")
Back to Journals » International Journal of Nanomedicine » Volume 14
Di-O-lauroyl-decitabine-lipid nanocapsules: toward extending decitabine activity
Authors Briot T , Roger E , Bou Haidar N , Bejaud J, Lautram N, Guillet C, Thépot S, Legeay S, Lagarce F
Received 11 October 2018
Accepted for publication 4 December 2018
Published 26 March 2019 Volume 2019:14 Pages 2091—2102
DOI https://doi.org/10.2147/IJN.S190482
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Thomas Briot,1,2 Emilie Roger,1 Naila Bou Haidar,1 Jerome Bejaud,1 Nolwenn Lautram,1 Catherine Guillet,3 Sylvain Thépot,4,5 Samuel Legeay,1 Frederic Lagarce1,2
1Micro & Nanomédecines Translationelles – MINT, UNIV Angers, INSERM 1066, CNRS 6021, University of Angers, MINT IBS-CHU, Larrey, 49933 Angers, France; 2University Hospital of Angers, Pharmacy Department, 49933 Angers, France; 3University of Angers, Molecular and Cellular Analysis Platform, IBS-CHU, 49933 Angers, France; 4University Hospital of Angers, Hematology, 49933 Angers, France; 5INSERM CRCINA, University of Angers, 49933 Angers, France
Background: Acute myeloid leukemia mainly affects adult patients. Complete remission for patients younger than 60 years, who are candidates for standard induction therapy, is achieved in 60%–80% of cases. However, the prognosis is still poor for older patients, who are unfit for intensive chemotherapy, and only a few therapies are available. Hypomethylating agents, such as decitabine, are approved for such patients. The current dosing regimen consists of one administration per day, for 5 days, each 4 weeks.
Methods: Here, we present the synthesis of a decitabine prodrug, combined with its encapsulation into a lipid-based nanocapsule formulation. Decitabine (C12)2 was synthetized, then loaded into nanocapsules. Its stability in phosphate buffer ans human plasma was checked. Its activity was evaluated by Cell proliferation assays and cell-cycle analysis on human erythroleukemia cells. Then its pharmacokinetics was determined on a rat model.
Results: Decitabine (C12)2 was obtained with a yield of 50%. Drug loading into nanocarriers of 27.45±0.05 nm was 5.8±0.5 mg/mL. The stability of decitabine was improved and its activity on leukemia cells was not altered. Finally, pharmacokinetics studies showed a prolonged mean residence time of the drug.
Conclusion: Decitabine (C12)2 as a prodrug showed high encapsulation efficiency, a good stability in plasma with no impact on its activity on leukemia cells and improved pharmacokinetics.
Keywords: lipid nanocapsules, acute myeloid leukemia, decitabine, nanomedicines, prodrugs
Introduction
Acute myeloid leukemia (AML) is a heterogenous hematological malignancy caused by the uncontrolled clonal expansion of leukemic progenitor cells. The incidence of AML increases with age, with a median of 64 years at presentation.1 Despite recent progress in the development of new therapeutics, the prognosis is still poor, with the 5-year relative survival estimated to be 19% in Europe.2
Induction treatment for AML is based on a chemotherapeutic regimen consisting of a combination of an anthracycline (daunorubicin, doxorubicin, or idarubicin) and cytarabine. Only younger patients, who can support this intensive regimen (cytarabine 100–200 mg/m2/day for 7 consecutive days and daunorubicin 60 mg/m2 or idarubicin 12 mg/m2 on days 1–3), receive such treatment. Such intensive chemotherapy leads to complete remission in 60%–80% of younger patients, whereas it is obtained in <30% in patients who are older than 60 years.3
For patients who are unfit to receive standard induction therapy, non-curative therapies are proposed, such as low-dose cytarabine, hypomethylating agents (azacytidine or decitabine), or best supportive care. The prognosis is poor for these patients, with a 1-year from diagnosis survival rate of <30%.4 For such patients, decitabine (Dacogen®, Janssen-Cilag, Beerse, Belgium) received European marketing authorization in 2012, whereas such authorization was not issued by the Food and Drug Administration (FDA) in the USA.
To be effective, azacytidine and decitabine need to be in contact with cells for a sufficient time to induce DNA hypomethylation.5,6 However, the half-life of decitabine in plasma is only 15–25 minutes.7 The drug, thus, needs to be administered once a day, for 5 days, each month. Longer exposure times (10-day schedules) appear to improve the response rate.8 Such a short half-life could be explained by rapid degradation due to cytidine deaminase. This degradation pathway is also encountered during cytarabine administration. A liposomal formulation of cytarabine and daunorubicin (Vyxeos®, Jazz Pharmaceuticals) has just been approved by the FDA to treat newly diagnosed therapy-related AML and AML with myelodysplasia-related changes. This formulation was proven to be capable of protecting cytarabine from cytidine deaminase degradation,9 demonstrating the promise of new formulations, such as nanocarriers, to improve AML treatment. Patients receiving Vyxeos® lived longer than those who received separate treatments of daunorubicin and cytarabine, with the median overall survival prolonged from 5.95 to 9.56 months.10 Further improvement of the formulation of decitabine is, thus, desirable to enhance its half-life and efficacy.
Various strategies have been proposed to overcome the short half-life of decitabine. For example, guadecitabine, a decitabine prodrug composed of a dinucleotide of decitabine linked to a deoxyguanosine with a phosphodiester bond, is currently being tested in clinical trials (NCT02348489 and NCT02920008). The phosphodiester bond present in guadecitabine is cleaved gradually, resulting in the slow release of decitabine, extending its half-life in vivo. Guadecitabine is also less subject to cytidine deaminase action than decitabine.11 Loading decitabine in a nanogel formulation has also been tested. The antiproliferative activity of decitabine, evaluated on leukemia cells, was enhanced by >2-fold. This could be due to the increased proportion of cells arrested in the G2/M phase for nanogel formulations relative to that observed for decitabine in solution.12
Lipid-core nanocapsules (LNCs), with a structure close to that of lipoproteins, were developed by our laboratory >15 years ago. Their production is based on a phase-inversion temperature process.13 These nanoparticles have demonstrated great potential for the encapsulation of lipophilic drugs14 and biopolymers.15 Such nanomedicines show a good toxicity profile16,17 and can be produced in large batches.18 We were, thus, interested in encapsulating decitabine in these nanocarriers.19 However, the encapsulation of decitabine alone has only resulted in low-encapsulation yields.
Our aim was to develop a decitabine prodrug designed to be entrapped in a lipid-based nanoparticle formulation to enhance the stability of decitabine in blood and prolong contact time with the target cells.
Materials and methods
Chemicals
Decitabine was purchased from LC laboratories (Woburn, MA, USA). Labrafac® WL1349 (caprylic-capric acid triglycerides) and Transcutol® HP (highly purified diethylene glycol monoethyl ether) were kindly provided by Gattefossé (Saint-Priest, France). Kolliphor® HS15 (a mixture of free polyethylene glycol [PEG] 660 and PEG 660 hydroxystearate) was kindly supplied by BASF (Angerville, France). Sodium chloride was purchased from Prolabo (Fontenay-Sous-Bois, France). Formic acid, Tween® 20 (sorbitan monolaurate), potassium chloride, sodium hydroxide, disodium hydrogen phosphate, dimethylsulfoxide (DMSO), potassium dihydrogen phosphate, dicyclohexylcarbodiimide (DCC), hydrochloric acid, dimethylformamide (DMF), deuterated DMSO, lauric acid, 4-(dimethylamino) pyridine (DMAP), potassium chloride, and propidium iodide were purchased from Sigma-Aldrich (St Quentin-Fallavier, France). RNAse A was obtained from Fisher Scientific (Illkirch, France). Deionized water was obtained from a Milli-Q plus system (Merck-Millipore, Darmstadt, Germany). Absolute ethanol, acetonitrile LC/MS grade, and methanol LC/MS grade were purchased from Biosolve (Dieuze, France). Methanol and dichloromethane (DCM) laboratory reagent grades were obtained from Fisher Chemical (Illkirch, France).
Decitabine (C12)2 synthesis
Decitabine (C12)2 was obtained after magnetic stirring at room temperature of lauric acid (7.02 g), DCC (5.42 g), and DMAP (107 mg) in 50 mL DMF. After total dissolution, decitabine (1 g) was added to the mixture and magnetic stirring was continued for 24 hours at room temperature. The reaction was monitored by thin-layer chromatography (DCM-methanol 90:10 v/v). A white precipitate was obtained (Figure 1).
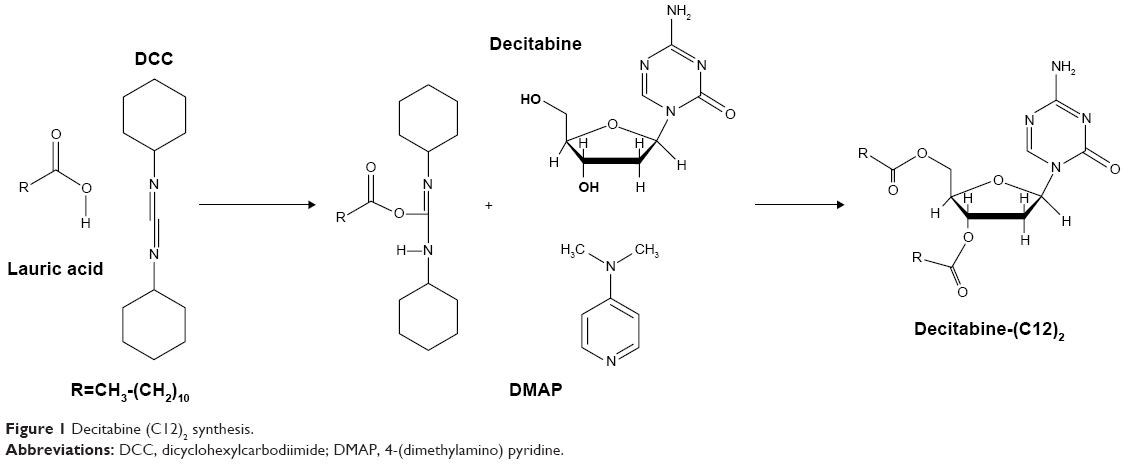 | Figure 1 Decitabine (C12)2 synthesis. |
After filtration on cotton wool, the precipitate was washed twice with 10 mL DMF. DMF present in the filtrate was evaporated under vacuum (Rotavapor® R-300, Buchi, Suisse) at 30°C, and the residues purified on a silica gel column with a first elution in DCM (2 L), followed by a second elution in DCM-methanol 99:1 v/v (3 L). The elution of decitabine (C12)2 was followed by thin-layer chromatography (DCM-methanol 90:10 v/v).
Pure fractions were then collected, and the solvents evaporated under vacuum to obtain a white product. The molecular weight of the obtained product was determined using a microToFQII apparatus (Bruker Daltonics GmbH, Bremen, Germany) by electrospray positive ionization. 1H-NMR spectra were recorded on an Avance DRX 500 MHz (Bruker Daltonics GmbH, Bremen, Germany) in deuterated DMSO.
LNC formulation
LNCs were obtained according to a phase-inversion process previously developed by Heurtault et al13 with minor modifications. Briefly, blank LNCs were obtained after mixing Labrafac® WL1349 (14.3% w/w), sodium chloride (1.5% w/w), Kolliphor® HS15 (32.7%), and water (34.7%). Three heating and cooling cycles between 65°C and 85°C were performed with magnetic stirring. During the last cooling cycle, at 85°C, a mixture composed of Span® 20 and Transcutol® HP (50:50 w/w) was added (16.8% w/w). Finally, tempering was induced at 55°C with 5 mL 4°C water. Magnetic stirring was then maintained for 5 minutes at room temperature and the formulations stored at 4°C for further evaluation.
The same procedure was used for decitabine (C12)2-loaded LNCs, except prior solubilization of decitabine (C12)2 was performed at 50°C for 1 hour under magnetic stirring in the mixture composed of Span® 20 and Transcutol® HP at 40 mg/g (Figure 2).
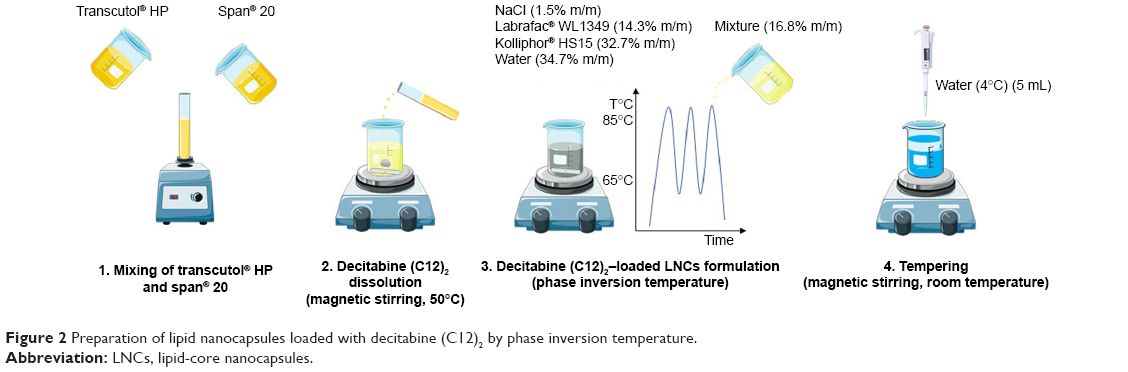 | Figure 2 Preparation of lipid nanocapsules loaded with decitabine (C12)2 by phase inversion temperature. |
Chromatographic methods
Determination of decitabine (C12)2 concentrations
A liquid chromatography method using tandem mass spectrometry (LC-MS/MS) was developed to quantify decitabine (C12)2. It was performed on an Alliance® 2695 system (Waters, Saint-Quentin-en-Yvelines, France) with an Uptisphere C18-ODB 150×2.0 mm, 5 μm column (Interchrom, Montluçon, France). The mobile phase consisted of a phase A (0.1% formic acid in acetonitrile) and a phase B (0.1% formic acid in methanol) in an isocratic mode (A:B 70:30 v/v). The flow rate was set to 0.4 mL/min for a run time of 6 minutes. The injection volume was 10 μL. The total HPLC effluent was directed into a Quattro Micro® triple quadrupole mass spectrometer (Waters). Ionization was achieved with an electrospray in the positive ion mode. The mass spectrometer was operated in the multiple reaction monitoring mode. The (M–H)+m/z transition for decitabine (C12)2 was 593.3>113.1. The entire system was controlled by Masslynx® software (Waters).
Calibration curves were obtained after solubilizing decitabine (C12)2 in methanol to obtain a stock solution of 0.5 mg/mL. A sufficient quantity of the stock solution was then diluted in methanol to obtain a calibration curve between 2.5 and 500 ng/mL (seven plots were used). A calibration curve was performed on each day of the analysis.
Determination of decitabine concentrations
The apparatus and software were the same as those used for decitabine (C12)2 quantification. An LC-MS/MS method previously published was used.19 The mobile phase consisted of a mixture of A) 0.1% formic acid in 10 mM ammonium acetate and B) 0.1% formic acid in acetonitrile, in gradient mode (Table 1). A 3-μm Nucleodur® HILIC column (Macherey Nagel, Hoerdt, France), containing an ammonium-sulfonic-acid-modified silica, was used with the temperature fixed at 25°C. The injection volume was set at 6 μL. The mass spectrometer was operated in the multiple reaction monitoring mode. The (M–H)+m/z transition for decitabine was 229.0>113.0.
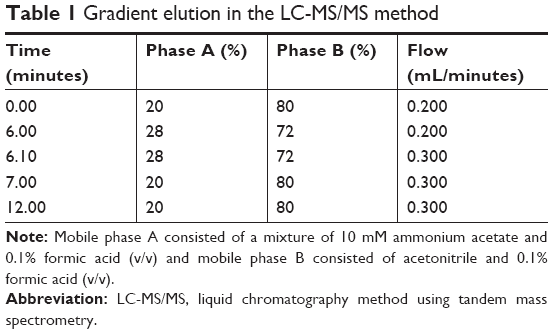 | Table 1 Gradient elution in the LC-MS/MS method |
Physiochemical characterization of the LNC formulations
Size and polydispersity index measurements
The size and polydispersity index (PDI) of blank LNCs and decitabine (C12)2-loaded LNCs were determined by dynamic light scattering on a Zetasizer® Nano series DTS 1060 (Malvern Instruments S.A., Worcestershire, UK) after dilution in deionized water (1:60 v/v). PDI measurements were used to evaluate the size distribution. Three consecutive measurements were systematically performed.
Drug encapsulation efficiency and drug loading
LNCs were filtered through 0.22 μm polyethersulfone syringe filters (Clearline®, Issy-les-Moulineaux, France). Decitabine (C12)2-filtrate concentrations, corresponding to the loaded drug, were determined after dilution in methanol to destabilize the LNCs and release the decitabine (C12)2. Decitabine (C12)2 concentrations were then determined by LC-MS/MS according to the method described above. The efficiency of drug loading, corresponding to the proportion of the drug encapsulated over the total drug concentration, was then determined.
Stability studies
In phosphate-buffered saline solution
A volume of decitabine (C12)2-loaded LNCs (1 mL) was diluted in 59 mL PBS adjusted to pH 7.4, containing disodium hydrogen phosphate (10 mM), potassium dihydrogen phosphate (1.8 mM), sodium chloride (137 mM), potassium chloride (2.7 mM), and hydrochloric acid to adjust the pH (experiments were performed in triplicate). Beakers were placed at 37°C under agitation. At pre-defined time points (T0, 2, 4, 8, 12, 24, and 48 hours), the efficiency of decitabine (C12)2 encapsulation and drug loading were determined by LC-MS/MS after filtration. The size and PDI of the LNCs were also evaluated over time.
Residual decitabine (C12)2 concentrations in PBS were determined. The decitabine (C12)2 concentration in PBS at the initial timepoint (T0) was considered to be 100%.
In human plasma
The stability of decitabine (C12)2-loaded LNCs in human plasma taken from healthy volunteers (provided by the Etablissement Français du Sang, Nantes, France) was evaluated. Formulations were first diluted 10-fold in water. A volume of 125 μL of the formulation was then mixed with 2 mL plasma (experiments were performed in triplicate). Samples were then placed in a beaker and the temperature was maintained at 37°C. The experiment was conducted for 24 hours. At each time point (0, 2, 4, 6, 9, and 24 hours), 40 μL plasma was removed, diluted 10 times in acetonitrile, and centrifuged 10 minutes at 9,500×g (5810 R, Eppendorf, Montesson, France). Supernatants were then collected and diluted 10-fold in methanol, and the decitabine (C12)2 concentrations determined.
Residual decitabine (C12)2 concentrations were determined in human plasma. The decitabine (C12)2 concentration at the initial timepoint (T0) was considered to be 100%.
Decitabine concentrations were also evaluated to detect the appearance of decitabine over time from decitabine (C12)2 transformation. The same protocol was applied, except the last dilution was performed in methanol and by a factor of two instead of 10.
Cell experiments
Cell culture
The human erythroleukemia cell line (HEL) (LGC Standard, Molsheim, France) was maintained in Roswell Park Memorial Institute medium 1640 (RPMI), supplemented with 10% FBS (Gibco, Fisher Scientific France, Illkirch, France) and 1% antibiotics, in a humidified incubator with an atmosphere containing 5% CO2 at 37°C.
In vitro cell proliferation
The antiproliferative capacity of decitabine (C12)2-loaded LNCs was compared to that of blank LNCs or decitabine or decitabine (C12)2 in solution.
Cells were plated at 5×103 cells/well in 96-well plates. After 24 hours, cells were treated with free-decitabine (C12)2, decitabine (C12)2-loaded LNCs, blank LNCs, or a solution of decitabine. Various concentrations of decitabine (C12)2 from 12.5 to 2,500 nM were tested. For the decitabine solution, the concentration ranged from 5 to 1,000 nM. For blank LNCs, the same range of excipient concentrations as those for decitabine (C12)2-loaded LNCs was tested. Cells cultured with medium alone were used as the 100% viability control. Three independent experiments were conducted, each with quadruplicate samples.
After 72 hours, the plates were centrifuged, culture medium removed, and cell pellets frozen at −80°C for at least 48 hours. Cell proliferation was then evaluated with the Cyquant® cell-proliferation assay kit, according to the manufacturer’s instructions (Fisher Scientific).
Cell-cycle analysis
Cells (HEL) were cultured in 12-well plates at 5×104 cells/well. After 24 hours, cells were treated with decitabine in solution (1,000 nM), decitabine (C12)2-loaded LNCs (2,500 nM), or blank LNCs with an equivalent excipient concentration as that of the decitabine (C12)2-loaded LNCs.
After 72 hours, cells were collected and centrifuged at 200×g for 5 minutes. Pellets were then washed in PBS and fixed in 70% cold ethanol. Fixed cells were washed in PBS and incubated for 15 minutes at room temperature in the dark in a PBS staining solution containing 40 μg/mL propidium iodide and 100 μg/mL RNAse A.
Samples were then analyzed on a BD FACSCANTO II system (BD Biosciences, Le Pont de Claix, France), and propidium iodide incorporation estimated using BD FACSDiva software (BD Biosciences, Le Pont de Claix, France). Cell-cycle analysis data were processed using Flowlogic V7.2 software (Inivai Technologies, Victoria, Australia) and the Dean Jett Fox model.
In vivo study
Animals
The study was approved by the ethical committee of Pays de la Loire, France, under the number APAFiS 10751. Experiments were carried out in strict accordance with the regulations of the French Ministry of Agriculture and were in accordance with the Principles of Laboratory Animal Care. Experiments were performed on male Wistar rats weighing between 222 and 410 g, aged 8–11 weeks, obtained from the Service Commun d’Animalerie Hospitalo-Universitaire (Angers, France). The rats had access to water and food ad libitum prior to the experimental procedure and were fasted 3 hours before experiments.
Pharmacokinetics analysis
Each group was composed of five animals. Animals were slowly injected via the penis vein under inhalational anesthesia (isoflurane-oxygen). Decitabine (C12)2-loaded LNCs (12.5 mg/kg) or decitabine (5 mg/kg) in a saline solution were injected. Doses were adapted with respect to the 2.5 molar ratio between decitabine (C12)2 and decitabine and corresponded to 21 μmol/kg. Blood samples were then collected via the tail vein at 2, 30, and 60 minutes and 2, 5, and 6 hours after injection, into heparinized tubes. Samples were centrifuged at 150×g for 5 minutes at room temperature and the supernatants (plasma) diluted in acetonitrile (5-fold for decitabine samples and 10-fold for decitabine (C12)2 samples) and centrifuged at 15,000×g for 10 minutes. Supernatants were collected and diluted in methanol (2-fold for decitabine samples and 20-fold for decitabine (C12)2 samples) and analyzed by LC-MS/MS.
Calibration curves were performed in rat plasma from 25 to 850 ng/mL decitabine or 50 to 1,000 ng/mL decitabine (C12)2.
The main pharmacokinetic parameters of decitabine were determined using Kinetica® 5.1 software (Thermo Scientific, Philadelphia, PA, USA). Intravenous non-compartmental analysis was used to evaluate these parameters for decitabine and decitabine (C12)2, and extravascular non-compartmental analysis was used to evaluate those of decitabine after decitabine (C12)2-loaded LNC injection. The maximal concentration (Cmax), time to reach the maximum concentration (Tmax), area under the plasma concentration-time curve (AUC), and mean residence time (MRT) were determined.
Statistical analysis
Results are expressed as the mean±standard deviation (SD) or standard error of the mean (SEM) for in vivo experiments. Group comparisons were performed using the Mann–Whitney test, P<0.05 was considered to be statistically significant.
Results
Decitabine (C12)2 synthesis
We obtained a white dried product. The yield of the chemical synthesis was estimated to be 50%. After elemental analysis, we determined the molecular weight of the obtained purified product to be 592 g/mol, confirming the chemical structure of decitabine (C12)2 (calculated molecular weight=592.81 g/mol).
Elemental analysis showed the purity to be 98%: 1H NMR (300 MHz, deuterated DMSO): δ 0.75 (6 H, t), 1.15 (40 H, m), 1.5 (4 H, m), 2.25 (4 H, m), 2.30 (2 H, dd), 4.10 (1 H, m), 4.20 (2 H, m), 5.20 (1 H, m), 5.90 (1 H, t), 7.60 (−NH2, 2 hours, m), 8.25 (1 H, s).
Characterization of the LNCs
We determined the efficiency of encapsulation and drug loading following the formulation of blank LNCs or decitabine (C12)2-loaded LNCs. The size, PDI, encapsulation efficiency, and drug loading are presented in Table 2. The drug loading is 5.8 mg/mL of formulation. The drug loading presented as the percentage of loaded drug amount relative to the drug and nanocarrier material is 1.63% w:w.
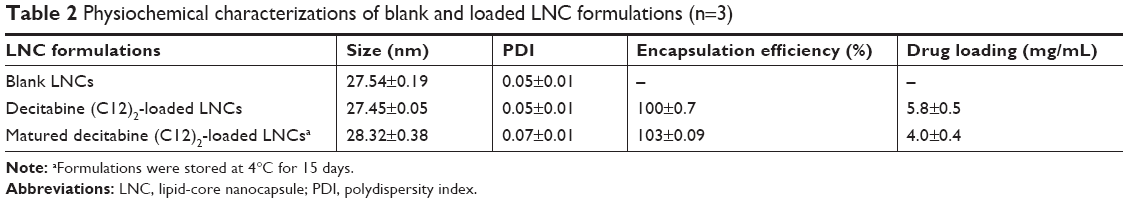 | Table 2 Physiochemical characterizations of blank and loaded LNC formulations (n=3) |
Encapsulation of decitabine (C12)2 into the LNCs did not modify the size of the nanoparticles relative to that of blank LNCs. The drug encapsulation efficiency was unchanged after 15 days (called “matured LNCs” in Table 2). However, drug loading decreased by a factor of 1.5, due to the degradation of decitabine (C12)2 over time.
Stability studies
In PBS
We evaluated the encapsulation efficiency and drug loading of decitabine (C12)2-loaded LNCs for 48 hours in PBS (Figure 3A). The encapsulation efficiency was unaltered after 48 hours at 37°C. However, decitabine (C12)2 slowly degraded over time. After 48 hours, half of the initial drug concentration was recovered. Thus, drug loading was reduced to half of that initially. The formulations were stable for the duration of the stability study, without modification of size or PDI (Figure 3B).
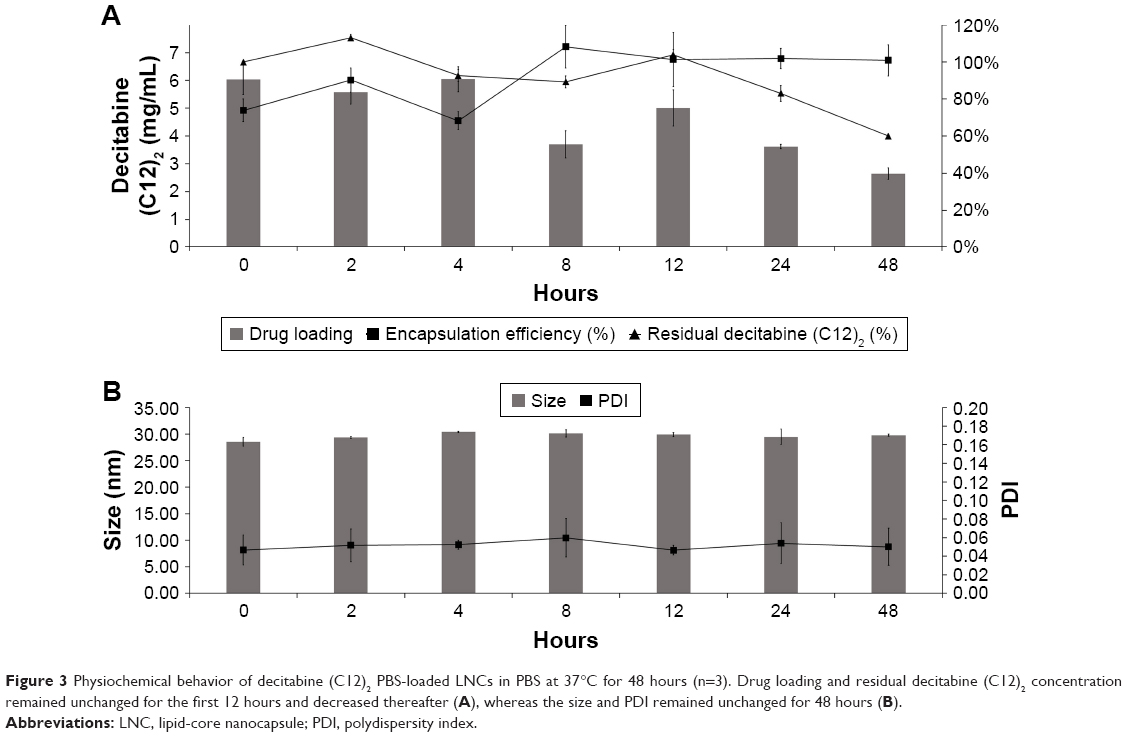 | Figure 3 Physiochemical behavior of decitabine (C12)2 PBS-loaded LNCs in PBS at 37°C for 48 hours (n=3). Drug loading and residual decitabine (C12)2 concentration remained unchanged for the first 12 hours and decreased thereafter (A), whereas the size and PDI remained unchanged for 48 hours (B). |
In human plasma
We evaluated the stability of decitabine (C12)2-loaded LNCs in human plasma (Figure 4). Decitabine (C12)2 slowly degraded over time, whereas the decitabine concentration increased, reaching a maximum after 6 hours. A plateau was then maintained for 3 hours before the decitabine started to degrade. This experiment confirmed the expected transformation of decitabine (C12)2 to decitabine. However, only 10% of the maximal theoretical decitabine concentration was reached, indicating that not all decitabine (C12)2 was transformed into decitabine over time.
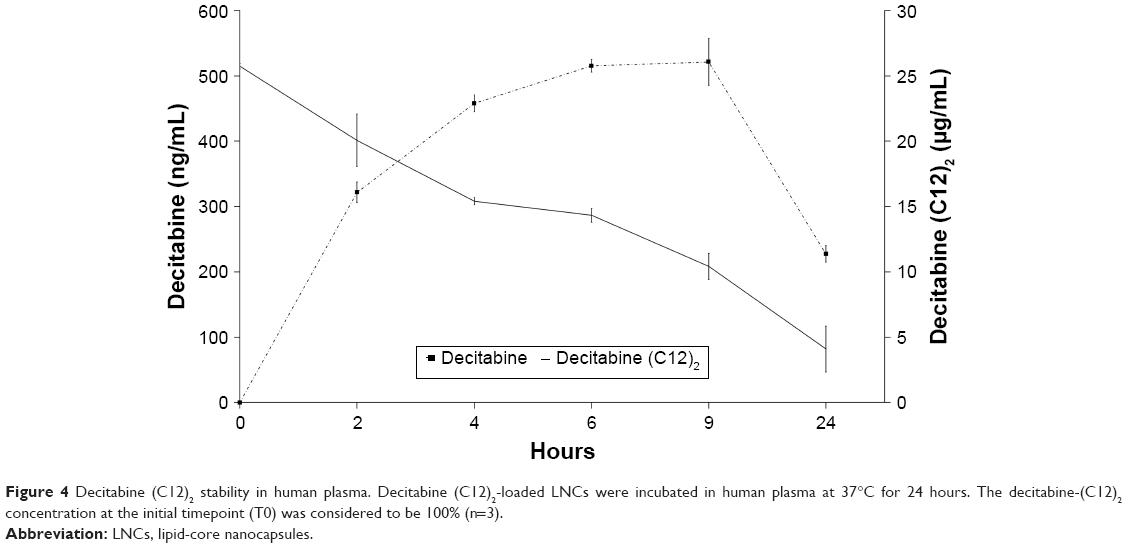 | Figure 4 Decitabine (C12)2 stability in human plasma. Decitabine (C12)2-loaded LNCs were incubated in human plasma at 37°C for 24 hours. The decitabine-(C12)2 concentration at the initial timepoint (T0) was considered to be 100% (n=3). |
Cell culture experiments
Cell proliferation study
We compared the antiproliferative activity of decitabine (C12)2-loaded LNCs to that of decitabine (C12)2 in solution, decitabine in solution, and blank LNCs by measuring cell proliferation after 3 days of contact with various concentrations of the compounds/LNCs (Figure 5). Blank LNCs were non-toxic at concentrations between 26 and 105 μg/mL, whereas there was a trend toward antiproliferative activity above 200 μg/mL. Exposure to above 50 nM decitabine or 125 nM decitabine (C12)2, either free or loaded in LNCs, decreased cell viability. Cell viability was systematically and significantly lower when the decitabine (C12)2 was loaded than when it was in solution at concentrations above 250 nM. Finally, the antiproliferative activity of decitabine was greater than that of decitabine (C12)2, free or loaded into LNCs.
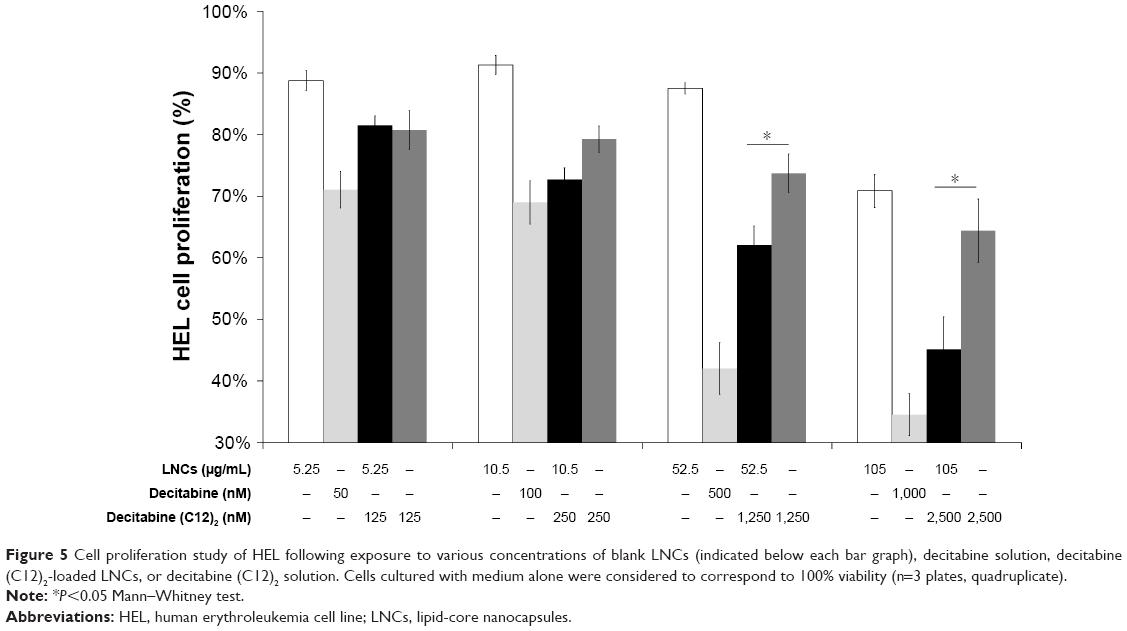 | Figure 5 Cell proliferation study of HEL following exposure to various concentrations of blank LNCs (indicated below each bar graph), decitabine solution, decitabine (C12)2-loaded LNCs, or decitabine (C12)2 solution. Cells cultured with medium alone were considered to correspond to 100% viability (n=3 plates, quadruplicate). |
Cell-cycle analysis
We then performed cell-cycle analysis on HEL cells (Figure 6). Untreated cells (RPMI alone) or those treated with blank LNCs showed <10% of the cells in the G2/M phase of the cell cycle. As expected, cells treated with decitabine showed a higher proportion of cells arrested in G2/M (17.2%). Treatment of the cells with decitabine (C12)2-loaded LNCs (2,500 nM) gave similar results, with 14.6% of the cells arrested in G2/M, which was not significantly different from that of treatment with decitabine (1,000 nM) (P=0.25, Mann–Whitney test).
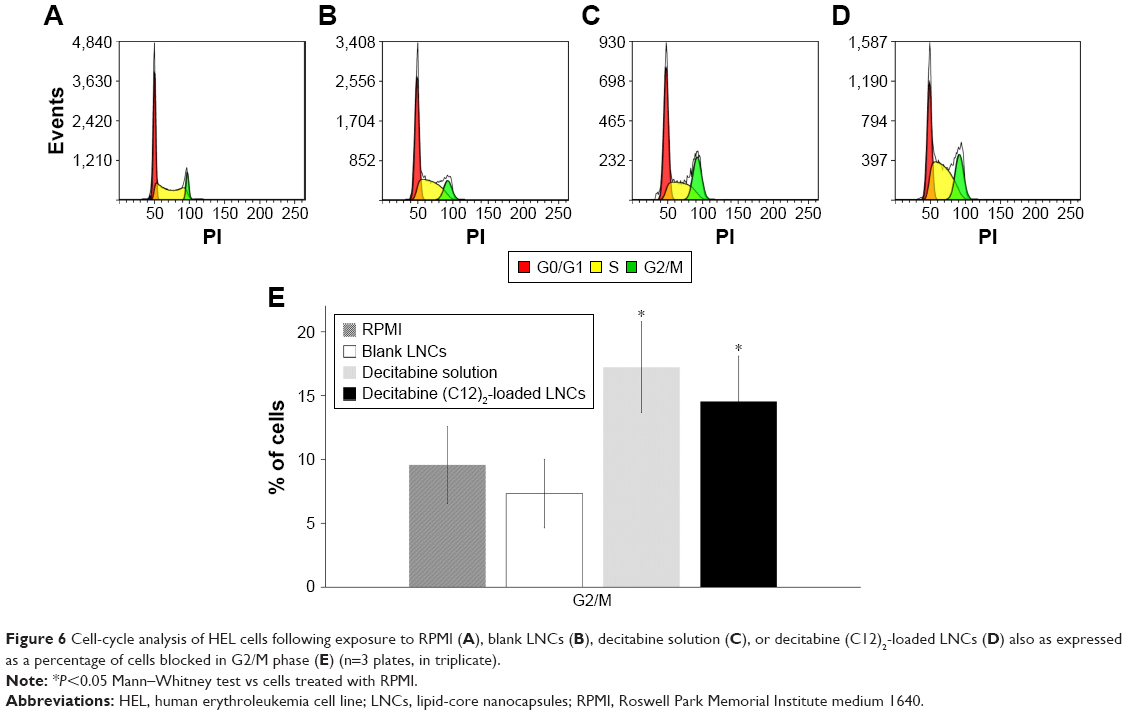 | Figure 6 Cell-cycle analysis of HEL cells following exposure to RPMI (A), blank LNCs (B), decitabine solution (C), or decitabine (C12)2-loaded LNCs (D) also as expressed as a percentage of cells blocked in G2/M phase (E) (n=3 plates, in triplicate). |
Pharmacokinetic analysis
We intravenously administered the same molar concentrations of decitabine and decitabine (C12)2 to rats. Plasma concentrations of decitabine (C12)2 were significantly higher (8-fold) than those of decitabine after 2 minutes (Figure 7). The plasma concentration of both then decreased rapidly, corresponding to a very short MRT (Table 3). The MRT of decitabine (C12)2 was significantly shorter than that of decitabine (P=0.008). However, the MRT of decitabine was significantly prolonged when the decitabine (C12)2-LNC formulation was injected (P=0.008). An increase in decitabine concentrations over time (Figure 7) confirmed the metabolism of decitabine (C12)2 to decitabine; the maximal concentration of decitabine was reached 84±33 minutes after injection. The Cmax of decitabine was lower when it was administered as a solution than in its encapsulated form (Table 3). The terminal half-lives (T1/2) were calculated from the three last points of the curves and were as follows: T1/2 of decitabine: 146±22 minutes; T1/2 of decitabine (C12)2: 64±48 minutes, and T1/2 of decitabine after decitabine (C12)2 transformation: 171±14 minutes. This last value does not significantly differ from the half-life of free decitabine (P>0.05).
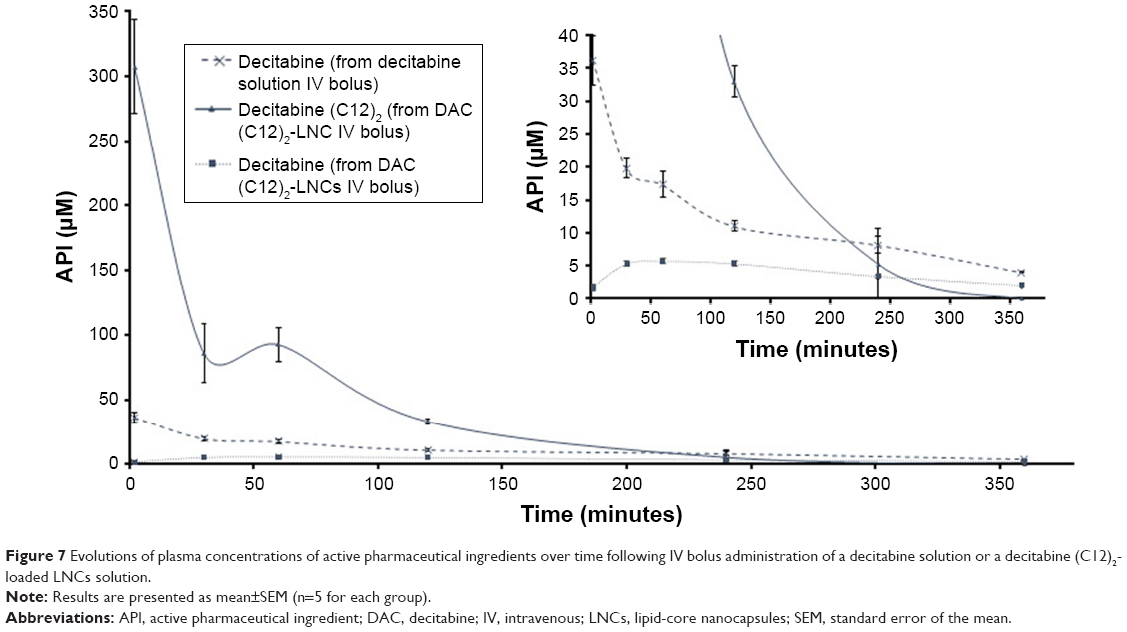 | Figure 7 Evolutions of plasma concentrations of active pharmaceutical ingredients over time following IV bolus administration of a decitabine solution or a decitabine (C12)2-loaded LNCs solution. |
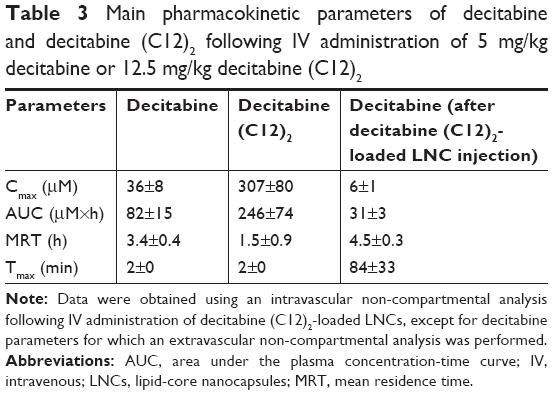 | Table 3 Main pharmacokinetic parameters of decitabine and decitabine (C12)2 following IV administration of 5 mg/kg decitabine or 12.5 mg/kg decitabine (C12)2 |
Discussion
In clinical trials, decitabine modestly increased the overall survival of older AML patients over that of supportive care.20 The blood residence time of the drug after intravenous administration is from 15 to 25 minutes due to its chemical instability and rapid degradation by cytidine deaminase.7 Here, we synthesized a prodrug to overcome such degradation. Lauric acid was selected to form an ester bond with decitabine. Modified decitabine was obtained by adding two alkyl chains to the 3′ and 5′ hydroxyl groups of the decitabine molecule. Finally, we obtained a decitabine (C12)2 molecule, with a molecular weight of 592 g/mol. The yield of the chemical synthesis was limited to 50%, probably due to the well-known chemical instability of decitabine under various conditions (acid, alkaline, and heat).6,21–23 This chemical modification confers a more lipophilic structure to the molecule than that of decitabine, allowing its encapsulation into LNCs, which are organic solvent-free nanoparticles.24 Gemcitabine, another deoxycytidine analog, has been largely chemically modified with numerous different chemical modifications.25 Among them, esterification of fatty acids on the 5′ position prolonged the cell retention time, rendered cellular internalization independent of the human equilibrative nucleoside transporters (hENT), and improved efficacy in gemcitabine-resistant cell lines.26,27 The activity of decitabine inversely correlates with hENT expression, demonstrating its cellular internalization by those transporters.28 Thus, if the cell penetration pathway of the 5′ fatty acid ester of decitabine designed in this study is hENT independent, as shown for the 5′ fatty acid ester of gemcitabine, then resistance to chemotherapy could be overcome.
The most advanced chemically modified form of decitabine in clinical trials is guadecitabine. Decitabine is released after gradual cleavage of the phosphodiester bond present in the structure and the in vivo exposure time was prolonged.29 Similarly, decitabine (C12)2, developed here, with two ester linkages, may gradually release decitabine in the blood once it is introduced into the blood stream. However, the modified decitabine was not soluble in water, and drug formulation was necessary for its administration. One possibility was surfactant adjunction or encapsulation into nanoparticles. Nanoparticles have several advantages, such as protecting the drug from several degradation pathways. Our group has previously developed LNCs that can encapsulate lipophilic drugs.14,30,31 The calculated log P of decitabine (C12)2 is 9.7, which explains its capacity to be well encapsulated in lipid-core nanocapsules. Indeed, drug loading of the prodrug improved over that of a previous formulation with no chemical modification of decitabine (log P=−2.2), for which drug loading of only ~0.5 mg/mL was obtained vs 2.3 mg/mL in the present study.19 In AML, decitabine is administered at a dose of 20 mg/m2 body surface area. With such a formulation, a volume of <9 mL/m2 body surface area is required to obtain a potential clinical effect. Lipid-based nanocarriers were also developed by Neupane et al32 using a high-pressure homogenizer technique. The encapsulation efficiency was over 80%, but lower than that of our prodrug strategy. Vijayaraghavalu and Labhasetwar12 developed another nanocarrier formulation, corresponding to a nanogel. Drug loading of ~6% w/w was obtained, but without considering the water content in the formulation. Our shelf-stability studies of decitabine (C12)2-loaded LNCs in suspension demonstrated a decrease of the relative decitabine (C12)2 concentration after 15 days, with stable encapsulation efficiency. A freeze-drying process may be used to allow the storage of formulations for several months, as previously described for LNCs.33
We then studied the stability of the formulation in several media. We first determined the stability in PBS maintained at 37°C. The decitabine (C12)2 concentration was stable; >75% of the initial concentration was recovered after 24 hours. Decitabine degradation in phosphate buffer was previously studied by Lin et al,21 who reported recovery of <50% of the initial concentration after 24 hours at 37°C. The chemical modification of decitabine associated with its encapsulation into LNCs led to its reduced degradation. This is in accordance with previous studies demonstrating that decitabine prodrugs enhance the stability of the drug in buffers, even if instability is generally observed after several hours.34–36
Several studies using cytidine deaminase inhibitors to increase the time of exposure to decitabine have also been published.37,38 We chose an alternative strategy using nanocarriers, as previously performed to protect cytarabine using a liposomal formulation.9 We performed a stability study of the decitabine (C12)2-loaded LNCs in human plasma to demonstrate the protective activity of the nanocarrier. The decitabine (C12)2 concentration decreased, but the decrease was slower than that found in a previous study using a decitabine solution under the same conditions, for which <40% of the initial concentrations were recovered after 6 hours.19 In addition, decitabine (C12)2 was cleaved to decitabine in human plasma, increasing the MRT. Stresemann and Lyko6 demonstrated that extended exposure to a hypomethylating agent enhanced DNA demethylation. The increased MRT of decitabine in plasma may, thus, enhance decitabine activity in vivo.
We then performed in vitro experiments on an acute myeloid leukemia cell line (HEL). We first performed cell proliferation assays and demonstrated that free decitabine (C12)2 decreased cell proliferation significantly less than a solution of decitabine. The antiproliferative activity of decitabine (C12)2 may require its transformation to decitabine. This effect was previously observed with a gemcitabine derivative.39 Blank LNCs were non-toxic at the highest concentrations tested, whereas cell proliferation was significantly decreased by treatment with a free decitabine solution or decitabine (C12)2-loaded LNCs. Moreover, the antiproliferative activity of decitabine (C12)2-loaded LNCs was significantly higher than that of decitabine (C12)2 alone, confirming the potential of LNCs to increase drug activity.
This increased activity of the decitabine (C12)2-loaded LNCs relative to that of free decitabine (C12)2 could be due to increased cellular penetration of the drug via the active uptake of LNCs mediated by clathrin/caveolae-independent pathways.40,41
Decitabine arrests leukemic cells in the G2/M phase.42–44 Although decitabine was modified, cell-cycle analysis demonstrated a trend toward a higher proportion of cells in G2/M after treatment with decitabine or decitabine (C12)2-loaded LNCs. The capacity of decitabine to arrest cells in the G2/M phase was, thus, not altered by chemical modification.
We finally compared the pharmacokinetic parameters of decitabine (C12)2-loaded LNCs to those of decitabine. The observed pharmacokinetic parameters of decitabine are in accordance with those previously published.45,46 The Cmax of the two drugs were significantly different 2 minutes after intravenous administration, with 8-fold more decitabine (C12)2 available than decitabine. However, decitabine (C12)2-loaded LNCs were then rapidly eliminated, and the MRT of decitabine (C12)2 was significantly lower than that of decitabine. This rapid elimination of the drug is probably due to the rapid accumulation of particles in the liver.47 Nevertheless, the MRT of decitabine was significantly prolonged after decitabine (C12)2-loaded LNC administration. However, the AUC of decitabine remained lower with LNC administration than when administered as a free solution. PEGylation of the particles could increase the MRT and AUC of decitabine by delaying the transformation of decitabine (C12)2 to decitabine. Currently, the PEGylation of LNCs induced by Kolliphor® HS15 is insufficient and the MRT of decitabine (C12)2-loaded LNCs has to be enhanced by the PEGylation of LNCs with a larger PEG chain.48 Indeed, Groo et al49 have already demonstrated that such PEGylation of LNCs enhances the MRT by a factor of 1.5.
Conclusion
We chemically modified decitabine and combined this with its encapsulation into LNCs. This strategy increased the shelf stability of decitabine and reduced its sensitivity to cytidine deaminase in vitro. In vivo pharmacokinetics evaluations confirmed the potential of such a strategy to enhance the MRT of decitabine. Further modifications of the LNCs may improve their pharmacokinetic parameters, especially the maximum blood concentrations and AUC. This would be useful for future pharmacological and biodistribution evaluations.
Acknowledgment
The authors are very grateful to the Ligue Contre le Cancer, particularly its Maine et Loire Committee for its financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Rodriguez-Abreu D, Bordoni A, Zucca E. Epidemiology of hematological malignancies. Ann Oncol. 2007;18(Suppl 1):i3–i8. | ||
Visser O, Trama A, Maynadié M, et al. Incidence, survival and prevalence of myeloid malignancies in Europe. Eur J Cancer. 2012;48(17):3257–3266. | ||
Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. | ||
Shah A, Andersson TM-L, Rachet B, Björkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971–2006: a population-based study. Br J Haematol. 2013;162(4):509–516. | ||
Lu LJ, Randerath K. Long term instability and molecular mechanism of 5-azacytidine-induced DNA hypomethylation in normal and neoplastic tissues in vivo. Mol Pharmacol. 1984;26(3):594–603. | ||
Stresemann C, Lyko F. Modes of action of the DNA Methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123(1):8–13. | ||
Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenetics. 2013;5(1):3. | ||
Blum W, Garzon R, Klisovic RB, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci. 2010;107(16):7473–7478. | ||
Lancet JE, Cortes JE, Hogge DE, et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood. 2014;123(21):3239–3246. | ||
Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML. J Clin Oncol. 2016;34(15_suppl):7000–7000. | ||
Griffiths EA, Choy G, Redkar S, Taverna P, Azab M, Karpf AR. SGI-110: DNA methyltransferase inhibitor oncolytic. Drugs Future. 2013;38(8):535–543. | ||
Vijayaraghavalu S, Labhasetwar V. Efficacy of decitabine-loaded nanogels in overcoming cancer drug resistance is mediated via sustained DNA methyltransferase 1 (DNMT1) depletion. Cancer Lett. 2013;331(1):122–129. | ||
Heurtault B, Saulnier P, Pech B, Proust J-E, Benoit J-P. A novel phase inversion-based process for the preparation of lipid nanocarriers. Pharm Res. 2002;19(6):875–880. | ||
Huynh NT, Passirani C, Saulnier P, Benoit JP. Lipid nanocapsules: a new platform for nanomedicine. Int J Pharm. 2009;379(2):201–209. | ||
Lagarce F, Passirani C. Nucleic-acid delivery using lipid nanocapsules. Curr Pharm Biotechnol. 2016;17(8):723–727. | ||
Hureaux J, Lagarce F, Gagnadoux F, et al. Toxicological study and efficacy of blank and paclitaxel-loaded lipid nanocapsules after i.v. administration in mice. Pharm Res. 2010;27(3):421–430. | ||
Hureaux J, Lacoeuille F, Lagarce F, et al. Absence of lung fibrosis after a single pulmonary delivery of lipid nanocapsules in rats. Int J Nanomedicine. 2017;12:8159–8170. | ||
Thomas O, Lagarce F. Lipid nanocapsules: a nanocarrier suitable for scale-up process. J Drug Delivery Sci Technol. 2013;23(6):555–559. | ||
Briot T, Roger E, Lautram N, Verger A, Clavreul A, Lagarce F. Development and in vitro evaluations of new decitabine nanocarriers for the treatment of acute myeloid leukemia. Int J Nanomedicine. 2017;12:8427–8442. | ||
Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. JCO. 2012;30(21):2670–2677. | ||
Lin KT, Momparler RL, Rivard GE. High-performance liquid chromatographic analysis of chemical stability of 5-aza-2′-deoxycytidine. J Pharm Sci. 1981;70(11):1228–1232. | ||
Tománková H, Zýka J. Study of the time dependence of the stability of 5-aza-2′-deoxycytidine in acid medium. Microchem J. 1980/09/01/ 1980;25(3):281–288. | ||
Kim Sun H, Heeb Rita M, Krämer I. Physicochemical stability of reconstituted decitabine (Dacogen®) solutions and ready-to-administer infusion bags when stored refrigerated or frozen. Pharmaceutical Technology in Hospital Pharmacy. Vol 22017:145. | ||
Hureaux J, Lagarce F, Gagnadoux F, et al. Lipid nanocapsules: ready-to-use nanovectors for the aerosol delivery of paclitaxel. Eur J Pharm Biopharm. 2009;73(2):239–246. | ||
Moysan E, Bastiat G, Benoit J-P. Gemcitabine versus modified gemcitabine: a review of several promising chemical modifications. Mol Pharm. 2013;10(2):430–444. | ||
Bergman AM, Kuiper CM, Voorn DA, et al. Antiproliferative activity and mechanism of action of fatty acid derivatives of arabinofuranosylcytosine in leukemia and solid tumor cell lines. Biochem Pharmacol. 2004;67(3):503–511. | ||
Bergman AM, Adema AD, Balzarini J, et al. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest New Drugs. 2011;29(3):456–466. | ||
Qin T, Jelinek J, Si J, Shu J, Issa J-PJ. Mechanisms of resistance to 5-aza-2′-deoxycytidine in human cancer cell lines. Blood. 2009;113(3):659–667. | ||
Issa J-PJ, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015;16(9):1099–1110. | ||
Saliou B, Thomas O, Lautram N, et al. Development and in vitro evaluation of a novel lipid nanocapsule formulation of etoposide. Eur J Pharm Sci. 2013;50(2):172–180. | ||
Roger E, Lagarce F, Benoit J-P. Development and characterization of a novel lipid nanocapsule formulation of SN38 for oral administration. Eur J Pharm Biopharm. 2011;79(1):181–188. | ||
Neupane YR, Srivastava M, Ahmad N, Kumar N, Bhatnagar A, Kohli K. Lipid based nanocarrier system for the potential oral delivery of decitabine: formulation design, characterization, ex vivo, and in vivo assessment. Int J Pharm. 2014;477(1–2):601–612. | ||
Dulieu C, Bazile D. Influence of lipid nanocapsules composition on their aptness to freeze-drying. Pharm Res. 2005;22(2):285–292. | ||
Tao W, Zhao D, Sun M, et al. Intestinal absorption and activation of decitabine amino acid ester prodrugs mediated by peptide transporter PEPT1 and enterocyte enzymes. Int J Pharm. 2018;541(1–2):64–71. | ||
Zhang Y, Sun J, Gao Y, et al. A carrier-mediated prodrug approach to improve the oral absorption of antileukemic drug decitabine. Mol Pharm. 2013;10(8):3195–3202. | ||
Clouser CL, Bonnac L, Mansky LM, Patterson SE. Characterization of permeability, stability and anti-HIV-1 activity of decitabine and gemcitabine divalerate prodrugs. Antivir Chem Chemother. 2014;23(6):223–230. | ||
Garcia-Manero G, Odenike O, Amrein PC, et al. Successful emulation of IV decitabine pharmacokinetics with an oral Fixed-dose combination of the oral cytidine deaminase inhibitor (CDAi) E7727 with oral decitabine, in subjects with myelodysplastic syndromes (MDS): final data of phase 1 study. Am Soc Hematology. 2016. | ||
Lavelle D, Vaitkus K, Ling Y, et al. Effects of tetrahydrouridine on pharmacokinetics and pharmacodynamics of oral decitabine. Blood. 2012;119(5):1240–1247. | ||
Sloat BR, Sandoval MA, Li D, et al. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int J Pharm. 2011;409(1–2):278–288. | ||
Lainé A-L, Clavreula, Rousseaua, et al. Inhibition of ectopic glioma tumor growth by a potent ferrocenyl drug loaded into stealth lipid nanocapsules. Nanomedicine. 2014;10(8):1667–1677. | ||
Paillard A, Hindré F, Vignes-Colombeix C, Benoit J-P, Garcion E. The importance of endo-lysosomal escape with lipid nanocapsules for drug subcellular bioavailability. Biomaterials. 2010;31(29):7542–7554. | ||
Hollenbach PW, Nguyen AN, Brady H, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One. 2010;5(2):e9001. | ||
Vijayaraghavalu S, Dermawan JK, Cheriyath V, Labhasetwar V. Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm. 2013;10(1):337–352. | ||
Thépot S, Lainey E, Cluzeau T, et al. Hypomethylating agents reactivate FOXO3a in acute myeloid leukemia. Cell Cycle. 2011;10(14):2323–2330. | ||
Liu Z, Marcucci G, Byrd JC, Grever M, Xiao J, Chan KK. Characterization of decomposition products and preclinical and low dose clinical pharmacokinetics of decitabine (5-aza-2′-deoxycytidine) by a new liquid chromatography/tandem mass spectrometry quantification method. Rapid Commun Mass Spectrom. 2006;20(7):1117–1126. | ||
Xu H, Lv S, Qiao M, et al. Development and validation of a liquid chromatography–tandem mass spectrometry method for quantification of decitabine in rat plasma. Journal of Chromatography B. 2012;899:81–85. | ||
Sasso MS, Lollo G, Pitorre M, et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials. 2016;96:47–62. | ||
Lainé A-L, Gravier J, Henry M, et al. Conventional versus stealth lipid nanoparticles: formulation and in vivo fate prediction through FRET monitoring. J Control Release. 2014;188:1–8. | ||
Groo AC, Bossiere M, Trichard L, Legras P, Benoit JP, Lagarce F. In vivo evaluation of paclitaxel-loaded lipid nanocapsules after intravenous and oral administration on resistant tumor. Nanomedicine. 2015;10(4):589–601. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.