induction of pancreatic stem/progenitor cells into insulin-producing cells by adenoviral-mediated gene transfer technology. Cell Transplant. 2006;15(10):929–938. | |
Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to b-cells. Nature. 2008;455(7213):627–632. | |
Sun J, Yang Y, Wang X, Song J, Jia Y. Expression of Pdx-1 in bone marrow mesenchymal stem cells promotes differentiation of islet-like cells in vitro. Sci China C Life Sci. 2006;49(5):480–489. | |
Limbert C, Path G, Ebert R, et al. PDX1- and NGN3-mediated in vitro reprogramming of human bone marrow-derived mesenchymal stromal cells into pancreatic endocrine lineages. Cytotherapy. 2011;13(7):802–813. | |
Moriscot C, de Fraipont F, Richard M-J, et al. Human bone marrow mesenchymal stem cells can express insulin and key transcription factors of the endocrine pancreas developmental pathway upon genetic and/or microenvironmental manipulation in vitro. Stem Cells. 2005;23(4):594–603. | |
Li Y, Zhang R, Qiao H, et al. Generation of insulin-producing cells from PDX-1 gene-modified human mesenchymal stem cells. J Cell Physiol. 2007;211(1):36–44. | |
Li L, Li F, Qi H, et al. Coexpression of Pdx1 and betacellulin in mesenchymal stem cells could promote the differentiation of nestin-positive epithelium-like progenitors and pancreatic islet-like spheroids. Stem Cells Dev. 2008;17(4):815–823. | |
He D, Wang J, Gao Y, Zhang Y. Differentiation of PDX1 gene-modified human umbilical cord mesenchymal stem cells into insulin-producing cells in vitro. Int J Mol Med. 2011;28(6):1019–1024. | |
Baer PC. Adipose-derived stem cells and their potential to differentiate into the epithelial lineage. Stem Cells Dev. 2011;20(10):1805–1816. | |
Lin G, Wang G, Liu G, et al. Treatment of type 1 diabetes with adipose tissue-derived stem cells expressing pancreatic duodenal homeobox 1. Stem Cells Dev. 2009;18(10):1399–1406. | |
Miyazaki S, Yamato E, Miyazaki J-I. Regulated expression of pdx-1 promotes in vitro differentiation of insulin-producing cells from embryonic stem cells. Diabetes. 2004;53(4):1030–1037. | |
Lavon N, Yanuka O, Benvenisty N. The effect of overexpression of Pdx1 and Foxa2 on the differentiation of human embryonic stem cells into pancreatic cells. Stem Cells. 2006;24(8):1923–1930. | |
Vincent R, Treff N, Budde M, Kastenberg Z, Odorico J. Generation and characterization of novel tetracycline-inducible pancreatic transcription factor-expressing murine embryonic stem cell lines. Stem Cells Dev. 2006;15(6):953–962. | |
Raikwar SP, Zavazava N. PDX1-engineered embryonic stem cell-derived insulin producing cells regulate hyperglycemia in diabetic mice. Transplant Res. 2012;1(1):19. | |
Kaneto H, Nakatani Y, Miyatsuka T, et al. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes. 2005;54(4):1009–1022. | |
Song YD, Lee EJ, Yashar P, Pfaff LE, Kim SY, Jameson JL. Islet cell differentiation in liver by combinatorial expression of transcription factors neurogenin-3, BETA2, and RIPE3b1. Biochem Biophys Res Commun. 2007;354(2):334–339. | |
Heremans Y, Van De Casteele M, in’t Veld P, et al. Recapitulation of embryonic neuroendocrine differentiation in adult human pancreatic duct cells expressing neurogenin 3. J Cell Biol. 2002;159(2):303–312. | |
Yechoor V, Liu V, Espiritu C, et al. Neurogenin3 is sufficient for transdetermination of hepatic progenitor cells into neo-islets in vivo but not transdifferentiation of hepatocytes. Dev Cell. 2009;16(3):358–373. | |
Yatoh S, Akashi T, Chan PP, et al. NeuroD and reaggregation induce β-cell specific gene expression in cultured hepatocytes. Diabetes Metab Res Rev. 2007;23(3):239–249. | |
Simpson AM, Tao C, Swan MA, Ren B, O’Brien BA. An engineered rat liver cell line H4IIEins/ND reverses diabetes in mice. In: International Diabetes Federation World Diabetes Congress; 2009; Montreal. Abstract no MT-0996. | |
Liew CG, Shah NN, Briston SJ, et al. PAX4 enhances beta-cell differentiation of human embryonic stem cells. PLoS One. 2008; 3(3):e1783. | |
Blyszczuk P, Czyz J, Kania G, et al. Expression of Pax4 in embryonic stem cells promotes differentiation of nestin-positive progenitor and insulin-producing cells. Proc Natl Acad Sci U S A. 2003;100(3):998–1003. | |
Stachelscheid H, Wulf-Goldenberg A, Eckert K, et al. Teratoma formation of human embryonic stem cells in three-dimensional perfusion culture bioreactors. J Tissue Eng Regen Med. 2013;7(9):729–741. | |
Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2009;2(3):198–210. | |
Gefen-Halevi S, Rachmut IH, Molakandov K, et al. NKX6.1 promotes PDX-1-induced liver to pancreatic beta-cells reprogramming. Cell Reprogram. 2010;12(6):655–664. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.