")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Diabetes Mellitus Promotes Atrial Structural Remodeling and PARP-1/Ikkα/NF-κB Pathway Activation in Mice
Authors Meng T , Wang J , Tang M, Liu S, Ding L, Yan Y
Received 13 January 2021
Accepted for publication 2 April 2021
Published 17 May 2021 Volume 2021:14 Pages 2189—2199
DOI https://doi.org/10.2147/DMSO.S300616
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ming-Hui Zou
Tianyu Meng,1,* Jie Wang,2,3,* Manyun Tang,2 Shangyu Liu,4 Ligang Ding,4 Yang Yan5
1Department of Geriatrics, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, 710061, People’s Republic of China; 2Department of Cardiovascular Medicine, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, 710061, People’s Republic of China; 3Department of Hematology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, 710061, People’s Republic of China; 4Clinical EP Lab & Arrhythmia Center, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, People’s Republic of China; 5Department of Cardiovascular Surgery, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, 710061, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ligang Ding
Clinical EP Lab & Arrhythmia Center, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, People’s Republic of China
Email [email protected]
Yang Yan
Department of Cardiovascular Surgery, The First Affiliated Hospital of Xi’an Jiaotong University, No. 277 Yanta West Road, Xi’an, Shaanxi, 710061, People’s Republic of China
Tel +86-2985323869
Fax +86-2985323869
Email [email protected]
Background: Diabetes mellitus (DM) has been demonstrated to be linked to atrial fibrillation (AF). However, the underlying mechanisms of the DM-associated increase in AF susceptibility and the potential effects of DM on atrial remodeling remain unclear.
Methods and Results: Twenty-five C57BL/6 mice were randomly assigned to the normal/control group (Con, n=10) and model group (n=15). Mice in the model group were administered a high-fat diet combined with multiple injections of low-dose streptozocin (STZ) (35 mg/kg). Eleven mice were ultimately included in DM group. Left atrial tissue structural and inflammatory alterations were assessed. In our study, the atrial weights of DM mice were markedly heavier than those of mice in the Con group. DM mice exhibited significantly increased fasting plasma glucose, fasting insulin, and dyslipidaemia. Furthermore, H&E and Masson’s staining revealed broadened interstitial spaces, myocyte disarray and atrial fibrosis in DM mice. The expression levels of the atrial inflammation-associated factor nuclear factor κB (NF-κB) and its pathway were significantly altered in the atria of DM mice.
Conclusion: DM could induce atrial structural remodeling and inflammation in mice.
Keywords: diabetes mellitus, inflammation, atrial remodeling, PARP-1, NF-κB
Introduction
Diabetes mellitus (DM) is one of the most common non-infectious chronic diseases and seriously affects human health and longevity. Patients with DM often suffer from obesity, hypercholesterolemia, atherosclerosis, microcirculation disorders and hypertension, which significantly increase the risk of heart damage, and cardiovascular complications are the main cause of death in DM patients.1,2
Atrial fibrillation (AF), the most common tachyarrhythmia, is considered to be a growing cardiovascular epidemic. Previous studies have shown that DM, as an independent risk factor for AF, is associated with an increased risk of AF.3–5 The mechanism of the increased risk of AF caused by DM has not been fully elucidated, and existing evidence suggests that oxidative stress and inflammation are important causes of DM-induced AF.6
Oxidative stress plays an important role in atrial structural remodeling in DM.7,8 Reactive oxygen species (ROS) might activate the nuclear factor κB (NF-κB) pathway, increase the expression of tumour necrosis factor (TNF) α and β, and cause atrial structural remodeling and fibrosis.9 In addition, increased oxidative stress from chronic hyperglycaemia leads to DNA breakage, which renders the DNA unstable, thereby activating the nuclear enzyme poly (ADP-ribose) polymerase-1 (PARP-1) in an attempt to repair such damage.10 When PARP-1 is overactivated, which occurs in DM, intracellular NAD+ is depleted, creating a redox imbalance that further exacerbates the oxidative state in the cell. NF-κB activation can be regulated by PARP-1 via multiple mechanisms.11 The role of PARP activation has been shown to alter heart function in DM.10 However, the role of PARP-1 in the structural changes in the diabetic atria has not been studied. Hence, in this study, we investigated alterations in the PARP-1 and NF-κB pathways and structural remodeling in the atria of C57BL/6 mice by establishing a DM model that was induced by a consistent high-fat diet for 8 weeks and a subsequent low-dose streptozocin (STZ) injection, to determine the impact of DM on atria and identify the underlying mechanisms.
Materials and Methods
Experimental Animals
The C57BL/6 mice used in this study were obtained from the Laboratory Animal Center of Xi’an Jiaotong University (Xi’an, China). The mice were housed in an animal research facility with a 12-h light/dark cycle at a room temperature of 20-25°C with free access to food and water. Ethics approval for this study was obtained from the Ethics Committee of Xi’an Jiaotong University. Animal experiments in this study were performed according to the Guidelines of Animal Handling and Experimentation (Xi’an, China).
Type 2 DM Model and Groupings
C57BL/6 mice (n=25) were randomly divided into a control group (Con, n=10), which was provided with a normal diet, and a model group (n=15), which was provided with a high-fat diet consisting of 18% lard, 20% sucrose, 3% egg yolk and 59% basal feed (Research Diets, Laboratory Animal Center of Xi’an Jiaotong University), starting at 6–8 weeks of age for 8 weeks. After 8 weeks of feeding, DM group mice were administered weekly intraperitoneal injections of STZ (35 mg/kg) for 4 weeks, while control group mice received intraperitoneal injection with sodium citrate buffer. During these 4 weeks, the two groups were provided a normal diet. A 12-hour fast before each injection of STZ was required in the model group. Three days after STZ injections, fasting blood glucose was measured, and mice with blood glucose above 11.1 mmol/L were selected for formal inclusion in the DM group.
Fasting Plasma Glucose (FPG), Fasting Insulin (FIns) and Plasma Lipid Measurements
From the beginning of feeding, FPG, FIns and plasma lipids and body weight were measured and recorded every 2 weeks until week 12. The blood was collected through the tail vein. Samples were sent to the Clinical Laboratory Department of the First Affiliated Hospital of Xi’an Jiaotong University to measure the levels of FPG, FIns, total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C) and triglycerides (TGs). FPG was measured by a Roche blood glucometer. FIns and plasma lipid levels were measured by corresponding ELISA kits. Second, according to the measured blood glucose and insulin levels, the following indicator was calculated: homeostatic model assessment for insulin resistance (HOMA-IR) = FPG level (mmol/L) × fasting insulin level (mU/L)/22.5.
Tissue Harvesting and Processing
First, the mice were sedated with phenobarbital and fixed. Then, the heart was quickly exposed. After piercing the left ventricle and carving the right atrium, the heart was perfused with phosphate-buffered saline (PBS), which was injected into the left ventricle. After perfusion, the heart tissue was cut, and filter paper was used to remove excess water. The vessels were rinsed, and the atrial tissue was harvested after thoroughly removing the aorta and fat. Atrial tissue weight was later measured.
Haematoxylin and Eosin (H&E) and Masson’s Staining
Sections (5-µm thick) of atrial tissue were stained with H&E and Masson’s staining and then sealed (Con n=10, DM n=11). The areas of the blue-purple collagen fibers and the percentage of the area relative to the entire field of view were assessed using Image-Pro Plus 6.0 analysis software (Media Cybernetics, Inc., Rockville, MD, USA).
RNA Extraction and Reverse Transcription-Quantitative PCR (RT-qPCR)
Total RNA was extracted from mouse atrial tissue (Con n=5, DM n=6) using TRIzol reagent according to the manufacturer’s instructions. The purity of the isolated RNA was determined, and cDNA was synthesized by reverse transcription for use as a template in the subsequent PCR analysis. Quantitative PCR (qPCR) was then performed. The primer sequences used were as follows: PARP-1 (forward) 5ʹ-CGG AGT CTT CGG ATA AGC TCT-3ʹ and (reverse) 5ʹ-TTT CCA TCA AAC ATG GGC GAC-3ʹ; AMP-activated protein kinase (AMPK) (forward) 5ʹ-TTG AAA CCT GAA AAT GTC CTG CT-3ʹ and (reverse) 5ʹ-GGT GAG CCA CAA CTT GTT CTT-3ʹ; Sirtuin 1 (Sirt1) (forward) 5ʹ-TAG CCT TGT CAG ATA AGG AAG GA-3ʹ and (reverse) 5ʹ-ACA GCT TCA CAG TCA ACT TTG T-3ʹ; Akt (forward) 5ʹ-AGC GAC GTG GCT ATT GTG AAG-3ʹ and (reverse) 5ʹ-GCC ATC ATT CTT GAG GAG GAA GT-3ʹ; IKKα (forward) 5ʹ-GGC TTC GGG AAC GTC TGT C-3ʹ and (reverse) 5ʹ-TTT GGT ACT TAG CTC TAG GCG A-3ʹ; NF-κB (forward) 5ʹ-ATG TGG AGA TCA TGA GCA GC-3ʹ and (reverse) 5ʹ-CCT GGT CCT GTG TAG CCA TT-3ʹ; nucleotide-binding oligomerization domain-like receptors 3 (NLRP3) (forward) 5ʹ-GAT CTT CGC TGC GAT CAA CAG-3ʹ and (reverse) 5ʹ-CGT GCA TTA TCT GAA CCC CAC-3ʹ; and GAPDH (forward) 5ʹ-TGA TTC TAC CCA CGG CAA GTT-3ʹ and (reverse) 5ʹ-TGA TGG GTT TCC CAT TGA TGA-3ʹ. The target mRNAs were normalized to the GAPDH mRNA level.
Western Blotting
Protein was extracted from mouse atrial tissue (Con n=5, DM n=5) after sufficient grinding and lysis. The antibodies used in the experiment were as follows: anti-PARP-1 (1:3000, ab227244; Abcam, Cambridge, UK), anti-AMPK (1:3000, ab32047; Abcam, Cambridge, UK), anti-Sirt1 (1:1000, cst9475; Cell Signaling Technology Inc., Danvers, MA, USA), anti-pan-Akt (1:500, ab8805; Abcam, Cambridge, UK), anti-IKKα (1:10,000, ab32041; Abcam, Cambridge, UK), anti-NF-κB (1:1000, cst8242; Cell Signaling Technology Inc., Danvers, MA, USA), anti-NLRP3 (1:3000, ab214185; Abcam, Cambridge, UK), and anti-GAPDH (1:5000, sc-25778; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Statistical Analysis
We first verified whether the results exhibited a normal distribution. If not, logarithmic transformation and the variance homogeneity test were performed. Then, a t-test was used to compare the results between different groups. Statistical data are expressed as the mean ± standard deviation (SD). A P-value <0.05 was considered statistically significant.
Results
High-Fat Diet and STZ Induced Increased Glucose Levels in DM Mice
During 4 weeks of STZ injection, DM mice had weak spirit, dull fur, poor activity, and increased water consumption and urine output and lost weight in the later stage. Unfortunately, there were no detailed statistics on the poor activity and increased water consumption and urine output in DM mice. Two mice in the model group died during model induction with STZ injections. Finally, according to the blood glucose levels, 11 mice were ultimately included in the DM group. Before the dietary intervention, there was no significant difference in body weight or baseline metabolic indexes in the two groups, including FPG, FIns and HOMA-IR (Figure 1). After 8 weeks of dietary intervention, notable insulin resistance (IR) was observed in high-fat diet-fed mice (HOMA-IR: Con versus DM, 2.09±0.44 versus 6.73±1.35, respectively, P<0.001) (Figure 1). Beginning in the first 2 weeks of STZ injections, DM mice (n=11) displayed significantly higher FPG (Con versus DM, 4.52±0.91 mmol/L versus 10.93±1.65 mmol/L, respectively, P<0.001) and FIns (Con versus DM, 0.45±0.11 ng/mL versus 0.45±0.09 ng/mL, respectively, P<0.001) levels than Con mice (n=10). Moreover, the analysis revealed markedly increased atrial weights in DM mice (Con versus DM, 0.30±0.04 mg/g versus 0.89±0.24 mg/g, respectively, P<0.05) (Figure 2).
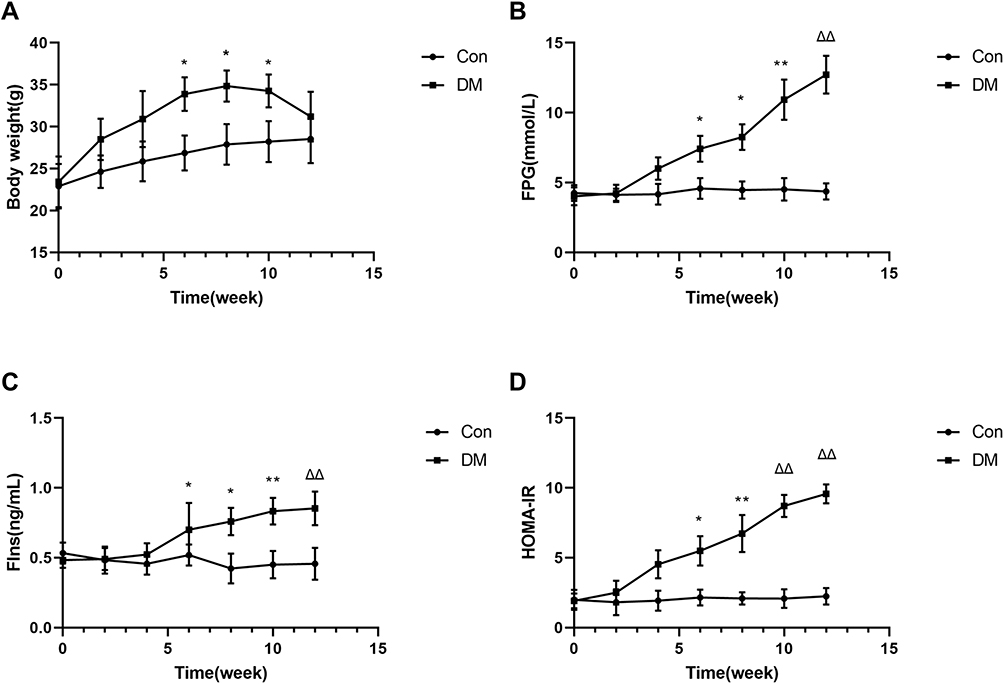 |
Figure 1 Comparison of body weight (A), FPG (B), FIns (C) and HOMA-IR(D) levels in the 2 groups. The data are expressed as the mean ± SD. Dietary intervention started at 6–8 weeks of age (time-point, 0). *P<0.05 vs Con; **P<0.01 vs Con; ΔΔP<0.001 vs Con. Abbreviations: FPG, fasting plasma glucose; FIns, fasting insulin; HOMA-IR, HOMA insulin resistance index. |
 |
Figure 2 Comparison of atrial weights in the 2 groups. Atrial weight was measured and normalized to body weight. The data are expressed as the mean ± SD. **P<0.001 vs Control after modelling. |
DM Mice Exhibited Dyslipidaemia
In this study, DM mice exhibited significantly upregulated levels of TGs (Con versus DM, 1.93±0.27 mmol/L versus 2.79±0.43 mmol/L, respectively, P<0.05), TC (Con versus DM, 1.64±0.33 mmol/L versus 1.93±0.50 mmol/L, respectively, P<0.05) and LDL-C (Con versus DM, 0.71±0.12 mmol/L versus 1.39±0.33 mmol/L, respectively, P<0.05) compared to those in the Con group (Figure 3), while there was no difference in HDL-C levels (Con versus DM, 1.24±0.21 mmol/L versus 1.05±0.33 mmol/L, respectively, P>0.05) between these two groups. These results indicated disturbed lipid homeostasis in DM mice.
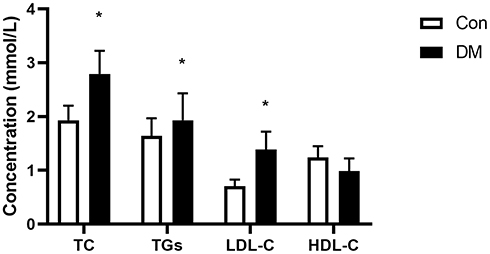 |
Figure 3 Diabetic mice exhibit dyslipidaemia. Plasma total cholesterol (TC), triglyceride (TG), low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) levels were determined from independent samples from Con and DM mice (means ± SD. *P<0.05 vs Con). |
DM Mice Exhibited More Extensive Atrial Fibrosis Than Con Mice
H&E staining of atrial paraffin sections revealed broadened interstitial spaces among the atrial myofibers, as well as a distinctly disordered layout of atrial myocytes, in DM mice instead of a normal myocardial structure (Figure 4). Significant fibrosis was observed by Masson’s staining in atrial sections from DM mice (Figure 5). Extensive fibrosis was particularly visible around atrial myocytes, which were disorderly distributed. In contrast, the small amounts of collagen fibers in the control mouse atria were continuous and complete. In addition, DM mice showed marked collagen proliferation compared to that of Con mice (Con versus DM, 5.34±0.81% versus 18.70±3.10%, respectively, P<0.05) (Figure 6). These results revealed that DM may induce and promote atrial fibrosis.
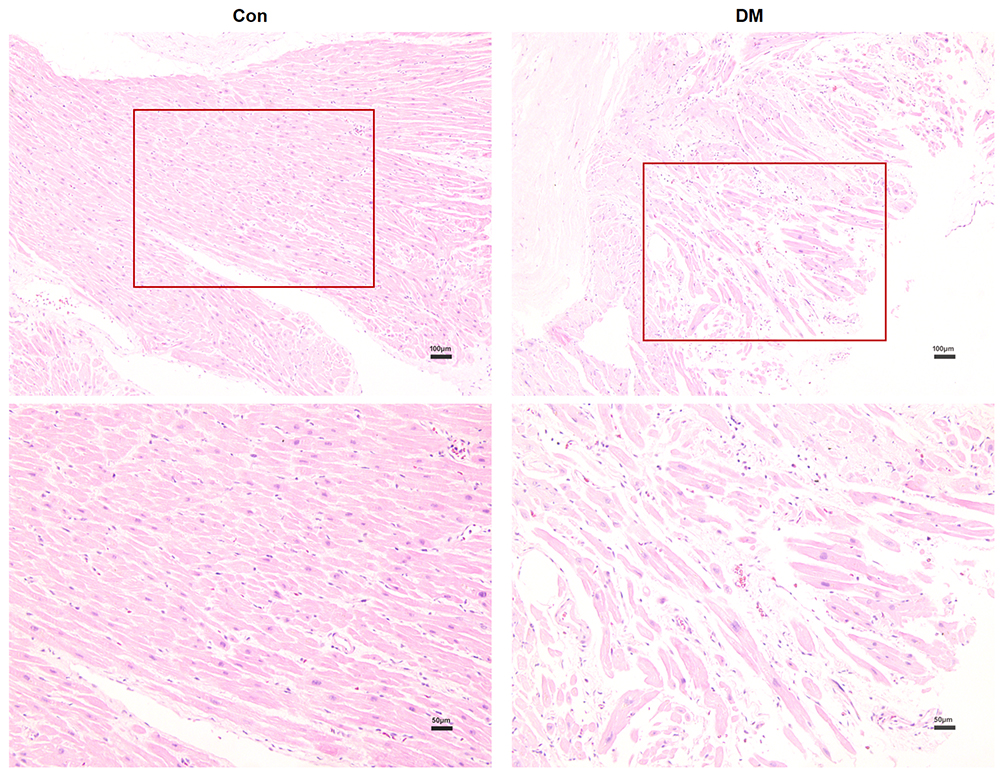 |
Figure 4 Haematoxylin and eosin (H&E) staining of atrial sections from mice in the 2 groups (Con n=10, DM n=11). Atrial myofibers were stained red, while nuclei were stained blue-purple. Con mice exhibited complete and regular atrial myocardial structures. In contrast, atrial myofibers were disorderly arrayed in DM mice. Representative images are shown at magnifications of ×100 (top panels) and ×200 (bottom panels). Red boxes show the partial magnification. |
 |
Figure 5 Masson’s staining of atrial sections from mice in the 2 groups (Con n=10, DM n=11). Atrial myocytes were stained red, while collagen fibers were stained blue-purple. DM mouse atria exhibited large amounts of disorderly collagen fibers compared to those of Con mouse atrial tissue. Representative images are shown at magnifications of ×100 (top panels) and ×200 (bottom panels). Red boxes show the partial magnification. |
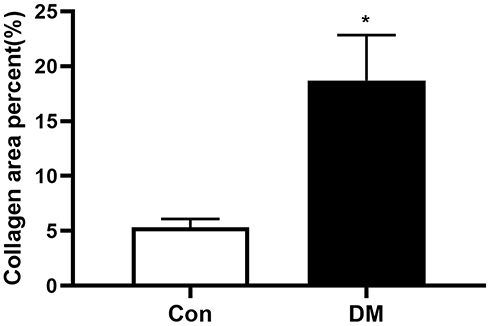 |
Figure 6 Semiquantitative assessment of collagen in atrial tissue in the 2 groups (Con n=10, DM n=11). Atrial tissue of DM mice exhibited larger collagen areas. The results are expressed as the means ± SD; *P<0.05 vs Con. |
DM Markedly Altered the mRNA and Protein Expression of NF-κB and Its Upstream Signaling Pathway Factors
RT-qPCR analysis revealed that the mRNA expression of NF-κB and factors in its upstream signaling pathways, including PARP-1, Akt, and IKKα and the downstream factor NLRP3, were significantly upregulated in atrial tissue from DM mice (Figure 7A and D–G), while the mRNA levels of AMPK and Sirt 1 were markedly downregulated (Figure 7B and C). Consistent with the RT-qPCR results, Western blotting showed significant alterations in the expression of NF-κB and its upstream signaling pathway proteins in the atria of DM mice (Figure 7). The marked alterations in the expression of NF-κB and its upstream signaling pathway proteins and mRNA further suggests that DM could activate the PARP-1/Ikkα/NF-κB pathway in the atrium.
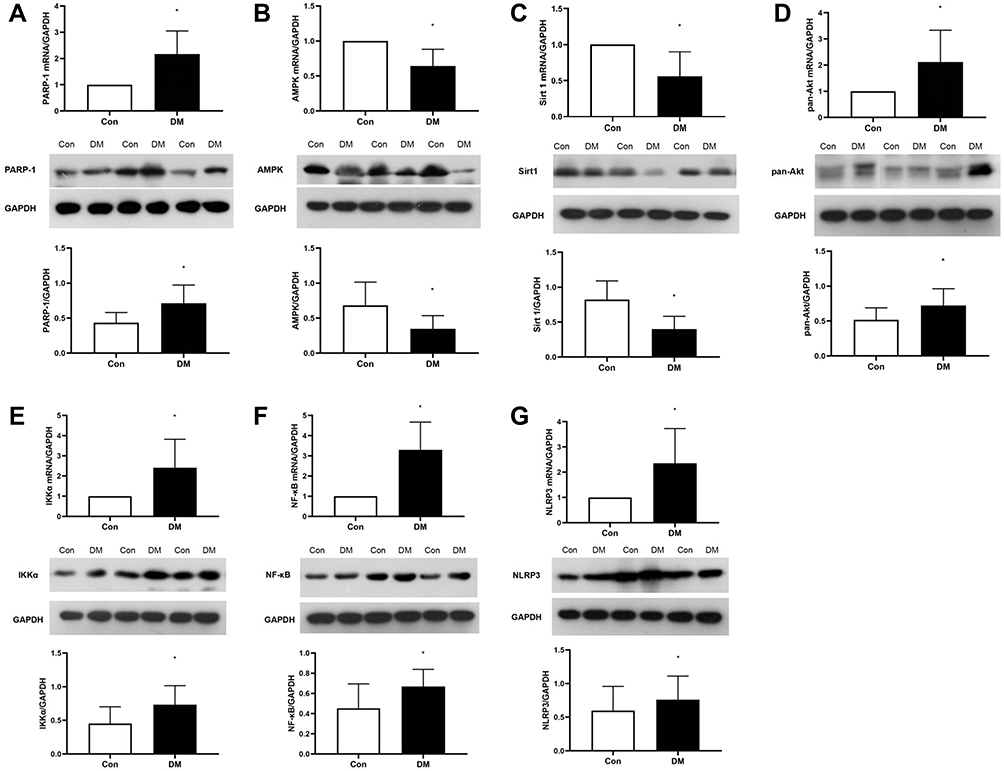 |
Figure 7 The mRNA (Con n=5, DM n=6) and protein (Con n=5, DM n=5) expression of PARP-1 (A), AMPK (B), Sirt1 (C), Akt (D), IKKα (E), NF-κB (F) and NLRP3 (G) in the atria of the 2 groups. The results are expressed as the means ± SD; *P<0.05 vs Con. |
Discussion
In this study, C57BL/6 mice underwent dietary intervention combined with STZ injection to establish a type 2 DM model, and the indicators of subsequent atrial structural remodeling and inflammation were analysed. We found that DM could affect IR and lipid metabolism, promote structural remodeling and alter the expression of mRNA and proteins involved in inflammation in mouse atrial tissue. The impact of DM on the atrial substrate could represent the underlying cause of the development of DM-induced atrial remodeling.
According to our results, after 8 weeks of a high-fat diet, DM mice showed significant weight increases and IR. Subsequently, multiple injections of low-dose STZ were performed for 4 weeks. DM mice showed increased water intake, polyuria and weight loss and had markedly higher levels of FPG, FIns and IR, which suggested successful DM modelling.
Previous studies have reported that atrial enlargement and fibrosis are the two major components of atrial structural remodeling.12,13 Atrial dimensions have been demonstrated to determine the persistence of AF maintaining re-entry.14 Although we could not perform echocardiography for these mice, the atrial weight/body weight ratios of DM mice were significantly higher than those of Con mice (Figure 2), which could indirectly reflect atrial enlargement. In addition, DM caused distinctly broadened interstitial spaces among atrial myofibers (Figure 4), leading to atrial enlargement. We also analysed Masson’s staining to determine whether DM induced atrial fibrosis in mice. In this study, increased collagen hyperplasia was observed in DM mouse atrial tissue (Figure 6), and the result was consistent with previously described studies. DM could induce marked fibrosis in the atrium. These findings indicated that DM could promote atrial structural remodeling, including atrial enlargement and fibrosis.
In recent years, increasing evidence has shown that the inflammatory response could induce atrial structural or electrical remodeling and further promote the genesis and maintenance of AF,15–17 while the underlying mechanism remains unknown. As an important upstream mediator of atrial remodeling, the inflammatory response can induce atrial remodeling through a series of inflammatory signaling pathways, leading to the occurrence and development of AF. NF-κB is a transcription factor and an important upstream regulator of inflammation and other biological processes. In fact, NF-κB can regulate target gene transcription, including that of many inflammatory cytokines, such as TNF-α, IL-6, IL-8 and IL-1, which are responsible for the occurrence and development of AF.15,16,18,19 After entering the nucleus, NF-κB can enhance the transcription of a number of inflammatory factors, including the NLRP3 inflammasome, which is one of the mediators of atrial remodeling.20 Previous studies have demonstrated that the NLRP3 inflammasome, a pattern recognition receptor expressed on the surface of macrophages, is activated by NF-κB signaling pathways, which in turn promotes the electrical activity of the atrium and produces the matrix of arrhythmia, which increases atrial remodeling. In this study, we observed significantly increased expression of NF-κB and NLRP3 in DM mice. In this context, previous studies have demonstrated that increased NLRP3 expression could further lead to the expression of IL-1β, which could result in prolonged myocyte action potential duration (APD) and atrial fibrosis and increase the risk of arrhythmogenesis in DM animals. However, the mechanism of NF-κB upstream pathways in the development of atrial remodeling, especially in the increased risk of diabetes-induced atrial remodeling, remains unclear.
PARP-1, a class of ribozymes associated with DNA injury repair, mediates apoptosis and regulates gene transcription; PARP-1 is closely associated with the pathogenesis of tumours, inflammatory injury, cardiac fibrosis, etc.21–24 PARP-1 can regulate NF-κB and other inflammatory signal transduction pathways,11 further promoting the expression of many inflammatory factors, such as TNF-α, IL-1β and IL-6. The NF-κB pathway is the central pathway by which PARP-1 regulates inflammation. Zhang25 showed that AF induced DNA damage and subsequent PARP-1 activation. PARP-1 activation resulted in the depletion of NAD+, and the consequent exhaustion of ATP led to irreversible structural remodeling and ultimately created substrates that impaired contractile function and AF persistence. This finding revealed that PARP-1 might be involved in the occurrence and persistence of AF. Our findings, in conjunction with the findings from previous studies, demonstrate a significant role for PARP-1 in the pathogenesis of diabetic atria.26 Downstream effects, such as PARP-1-regulated NF-κB activation, can detrimentally impair atrial function. This investigation was the first to show a mechanistic pathway for the effect of PARP-1 on DM-induced structural alterations in the atrium.
The AMPK/Sirt1 pathway has been shown to be involved in the regulation of proinflammatory cytokine release.27 Sirt1, a nuclear and cytoplasmic silencing information regulator 2 (SIR2)-related protein (sirtuin), could be downregulated by PARP-1 activation in DM mice.24 It has been shown that higher levels of ROS promote activation of the NAD-dependent DNA repair enzyme PARP-1, with subsequent NAD depletion and downregulation of Sirt1 activity.28 PARP-1 inhibition led to increased Sirt1 and attenuated oxidative stress, inflammation and fibrosis. Moreover, the mechanism of Sirt1 reduction in diabetic conditions is not completely understood, but a reduction in the phosphorylation of AMPK may play a role.29 Previous studies have suggested that AMPK and Sirt1 can promote each other and jointly promote downstream responses. Papadimitriou30 showed that in the context of DM, Sirt1 activity was reduced by PARP-1 activation and NAD+ depletion due to low AMPK, which increased NOX4 expression, leading to extracellular matrix (ECM) accumulation mediated by transforming growth factor (TGF)-beta 1 signaling. In addition, AMPK/Sirt1 may be involved in the pathogenesis of DM and diabetic cardiomyopathy and may play protective roles in the myocardial tissue of DM patients.31 However, the direct relationships between atrial-specific Sirt1, AMPK, and DM have not yet been elucidated. In our study, a Western blot analysis showed that DM led to decreases in AMPK and Sirt1 in atrial myocytes.
There was also an experiment32 suggesting that Sirt1 could inhibit Akt and IKK activity and then exert myocardial protective effects. IKK is the regulatory subunit of an NF-κB inhibitory complex. PARP-1 provides the scaffold needed for the SUMOylation of IKK, leading to the activation of IKK and NF-κB.33 This study showed notable activation of Akt and IKK in atrial myocytes in the context of DM.
Inflammation, which arises from IR, hyperglycaemia and glucose fluctuations, is largely responsible for DM-associated atrial proarrhythmic remodeling. Diabetes has long been acknowledged to be a proinflammatory disease, and inflammation is known to promote AF by increasing atrial electrical instability, promoting atrial fibrosis, and modulating the intrinsic autonomic nervous system of the heart.34
Taking into consideration the aforementioned hypotheses, it is reasonable to assume that NF-κB activation plays an important role in AF development and maintenance in DM via canonical and noncanonical pathways, such as inflammation, fibrosis, and atrial enlargement. NF-κB activation is induced by different factors, such as the AMPK/PARP-1/Sirt1 pathway and Akt/Ikk pathway. Increasing expression of NLRP3 and its downstream signal factors lead to atrial enlargement and fibrosis may serve as important substrates for the development of AF.
Besides inflammation, there are many other factors and pathways potentially involved in DM-AF relationship, such as oxidative stress,34 activation of renin-angiotensin system (RAS),34 and the Rock pathway.35
Therefore, we have come to the following assumption (Figure 8): DM can cause hyperglycemia, IR and oxidative stress, further induce PARP-1 activation, inhibit AMPK and Sirt 1, upregulate the NF-κB/NLRP3 pathway, lead to atrial inflammation, induce atrial structural remodeling which includes of atrial enlargement and fibrosis and electrical remodeling, and finally take part in the initiation and maintenance of AF.
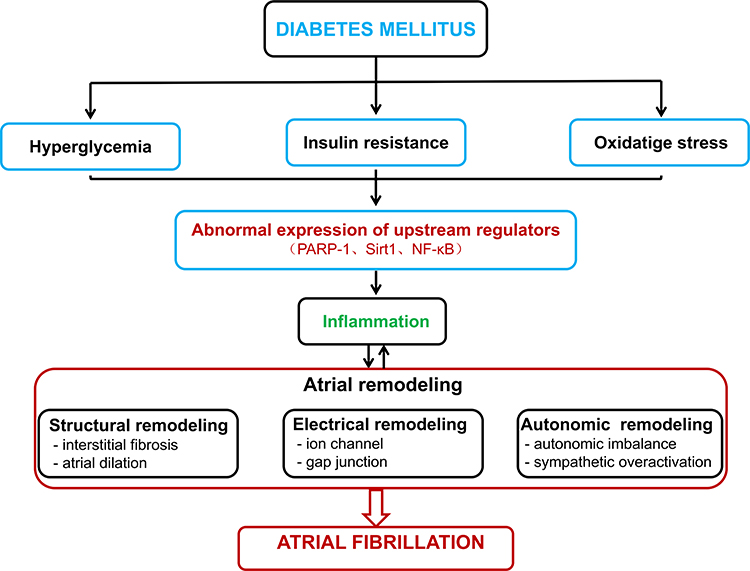 |
Figure 8 Potential mechanisms of DM-induced AF. Abbreviations: PARP-1, poly (ADP-ribose) polymerase-1; Sirt1, sirtuin 1; NF-κB, nuclear factor κB. |
There are several limitations to the present study. We measured atrial structural remodeling and inflammation, which are considered to be the main causes of AF. Although we determined the susceptibility to AF caused by DM, the initiation of AF was not examined. We will further investigate this aspect in the future. In addition, the specific mechanism of the PARP-1/Sirt1/NF-κB pathway during DM-induced AF needs to be explored. In the future, we plan to further validate our results by performing drug interventions and constructing transgenic models.
Conclusion
We conclude that DM can promote atrial structural remodeling, including atrial enlargement and atrial fibrosis, and alter the expression of factors involved in atrial inflammation, which can further contribute to the initiation and development of AF.
Acknowledgments
We are grateful to Dr Guoliang Li and Dr Chaofeng Sun for technical assistance and valuable comments on this manuscript. Tianyu Meng and Jie Wang share first authorship. This work was supported by the Key Research and Development Program of Shaanxi province (2020ZDLSF04-11).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was obtained to support this work.
Disclosure
The authors declare no conflicts of interests for this study.
References
1. Mozaffarian D, Benjamin Emelia J, Go Alan S, et al. Heart disease and stroke statistics—2016 update. Circulation. 2016;133(4):e38–e360. doi:10.1161/CIR.0000000000000350
2. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc Diabetol. 2018;17(1):83. doi:10.1186/s12933-018-0728-6
3. Chao TF, Suenari K, Chang SL, et al. Atrial substrate properties and outcome of catheter ablation in patients with paroxysmal atrial fibrillation associated with diabetes mellitus or impaired fasting glucose. Am J Cardiol. 2010;106(11):1615–1620. doi:10.1016/j.amjcard.2010.07.038
4. Huxley RR, Alonso A, Lopez FL, et al. Type 2 diabetes, glucose homeostasis and incident atrial fibrillation: the Atherosclerosis Risk in Communities Study. Heart. 2012;98(2):133–138. doi:10.1136/heartjnl-2011-300503
5. Aksnes TA, Schmieder RE, Kjeldsen SE, Ghani S, Hua TA, Julius S. Impact of new-onset diabetes mellitus on development of atrial fibrillation and heart failure in high-risk hypertension (from the VALUE trial). Am J Cardiol. 2008;101(5):634–638. doi:10.1016/j.amjcard.2007.10.025
6. Goudis CA, Korantzopoulos P, Ntalas IV, Kallergis EM, Liu T, Ketikoglou DG. Diabetes mellitus and atrial fibrillation: pathophysiological mechanisms and potential upstream therapies. Int J Cardiol. 2015;184:617–622. doi:10.1016/j.ijcard.2015.03.052
7. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54(20):1891–1898. doi:10.1016/j.jacc.2009.07.031
8. Duicu OM, Lighezan R, Sturza A, et al. Assessment of mitochondrial dysfunction and monoamine oxidase contribution to oxidative stress in human diabetic hearts. Oxid Med Cell Longev. 2016;2016:8470394. doi:10.1155/2016/8470394
9. Wu N, Shen H, Liu H, Wang Y, Bai Y, Han P. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc Diabetol. 2016;15(1):109. doi:10.1186/s12933-016-0427-0
10. Chiu J, Farhangkhoee H, Xu BY, Chen S, George B, Chakrabarti S. PARP mediates structural alterations in diabetic cardiomyopathy. J Mol Cell Cardiol. 2008;45(3):385–393. doi:10.1016/j.yjmcc.2008.06.009
11. Weaver AN, Yang ES. Beyond DNA repair: additional functions of PARP-1 in cancer. Front Oncol. 2013;3:290. doi:10.3389/fonc.2013.00290
12. Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol. 2014;63(22):2335–2345. doi:10.1016/j.jacc.2014.02.555
13. Thomas L, Abhayaratna WP. Left atrial reverse remodeling: mechanisms, evaluation, and clinical significance. JACC Cardiovasc Imaging. 2017;10(1):65–77. doi:10.1016/j.jcmg.2016.11.003
14. Zou R, Kneller J, Leon LJ, Nattel S. Substrate size as a determinant of fibrillatory activity maintenance in a mathematical model of canine atrium. Am J Physiol Heart Circ Physiol. 2005;289(3):H1002–H1012. doi:10.1152/ajpheart.00252.2005
15. Chen G, Chelu MG, Dobrev D, Li N. Cardiomyocyte inflammasome signaling in cardiomyopathies and atrial fibrillation: mechanisms and potential therapeutic implications. Front Physiol. 2018;9:1115. doi:10.3389/fphys.2018.01115
16. Liu Y, Shi Q, Ma Y, Liu Q. The role of immune cells in atrial fibrillation. J Mol Cell Cardiol. 2018;123:198–208. doi:10.1016/j.yjmcc.2018.09.007
17. Hu Y-F, Chen Y-J, Lin Y-J, Chen S-A. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. 2015;12(4):230–243. doi:10.1038/nrcardio.2015.2
18. Fu H, Li G, Liu C, et al. Probucol prevents atrial remodeling by inhibiting oxidative stress and TNF-α/NF-κB/TGF-β signal transduction pathway in alloxan-induced diabetic rabbits. J Cardiovasc Electrophysiol. 2015;26(2):211–222. doi:10.1111/jce.12540
19. Sun Z, Zhou D, Xie X, et al. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res Cardiol. 2016;111(6):63. doi:10.1007/s00395-016-0584-z
20. Jian XU, Yan HE, Bei-Bei L, et al. Correlation study between NLRP3 inflammasome and atrial fibrillation. Chin Circ J. 2017.
21. Muthurajan UM, Hepler MRD, Hieb AR, et al. Automodification switches PARP-1 function from chromatin architectural protein to histone chaperone. Proc Natl Acad Sci USA. 2014;111(35):12752–12757. doi:10.1073/pnas.1405005111
22. Feng FY, de Bono JS, Rubin MA, Knudsen KE. Chromatin to clinic: the molecular rationale for PARP1 inhibitor function. Mol Cell. 2015;58(6):925–934. doi:10.1016/j.molcel.2015.04.016
23. Ciccarone F, Zampieri M, Caiafa P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin Cell Dev Biol. 2017;63:123–134. doi:10.1016/j.semcdb.2016.11.010
24. Waldman M, Nudelman V, Shainberg A, et al. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1α axis. Exp Cell Res. 2018;373(1–2):112–118. doi:10.1016/j.yexcr.2018.10.003
25. Zhang D, Hu X, Li J, et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD depletion in experimental atrial fibrillation. Nat Commun. 2019;10(1):1307. doi:10.1038/s41467-019-09014-2
26. Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabó E, Szabó C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51(2):514–521. doi:10.2337/diabetes.51.2.514
27. Chen L, Lan Z. Polydatin attenuates potassium oxonate-induced hyperuricemia and kidney inflammation by inhibiting NF-κB/NLRP3 inflammasome activation via the AMPK/SIRT1 pathway. Food Funct. 2017;8(5):1785–1792. doi:10.1039/C6FO01561A
28. Pillai JB, Isbatan A, Imai S-I, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280(52):43121–43130. doi:10.1074/jbc.M506162200
29. Kim MY, Lim JH, Youn HH, et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1α axis in db/db mice. Diabetologia. 2013;56(1):204–217. doi:10.1007/s00125-012-2747-2
30. Papadimitriou A, Silva KC, Peixoto EBMI, Borges CM, Lopes de Faria JM, Lopes de Faria JB. Theobromine increases NAD⁺/Sirt-1 activity and protects the kidney under diabetic conditions. Am J Physiol Renal Physiol. 2015;308(3):F209–F225. doi:10.1152/ajprenal.00252.2014
31. Yuan Q, Zhou Q-Y, Liu D, et al. Advanced glycation end-products impair Na⁺/K⁺-ATPase activity in diabetic cardiomyopathy: role of the adenosine monophosphate-activated protein kinase/sirtuin 1 pathway. Clin Exp Pharmacol Physiol. 2014;41(2):127–133. doi:10.1111/1440-1681.12194
32. Busch F, Mobasheri A, Shayan P, Stahlmann R, Shakibaei M. Sirt-1 is required for the inhibition of apoptosis and inflammatory responses in human tenocytes. J Biol Chem. 2012;287(31):25770–25781. doi:10.1074/jbc.M112.355420
33. Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell. 2009;36(3):365–378. doi:10.1016/j.molcel.2009.09.032
34. Şerban RC, Scridon A. Data linking diabetes mellitus and atrial fibrillation-how strong is the evidence? From epidemiology and pathophysiology to therapeutic implications. Can J Cardiol. 2018;34(11):1492–1502. doi:10.1016/j.cjca.2018.08.018
35. Chen J, Li Q, Dong R, et al. The effect of the Ras homolog gene family (Rho), member A/Rho associated coiled-coil forming protein kinase pathway in atrial fibrosis of type 2 diabetes in rats. Exp Ther Med. 2014;8(3):836–840. doi10.3892/etm.2014.1843
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.