")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
DHA Protects Against Hepatic Steatosis by Activating Sirt1 in a High Fat Diet-Induced Nonalcoholic Fatty Liver Disease Mouse Model
Authors Luo X, He Z, Sun X, Gu X, Zhang W, Gao J, Li X, Jia R, Wei J, Yu Y, Luo X
Received 24 September 2019
Accepted for publication 20 December 2019
Published 22 January 2020 Volume 2020:13 Pages 185—196
DOI https://doi.org/10.2147/DMSO.S232279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Muthuswamy Balasubramanyam
Xiao Luo, 1, 2,* Zhangya He, 1, 3,* Xiaomin Sun, 1, 3 Xinqian Gu, 1, 3 Wanyu Zhang, 1, 3 Jiayi Gao, 1, 3 Xiaomin Li, 1, 3 Ru Jia, 4 Junxiang Wei, 5 Yan Yu, 1, 3 Xiaoqin Luo 1, 3
1Department of Nutrition and Food Safety, School of Public Health, Xi’an Jiaotong University, Xi’an 710061, People’s Republic of China; 2Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Xi’an Jiaotong University Health Science Center, Xi’an 710061, People’s Republic of China; 3Nutrition and Food Safety Engineering Research Center of Shaanxi Province, School of Medicine, Xi’an Jiaotong University, Xi’an 710061, People’s Republic of China; 4Department of Prosthodontics, Stomatological Hospital, College of Stomatology, Xi’an Jiaotong University, Xi’an 710061, People’s Republic of China; 5Department of Epidemiology and Health Statistics, School of Public Health, Xi’an Jiaotong University, Xi’an 710061, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoqin Luo
Department of Nutrition and Food Safety, School of Public Health, Xi’an Jiaotong University, Xi’an 710061, People’s Republic of China
Tel/Fax +86-29-82655111
Email [email protected]
Aim: Docosahexaenoic acid (DHA; C22; n-3) shows beneficial effects on Non-alcoholic fatty liver disease (NAFLD). Deacetylase Sirtuin1 (Sirt1) was reported to increase energy metabolism and decrease lipogenesis. Here, we investigated whether DHA plays a role in protecting against hepatic steatosis via Sirt1.
Main Methods: Both in vivo and in vitro hepatic steatosis models were used: diet-induced obesity (DIO) model (middle-aged C57BL/6 mice fed a high-fat diet (HFD)) and palmitic acid (PA)-induced lipid accumulation cell model (HepG2 cells).
Key Findings: In DIO mice, treatment with DHA (gavage supplementation) for 8 weeks not only inhibited the lipid accumulation, but also increased fatty acids (FA) oxidation and induced triglyceride export in liver. These changes were accompanied by attenuation of inflammation. Moreover, DHA reversed the HFD-induced reduction of Sirt1 in liver. Interestingly, the beneficial effects of DHA were reversed by lentivirus-mediated Sirt1 knockdown, accompanied with increased expression of markers of lipogenesis, inflammation and reduced FA oxidation. In HepG2 cells, DHA prevented the accumulation of PA-induced lipid droplets, the decrease of FA oxidation and the reduction of Sirt1 level. Inhibition of Sirt1 by sirtinol partially reversed the beneficial effects of DHA on PA-treated cells.
Significance: DHA alleviated hepatic steatosis and reduced inflammation of liver in obese middle-aged mice by mechanisms involving Sirt1 activation.
Keywords: non-alcoholic fatty liver disease, steatosis, docosahexaenoic acid, high-fat diet, Sirt1
Introduction
Obesity has been implicated as a severe public health challenge worldwide. Consequently, obesity-related diseases including type 2 diabetes, cardiovascular disease, and non-alcoholic fatty liver disease (NAFLD) will continue to escalate.1 Among them, NAFLD defined as the accumulation of fat in liver cells in the absence of excessive alcohol consumption has garnered much attention from researchers during the last decade. When having inefficiency in removing lipids through β-oxidation or efflux, the liver is overloaded with triglycerides in the form of lipid droplets in hepatocytes, leading to hepatic steatosis.2 The latter is the initial stage of NAFLD. Moreover, steatosis makes the liver more sensitive to a “second hit,” such as inflammatory cytokines, oxidative stress, endoplasmic reticulum (ER) stress and/or mitochondrial dysfunction. Mentioned above are hallmarks of the injury observed in the liver of obese patients and ultimately result in the development of nonalcoholic steatohepatitis (NASH), which is the advanced stage of NAFLD.3 In addition, excessive fat intake during obesity enhances liver uptake of fatty acids and glycerol from the circulation, which makes the liver exhibit a selective insulin resistance (IR). In this circumstance, the effect of insulin on hepatic de novo lipogenesis is enhanced, resulting in chronic hyperglycemia and hypertriglyceridemia.4 NAFLD has been implicated as one of the most prevalent chronic liver diseases worldwide.5 Meanwhile, ameliorating NAFLD/NASH, the most severe complication of this disease, eg, hepatocarcinoma could be possibly prevented in its onset.6 However, the effective ways for preventing NAFLD/NASH are still not available.
N-3 polyunsaturated fatty acids (n-3 PUFAs), including eicosapentaenoic acid and docosahexaenoic acid (DHA), are reported to have benefits against NAFLD.7 This antisteatotic effect of n-3 PUFAs appears to be attributable to the inhibition of de novo lipogenesis and the β-oxidation activation, very-low density lipoproteins secretion, and bile acid synthesis.8 Furthermore, n-3 PUFAs possess pleiotropic properties, which help attenuate hepatic steatosis-induced inflammation and oxidative stress.9 In line with these observations, a recent review by Delarue and Lalles concluded that n-3 PUFAs decrease liver steatosis, but do not reverse the histologic features of NASH already established.10 Albeit the benefits of n-3 PUFAs in NAFLD, the independent effects of DHA on NAFLD and the possible mechanisms are not clear.
Sirt1, belongs in a family of proteins which are purported to be involved in the regulation of glucose homeostasis and attenuates insulin resistance via reducing mitochondrial dysfunction.11 Sirt1 physically interacts with peroxisome proliferator-activated receptor-gamma coactivator (PGC1α), a key enzyme in hepatic mitochondrial β-oxidation, and deacetylates it, ultimately leading to the induction of liver gluconeogenic and β-oxidation genes transcription.12 Our previous study revealed that DHA reduces angiogenesis of adipose tissues and attenuates insulin resistance in HFD-induced obese mice via the activation of Sirt1.13 We also found that DHA attenuates palmitic acid (PA)-induced lipid accumulation and inflammation in hepatocytes through suppressing inflammasome activation and inflammatory cytokine maturation in vitro.14 However, a more comprehensive profile of physiological and genetic events under DHA treatment in NAFLD mice have not yet been documented. Further, it is unclear whether the beneficial effect of DHA on NAFLD is mediated by Sirt1. Therefore, the aim of the present study was to determine whether DHA supplementation is able to decrease hepatic lipid accumulation and inflammation in a NAFLD mouse model.
Materials and Methods
Chemical Sources
Animal diet was purchased from Research Diet, Inc. (New Brunswick, NJ, USA). DHA was obtained from Sigma-Aldrich (St. Louis, MO, USA) and the stock solution (1000 mg/mL) was prepared as follows: DHA powder (1 g) was dissolved in a small amount of ethanol (100–150 μL), dried under nitrogen and then dissolved in 100 μL NaOH (0.01N) with 900 μL PBS. The stock solution was aliquoted and stored in −80°C. Antibodies used are from Cell Signaling Technology, Inc. (Danvers, MA, USA). All the other reagents and materials were purchased from Sigma Aldrich and Invitrogen (Carlsbad, CA, USA) unless otherwise noted.
Animals and Diets
Middle-aged (9 months) male C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. In order to prevent mutual attacks, animals were housed separately under a controlled ambient temperature (24°C) and diurnal condition (12-h light-dark cycle) with access to food and water ad libitum and were given fresh diet every 3 days. After acclimatization, mice were randomly divided into five groups (n=10 per group). Except for the control group (CD group) which placed on a control diet (10% kcal fat, D12450), all mice from other groups were fed a HFD (60% kcal fat, D12492) for 20 weeks. One group of mice (HFD group) was not changed diet during the whole experiment. One group of mice (HFD+DHA group) was gavagely supplemented with DHA (100 mg/kg bw, twice a week) for the last 8 weeks of the experiment. The remaining two groups of mice were also given DHA supplementation and semiweekly intravenously injected of Sirt1 shRNA or Scr shRNA lentivirus solution produced as we previously described for the last 8 weeks of the experiment.13 Food consumption and body weight were monitored weekly. After the mice were anesthetized by inhalation of CO2, blood was collected from the orbit and stored at −80°C. Then, tissues were obtained and weighted. Fresh liver tissues either were fixed with 4% w/v paraformaldehyde for hematoxylin-eosin (H&E) staining or iced in refrigerator (−80°C) with optimal cutting temperature (OCT) compound for Oil Red O staining. The remaining liver tissue was stored at −80°C till analysis. The animals were cared for in accordance with the Guiding Principles in the Care and Use of Animals. The experiment was approved by the Institutional Animal Care and Use Committee of Xi’an Jiaotong University (No. XJTULAC-2013-024).
Measurement of Inflammatory Cytokines by ELISA Assay
Blood was centrifuged at 10,000 g for 15 min at 4°C and the supernatants were stored at −20°C before analysis. Serum levels of inflammatory cytokines, including IL-6 and TNF-α as well as adiponectin proteins were determined by Quantikine ELISA kit (R&D Systems Inc, Minneapolis, USA). The color generated was determined by measuring the OD value at 450 nm of spectrophotometric microtiter plate reader (Molecular Devices Corp., Sunnyvale, CA, USA).
Cell Cultures
Cell cultures were performed as previously described.14 In brief, HepG2 cells (ATCC, Manassas, VA, USA) were routinely cultured with Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin (100 IU/mL) and streptomycin (30 mg/mL). Cells were seeded into plates 24 h prior to treatments at approximately 80% confluence and exposed to different stimuli. For each assay, at least three independent experiments were conducted.
RNA Isolation and Quantitative PCR (qPCR) Analysis
Total RNA was isolated from liver and HepG2 cells using TRIzol reagent (Invitrogen, San Diego, CA, USA) and reversely transcribed into cDNA using a commercial RT-PCR Kit according to the manufacturer’s instructions (Thermo scientific, USA). qPCR was performed as previously described.13 Briefly, primers (Table 1) were designed using the sequence information of the NCBI database and synthesized by Beijing AUGCT Biological Technology Co., Ltd. (Beijing, China). The PCR conditions were as follows: initial denaturation: 95°C, 5 min; 40 cycles of denaturation (95°C, 30 s), annealing (58°C, 30 s), and elongation (72°C, 60 s). The fluorescent signals were collected during the extension phase; Ct values of the sample were calculated. The expression of Gapdh was assessed as a housekeeping gene. Relative expression of the gene of interest was therefore determined using the 2–△△CT method.
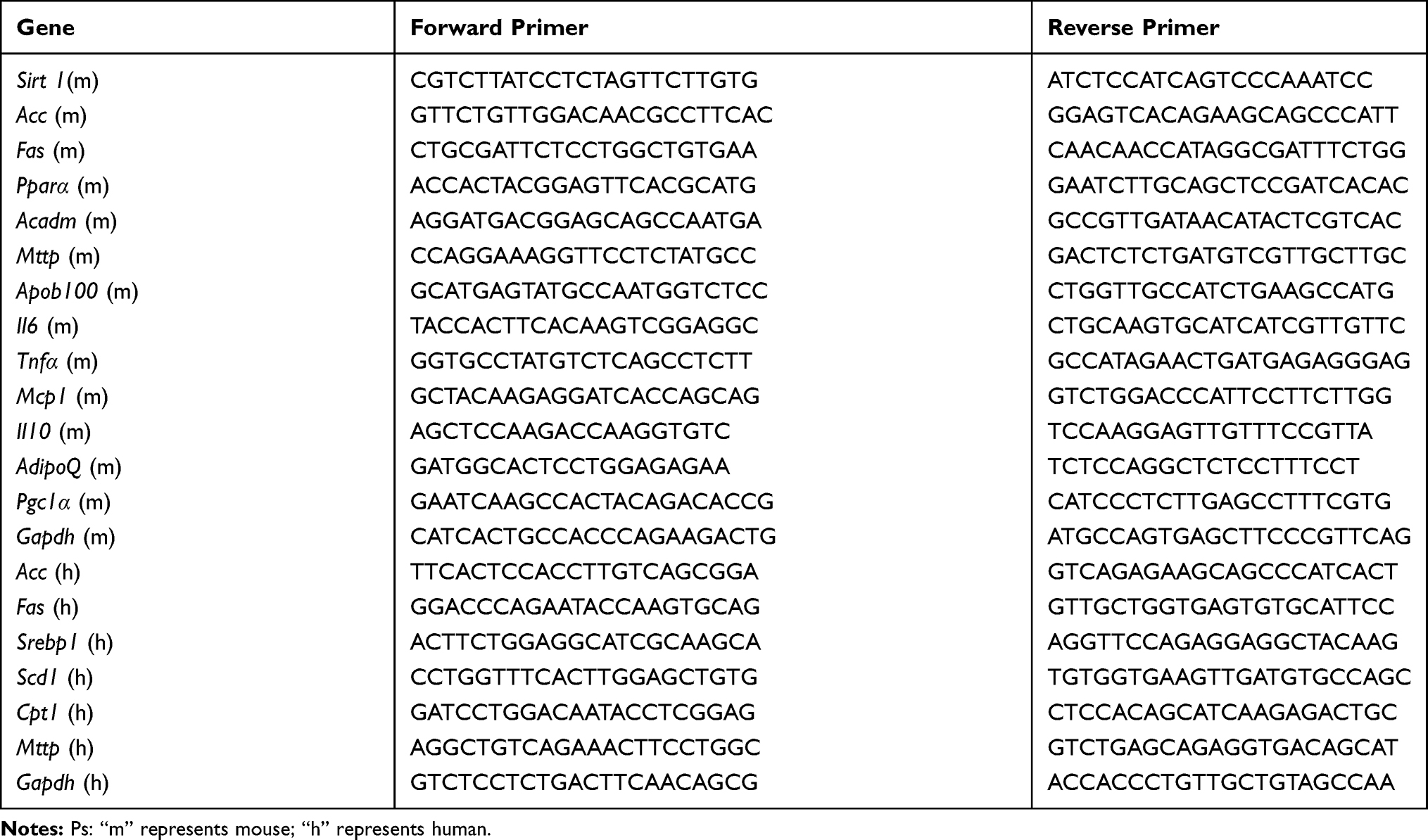 |
Table 1 Primers Used for Quantitative Real-Time PCR Analysis |
Western Blotting Analysis
Liver tissues were homogenized in RIPA buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and sodium orthovanadate, sodium fluoride, EDTA, leupeptin) with protease inhibitor cocktail and phosphatase inhibitor cocktail (Thermo scientific, USA). After lysis on ice, samples were centrifuged at 12,000 rpm at 4°C for 15 min. The protein concentration was determined using bovine serum albumin (BSA) as standard and then processed to Western blotting regularly. The bands of proteins were quantified using Image J software (National Institute of Health, Bethesda, MD, USA). The ratio of the intensity of the target protein to that of β-actin was calculated to represent the expression level of the protein.
Biochemical Measurements and HOMA-IR
Blood samples of fasted mice were collected to determine serum concentrations of total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and triacylglycerols (TAG) by enzymatic methods (BioAssay Systems, Haward, CA). In addition, liver tissues were homogenized in cold Tris–HCl (pH 7.4) (1:10, w/v) of 20 mM. The homogenate was centrifuged for 30 min at 2500 g. And then, hepatic TAG was measured by commercial kits from Randox Laboratories Ltd. Fasting blood glucose concentration and fasting plasma insulin concentration were evaluated as previously described.13 The homeostasis model assessment of insulin resistance (HOMA-IR) was calculated as fasting glucose (mmol/L) x fasting insulin level (mIU/L)/22.5.
Histology and Oil Red O Staining
Liver tissues were fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin wax. Sections (7 μm) were stained with H&E and assessed by light microscopy for morphology (BX53, Olympus, Japan). Data were collected from all mice in each group, five fields per mouse, using Image J software. To determine hepatic lipid accumulation, frozen liver sections (5 μm) were stained with 0.5% Oil Red O for 10 min, washed, and counterstained with Mayer’s hematoxylin for 45 s. Data were presented as the mean percentage of stained area to a total hepatic region in 10 fields from each liver section. Quantitative analysis was performed using analySIS-FIVE program (Olympus Soft Imaging System, Münster, Germany).
Statistical Analyses
Statistical analysis was performed using SPSS 16.0 statistical software (SPSS Inc., Chicago, IL, USA) and based on one-way ANOVA, followed by the LSD post hoc test if the overall differences were significant. p < 0.05 was considered statistically significant.
Results
Expression of Sirt1 in Steatotic Livers
Schematic flow of experiment was shown in Figure 1A. The injection of Scr shRNA lentivirus solution had no obvious extra effect on all aspects we concerned compared to HFD+DHA mice, so the data were not shown. The expression level of Sirt1 was studied by qPCR and Western blotting in livers from each group of animals. As depicted in Figure 1B and C, the expression of Sirt1 mRNA and protein was lower in steatotic livers from HFD-fed mice compared to those from animals on control diet (CD), but DHA supplementation significantly reversed the decrease of Sirt1 expression in HFD-fed mice livers. As expected, Sirt1 knockdown remarkably decreased Sirt1 mRNA and protein levels (Figure 1B and C).
 |
Figure 1 Sirt1 expression in steatotic livers. (A) Schematic flow of the experiment. Sirt1 mRNA (B) and protein expression (C) in liver from mice. β-Actin was used as a loading control. Data represent the means ± S.D. (standard deviation) (n=8, *p < 0.05 and **p < 0.01). |
Sirt1 Knockdown Diminishes the Protective Effects of DHA on HFD-Induced Hepatic Steatosis
As we previously described, body weight significantly increased responsive to a high-fat diet and there were no differences in body weight and daily food intake between HFD mice and HFD+DHA mice.13 Liver weight and its compositional proportion from each group of mice were conducted on the day of sacrifice. HFD caused a significant increase in liver weight and liver coefficient compared to CD group. Significant lower values were found after DHA supplementation but these were not sustained in Sirt1 knockdown mice (Table 2). The HOMA-IR index was significantly higher in HFD-fed mice compared to CD-fed mice. Interestingly, significantly lower values were found after DHA supplementation but these were not sustained in Sirt1 knockdown mice (Supplementary Figure 1). We also measured the levels of plasma TAG, TC, HDL-C and LDL-C. Within groups supplemented with the HFD, TAG and LDL-C levels were significantly lower whereas HDL-C level was higher in mice supplemented with DHA than in the other two groups. Regarding to TC, HFD induced an increase in TC values, which were not significantly different among HFD groups (Table 2). Liver morphological alterations are presented in Figure 2A. Compared to CD mice, animals subjected to HFD for 20 weeks showed liver steatosis, with more droplets being found in the studied fields in HFD group but less in HFD+DHA group (Figure 2A). However, systemic Sirt1 knockdown diminished the effect of DHA on steatosis of liver. Next, we investigated hepatic accumulation of lipids in each group of mice. Results from Oil Red O staining showed that small amounts of lipid droplets were found in liver sections of CD mice but massive accumulation of lipid droplets were observed in HFD group. As expected, 8 weeks of DHA supplementation almost entirely protected from NAFLD (Figure 2A). In agreement with the histological changes, hepatic TAG level of HFD+DHA group was significantly lower compared to those of the HFD group but Sirt1 knockdown attenuated the TAG-lowing effects of DHA (Figure 2B).
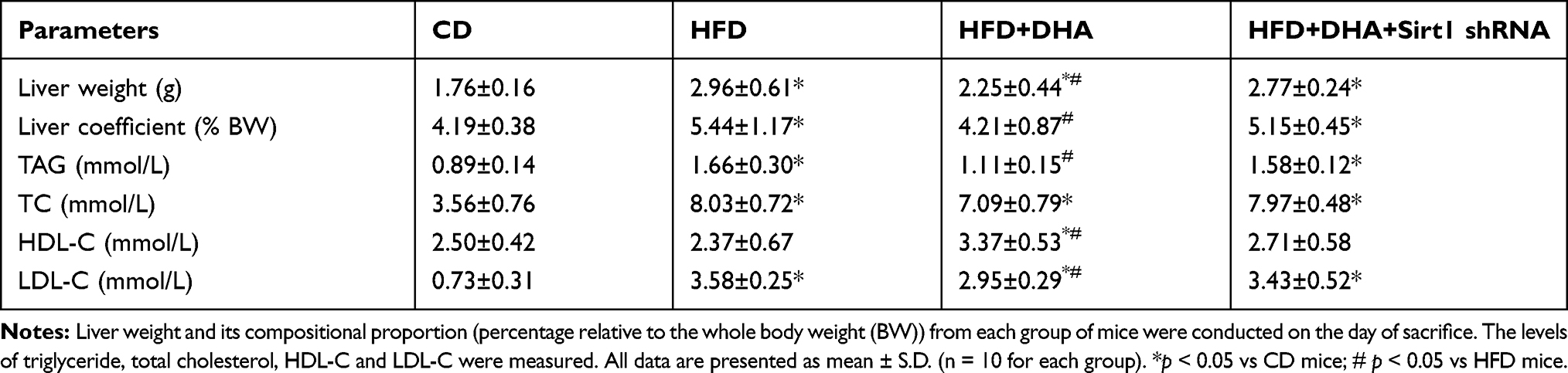 |
Table 2 Liver and Plasma Metabolic Parameters in Mice |
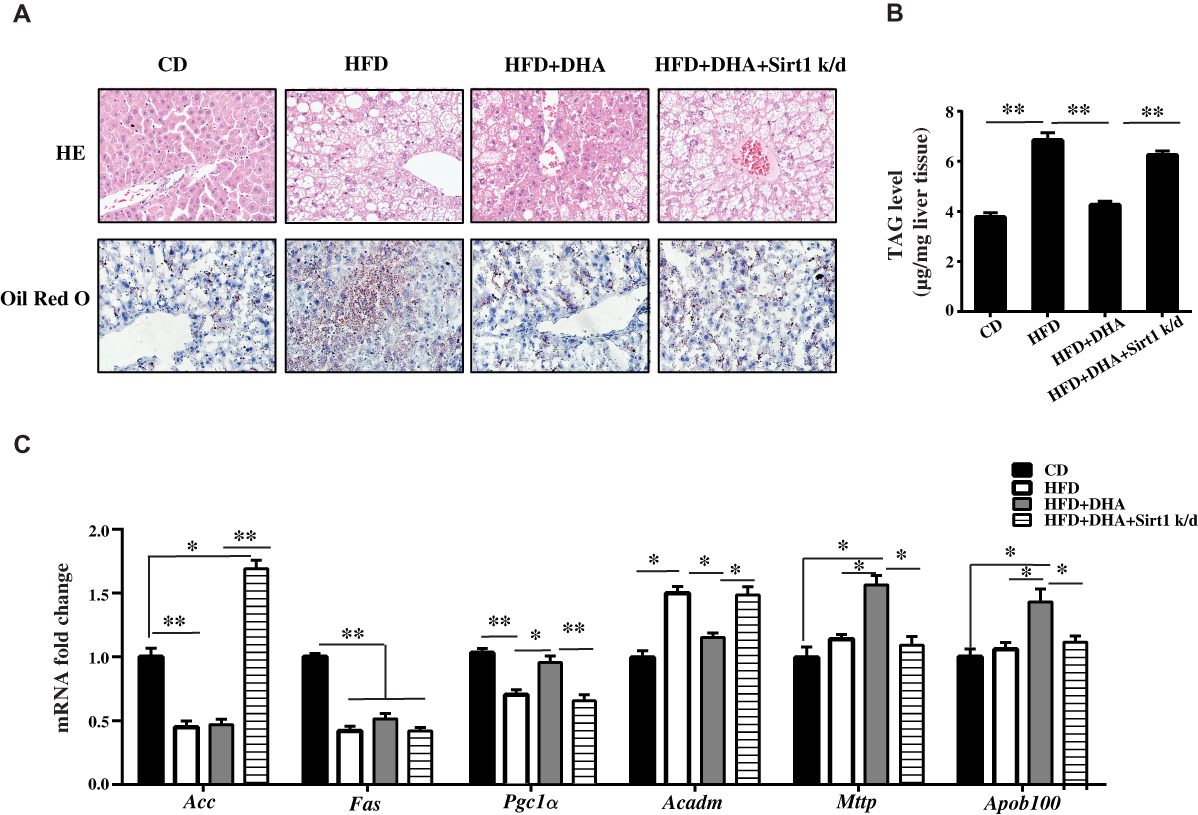 |
Figure 2 The effect of DHA on HFD-induced hepatic steatosis is blocked by Sirt1 knockdown. (A) Representative photomicrographs (original magnification 400×) of liver sections stained with H&E (upper panels) and Oil Red O (low panels) from all groups of mice. (B) TAG level was measured in livers from four groups of mice. (C) qPCR analysis of gene expression in liver of four groups of mice. Data are expressed as fold change compared with CD group. Values are means ± S.D. (n = 10, *p < 0.05; **p < 0.01). |
We further investigated the regulation of enzymes involved in fat metabolism (Figure 2C). Interestingly, HFD markedly decreased mRNA levels of key lipogenic gene fatty acid synthase (Fas) and DHA supplementation did not change its expression. However, DHA supplementation prominently upregulated Pgc1α mRNA level and Sirt1 knockdown reversed it. In addition, DHA remarkably induced the expression of Mttp, which is involved in the TAG export, and Sirt1 knockdown diminished these effect of DHA. These findings suggest that the activation of mitochondrial β-oxidation and TAG export may contribute to the antisteatotic properties of DHA, which is possibly mediated by Sirt1.
DHA Treatment Attenuates Inflammation via Sirt1 in Mice with NAFLD
We found that HFD treatment resulted in a significant increase in serum IL6 and TNFα concentration levels (Figure 3A and B) accompanied by a marked reduction in serum AdipoQ level compared to controls (Figure 3C). These changes were reversed by DHA administration but were comparable in Sirt1 knockdown group. We next analyzed the mRNA levels of pro-inflammatory and anti-inflammatory genes. In agreement with biochemical data, DHA showed significantly downregulated in the expression of the pro-inflammatory genes Il6, Tnfα and Mcp1 while upregulated gene expression of the anti-inflammatory cytokines Il-10 and AdipoQ in liver. Expectedly, Sirt1 knockdown only reversed the decrease of Mcp1 and the increase of AdipoQ mRNA levels compared to DHA group (Figure 3D).
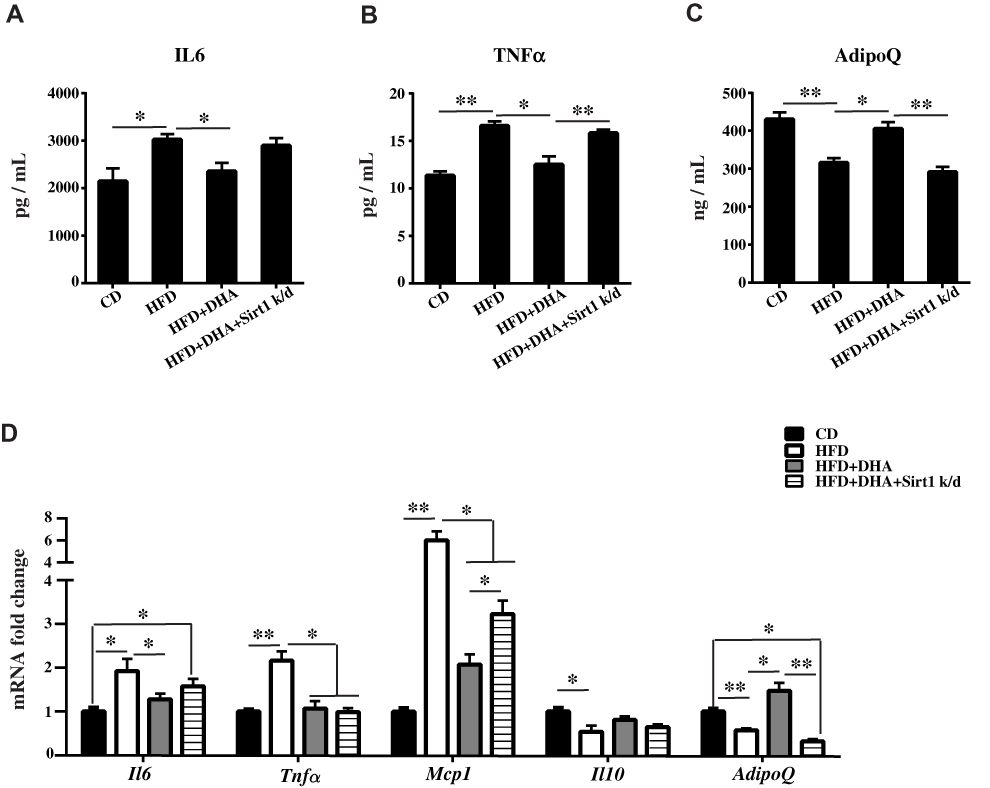 |
Figure 3 DHA attenuates inflammation via Sirt1 in mice. Serum levels of IL6 (A), TNFα (B) and AdipoQ (C) were measured. (D) qPCR analysis of Il6, Tnfα, Mcp1, Il10 and AdipoQ gene expression in liver of mice. All data are expressed as fold change compared with CD control group. Values are means ± S.D. (n = 10, *p < 0.05; **p < 0.01). |
DHA Blocks Lipogenesis and Stimulates FA Oxidation and TAG Export Through Sirt1 in HepG2 Cells
In cultured HepG2 cells incubated with PA (300 μM) for 24 h, we observed obvious lipid accumulation in cells. DHA treatment elicited a significant diminution of lipid drops, which was abrogated by sirtinol, an inhibitor of Sirt1 (Figure 4A, the left panel). We then measured the TAG levels in cells and found a consistent result with morphologic changes (Figure 4A, the right panel). The results from qPCR showed that mRNA levels of lipogenic gene Fas was downregulated in DHA-treated cells even those expression were comparable between PA only-treated cells and control cells (Figure 4B). In contrast, the expression of Pgc1α and Mttp were hampered in PA-treated cells. DHA significantly increased their mRNA levels and Sirt1 inhibition almost abolished these effects (Figure 4B).
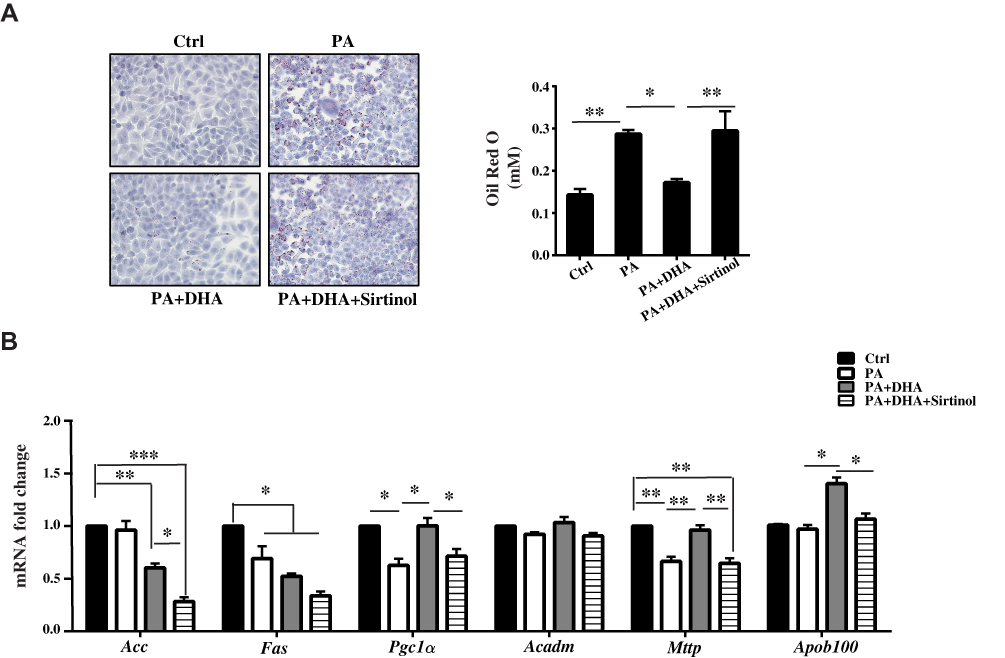 |
Figure 4 DHA decreases lipogenesis and stimulates FA oxidation through Sirt1 in HepG2 cells. (A) Micrographs showing Oil Red O staining (magnification 200X) and quantification of Oil Red O staining. (B) qPCR analysis of gene expression in liver of mice. All data are expressed as fold change compared with Ctrl cells. Values are mean±S.E.M. (standard error of the mean) from three independent experiments and expression of genes was corrected for β-actin (*p < 0.05; **p < 0.01; ***p < 0.001 versus Ctrl cells). |
DHA Inhibits NF-κB Activation Through Deacetylation of P65 Mediated by Sirt1
To further understand the molecular mechanism of DHA in the control of inflammation, we determined the expression level and activation status of p65 NF-κB under DHA treatment. As shown in Figure 5A, DHA treatment resulted in an increased expression of Sirt1 in PA-treated cells. Because Sirt1 is well known to deacetylate p65 specifically at lysine 310, we examined the roles of DHA and Sirt1 in the regulation of acetylated p65 NF-κB. Notably, the levels of acetylated p65 NF-κB at lysine 310 were predominantly decreased in DHA-treated cells and Sirt1 inhibition by sirtinol increased acetylated p65 protein level. However, the total p65 was not considerably altered among all treatments. We also found that phosphorylation of p65 NF-κB (S468) was increased in Sirt1 inhibition cells but comparable among other three groups of cells.
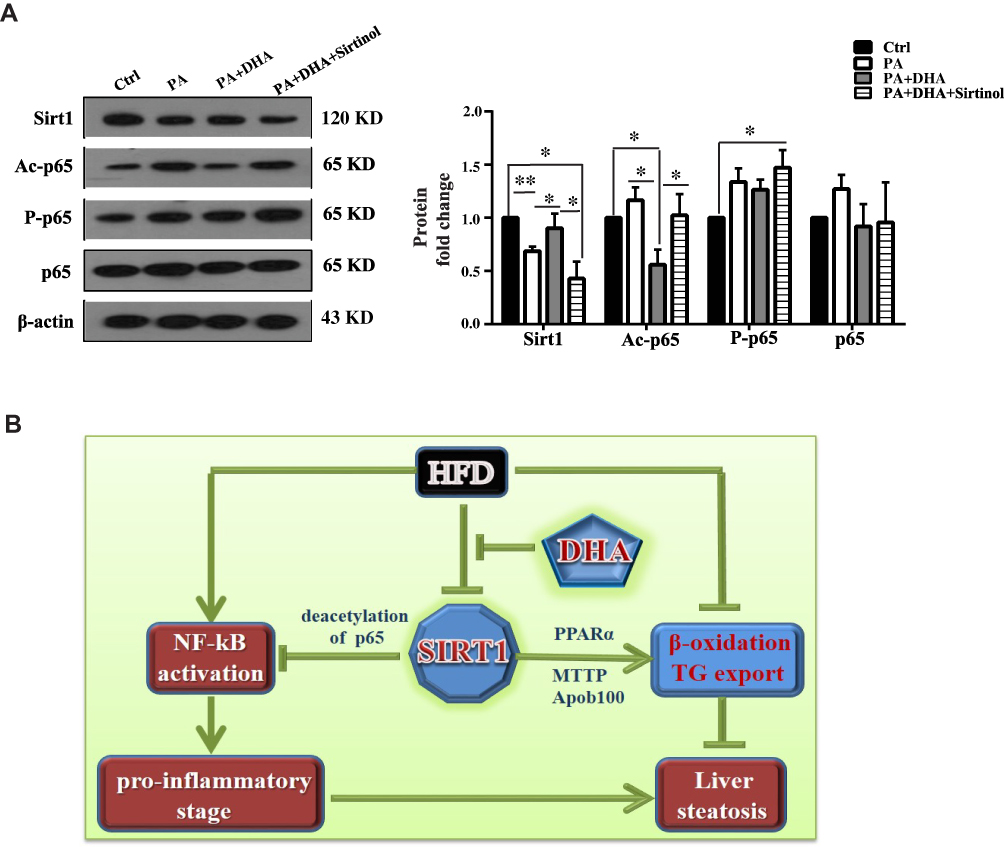 |
Figure 5 DHA inhibits NF-κB activation through deacetylation of p65 mediated by Sirt1. (A) A representative set of data from three independent experiments is illustrated (left panel) and quantification histograms (right panel) of Sirt1, Ac-p65, P-p65 and p65 expressions in HepG2 cells. Values are mean±S.E.M. (n = 3, *p < 0.05; **p < 0.01 versus Ctrl cells). (B) Proposed model for DHA regulating hepatic steatosis in liver. DHA inhibits HFD-induced downregulation of Sirt1. On the one hand, accumulation of Sirt1 inhibits NF-kB activation through deacetylating p65. Inactivated NF-kB reduces pro-inflammation, leading to ameliorated hepatic steatosis. On the other hand, increased Sirt1 promotes β-oxidation and TAG export resulting in decreased hepatic steatosis. |
Discussion
Long term of HFD exposure causes lipids accumulation in the liver, a process which leads to fatty liver disease known as NAFLD and finally to NASH. In the present study, we have demonstrated that DHA significantly decreased dyslipidemia and ameliorated hepatic steatosis, which was supported by a complex regulation of mitochondrial β-oxidation and TAG export in the liver; DHA markedly prevented liver inflammation in middle-aged mice with NAFLD supplemented with DHA. In HepG2 cells, DHA blocked lipogenesis but stimulated FA oxidation and TAG export. In addition, the anti-inflammatory effect of DHA is related to the deacetylation of p65 NF-κB. Importantly, these effects of DHA on NAFLD were mediated by Sirt1.
At present, three types of diets are commonly used to induce NAFLD: methionine and choline deficiency diet (MCD), choline deficiency diet (CD) and high-fat diet (HFD). Although rodents fed MCD will develop significant fatty liver in 2–4 weeks and then quickly evolve into hepatitis and fibrosis, they often lose weight and do not develop insulin resistance. This is in contrast to typical human NAFLD/NASH patients. On contrary, HFD can induce weight gain in rodent models and lead to insulin resistance. Hence, in this study, we chose HFD to establish NAFLD mouse model and would like to explore the effect of DHA on obesity-related NAFLD. Generally, TAG accumulation in hepatic steatosis caused by HFD mainly results from three pathways, including increased de novo FA synthesis, decreased fatty acid β-oxidation and impaired TAG β-oxidation from hepatocytes.15 FAS is one of the key enzymes that regulate fat synthesis. Some studies have showed that HFD up-regulated the expression or activity of FAS.16,17 In our study, we observed that a HFD for 20 weeks led to an obvious lipid accumulation in liver, but the hepatic Fas was down-regulated and even DHA supplementation had no effect on its expression, which was in line with our in vitro data (Figure 4A and B). This discrepancy might be attributed to compensatory response of hepG2 cells. Furthermore, these animals already had disorders of blood glucose metabolism and their liver FAS activity may be reduced.13 This was in agreement with a previous study in which hepatic FAS activity in diabetic mice fed a HFD was significantly reduced.18 How did HFD cause hepatic steatosis in our animal model? From the results of related gene expression, we found HFD reduced the expression of mitochondrial β-oxidation gene Pgc1α and TAG export gene Mttp. It seems like that in our specific model, reduced mitochondrial β-oxidation and TAG export rather than increased lipogenesis contributed to lipid accumulation finally.
Our data revealed that DHA supplementation to NAFLD mice effectively reduced hepatic lipid accumulation and concomitantly reduced liver weights and serum TAG levels. We found that although DHA supplementation had no effect on lipogenic gene expression, it was able to completely eliminate hepatic steatosis in HFD-fed mice. The possible reason is that this effect was accompanied by a significant induction of Mttp as well as Pgc1α, an important transcriptional factor that activates FA oxidation.19 Consistently, DHA was reported to upregulate mRNA levels of Pgc1α and stimulate PGC1α promoter activity in a dose- and time-dependent manner in C2C12 muscle cells.20 These results suggest that the anti-hepatosteatotic effect of DHA in our NAFLD model is partly mediated by the modulation of mitochondrial biogenesis and promotion of TAG export but not the reduction of hepatic lipogenesis. The protective effect of DHA on hepatic steatosis in vivo was also supported by in vitro data. Considering that DHA improved insulin sensitivity in these middle-aged obese mice as we previously described, we infer that the antisteatotic effects of DHA may contribute to augment its insulin-sensitizing effect in our animal model.
Emerging researches indicated that Sirt1 had a vital role in the regulation of lipid metabolism. Hepatocyte-specific deletion of Sirt1 results in hepatic steatosis and inflammation.21 However, both transgenic Sirt1 mice and over-expression of Sirt1 specifically in the liver show lower hepatic steatosis along with better glucose tolerance, which may represent a new strategy to counteract NAFLD and related diseases such as type 2 diabetes.22,23 Our previous study also revealed that DHA attenuates insulin resistance in HFD-induced obese mice via the activation of Sirt1.13 In the present study, DHA treatment increased the expression of Sirt1 in liver and systemic Sirt1 knockdown abrogated almost all the benefits of DHA on NAFLD. Basically, the explored mechanisms of Sirt1 counteracting hepatic steatosis is that Sirt1 upregulates the expression of genes that control β-oxidation and downregulates lipogenesis.24,25 In this study, Sirt1 knockdown had no effect on Fas gene expression. In the contrast, Sirt1 mediated other two pathways involving steatosis regulated by DHA treatment in vivo. Firstly, the dysfunction of liver mitochondria is very important in the “first and second hit” leading to NAFLD and PGC1α is regarded as a key and positive protein in regulating mitochondrial biogenesis.26 Sirt1 has been shown to deacetylate PGC1α and positively regulate it.27 In the present study, we found that Sirt1 knockdown reduced the mRNA expression of Pgc1α and the protective role of DHA in ameliorating hepatic steatosis was almost blocked. Secondly, the induction of TAG export-related genes by DHA was inhibited in Sirt1 knockdown group. This is consistent with a previous study in which lack of Sirt1 activity leads to reduced fat export in the liver of mice and causes steatosis.28 Hence, we supposed that the increased mitochondrial biogenesis by deacetylation of Sirt1 to PGC1α and promotion of TAG export are important molecular mechanisms of DHA to improve NAFLD.
Another common feature of NAFLD is inflammation.29 In agreement with the well-known anti-inflammation propriety of DHA mediated by Sirt1,30,31 here we demonstrated that the steatosis-induced inflammatory milieu in HFD-fed mice were all reversed by DHA treatment. Systemic Sirt1 knockdown can partially abrogate these effects. Inflammation is tightly controlled by NF-κB. Both deacetylation and phosphorylation of p65 NF-κB are known to influence its ability to bind target DNA sequences and then gene expression.32 Furthermore, an antagonistic crosstalk between NF-κB and Sirt1 is present in the regulation of inflammation.33 Sirt1 inhibits NF-κB signaling directly by deacetylating the p65 subunit of NF-κB complex, specifically at lysine 310.34 Consistently, we found that the acetylation of p65 NF-κB at lysine 310 was markedly decreased in DHA-treated cells and Sirt1 inhibition by sirtinol. Similar to our findings, resveratrol, a Sirt1 activator, was reported to upregulate the gene expression of Sirt1 and downregulate acetylated p65 NF-κB (lys 310) in BJAB B cells.35 However, the phosphorylation of p65 NF-κB (S468) was all comparably increased compared to control cells. A previous study indicated that the phosphorylation of p65 NF-κB on serine 536 was able to regulate the acetylation of lysine 310,36 but whether the acetylation of lysine 310 could conversely directly affect the phosphorylation of p65 NF-κB on serine 468 is not detected in our study. Nevertheless, these results suggest that the deacetylation of p65 NF-κB mediated by Sirt1 but not the phosphorylation of p65 NF-κB is associated with the inactivation of NF-κB by DHA, which contributes to DHA’s protective role in ameliorating NAFLD. These data point to a participation of this pathway in the protective role of DHA in the liver. The molecular interaction among the possible metabolic pathways involved in the inhibition of metabolic disorders and liver damage induced by a HFD in mice is depicted in Figure 5B.
Conclusion
In conclusion, this study demonstrates that DHA might be a potential dietary therapeutic tool against NAFLD and reveals a previously unrecognized role of Sirt1 in this process, which indicating Sirt1 may be a key molecular target by which DHA improves NAFLD.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. 81874263 and 81871190); the Fundamental Research Funds for the Central Universities (no.xjj2017186); and the Natural Science Foundation of Shaanxi Province (no.2018JQ8007). The abstract of this paper was presented at the Nutrition 2018 Conference as a poster presentation with interim findings. (https://www.eventscribe.com/2018/Nutrition2018/ajaxcalls/PosterInfo.asp?PosterID=146696&efp=UlhTRFpZVVI0ODYw&rnd=0.9920195).
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
1. Brewis A, SturtzSreetharan C, Wutich A. Obesity stigma as a globalizing health challenge. Global Health. 2018;14:20. doi:10.1186/s12992-018-0337-x
2. Yki-Jarvinen H. Diagnosis of non-alcoholic fatty liver disease (NAFLD). Diabetologia. 2016;59:1104–1111. doi:10.1007/s00125-016-3944-1
3. Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–264.
4. Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des. 2010;16:1941–1951. doi:10.2174/138161210791208875
5. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11–20. doi:10.1038/nrgastro.2017.109
6. Scalera A, Tarantino G. Could metabolic syndrome lead to hepatocarcinoma via non-alcoholic fatty liver disease? World J Gastroenterol. 2014;20:9217–9228. doi:10.3748/wjg.v20.i28.9217
7. Scorletti E, Byrne CD. Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu Rev Nutr. 2013;33:231–248. doi:10.1146/annurev-nutr-071812-161230
8. Shapiro H, Tehilla M, Attal-Singer J, Bruck R, Luzzatti R, Singer P. The therapeutic potential of long-chain omega-3 fatty acids in nonalcoholic fatty liver disease. Clin Nutr. 2011;30:6–19. doi:10.1016/j.clnu.2010.06.001
9. Jump DB, Depner CM, Tripathy S, Lytle KA. Potential for dietary omega-3 fatty acids to prevent nonalcoholic fatty liver disease and reduce the risk of primary liver cancer. Adv Nutr. 2015;6:694–702. doi:10.3945/an.115.009423
10. Delarue J, Lalles JP. Nonalcoholic fatty liver disease: roles of the gut and the liver and metabolic modulation by some dietary factors and especially long-chain n-3 PUFA. Mol Nutr Food Res. 2016;60:147–159. doi:10.1002/mnfr.201500346
11. Colak Y, Ozturk O, Senates E, et al. SIRT1 as a potential therapeutic target for treatment of nonalcoholic fatty liver disease. Med Sci Monit. 2011;17:HY5–9. doi:10.12659/MSM.881749
12. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi:10.1038/nature03354
13. Luo X, Jia R, Yao Q, Xu Y, Luo Z, Wang N. Docosahexaenoic acid attenuates adipose tissue angiogenesis and insulin resistance in high fat diet-fed middle-aged mice via a sirt1-dependent mechanism. Mol Nutr Food Res. 2016;60:871–885. doi:10.1002/mnfr.201500714
14. Luo X, Yang Y, Shen T, et al. Docosahexaenoic acid ameliorates palmitate-induced lipid accumulation and inflammation through repressing NLRC4 inflammasome activation in HepG2 cells. Nutr Metab (Lond). 2012;9:34. doi:10.1186/1743-7075-9-34
15. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–838. doi:10.1172/JCI34275
16. Rosado CP, Rosa VHC, Martins BC, et al. Resistant starch from green banana (Musa sp.) attenuates non-alcoholic fat liver accumulation and increases short-chain fatty acids production in high-fat diet-induced obesity in mice. Int J Biol Macromol. 2019. doi:10.1016/j.ijbiomac.2019.09.199
17. Miller CN, Morton HP, Cooney PT, et al. Acute exposure to high-fat diets increases hepatic expression of genes related to cell repair and remodeling in female rats. Nutr Res. 2014;34:85–93. doi:10.1016/j.nutres.2013.10.010
18. Choi RY, Ham JR, Lee MK. Esculetin prevents non-alcoholic fatty liver in diabetic mice fed high-fat diet. Chem Biol Interact. 2016;260:13–21. doi:10.1016/j.cbi.2016.10.013
19. Bagattin A, Hugendubler L, Mueller E. Transcriptional coactivator PGC-1alpha promotes peroxisomal remodeling and biogenesis. Proc Natl Acad Sci U S A. 2010;107:20376–20381. doi:10.1073/pnas.1009176107
20. Lee MS, Shin Y, Moon S, Kim S, Kim Y. Effects of eicosapentaenoic acid and docosahexaenoic acid on mitochondrial DNA replication and PGC-1alpha gene expression in C2C12 muscle cells. Prev Nutr Food Sci. 2016;21:317–322. doi:10.3746/pnf.2016.21.4.317
21. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–338. doi:10.1016/j.cmet.2009.02.006
22. Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105:9793–9798. doi:10.1073/pnas.0802917105
23. Vila L, Elias I, Roca C, et al. AAV8-mediated Sirt1 gene transfer to the liver prevents high carbohydrate diet-induced nonalcoholic fatty liver disease. Mol Ther Meth Clin Dev. 2014;1:14039. doi:10.1038/mtm.2014.39
24. Feige JN, Lagouge M, Canto C, et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi:10.1016/j.cmet.2008.08.017
25. Yamazaki Y, Usui I, Kanatani Y, et al. Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am J Physiol Endocrinol Metab. 2009;297:E1179–86. doi:10.1152/ajpendo.90997.2008
26. Pessayre D. Role of mitochondria in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2007;22 Suppl 1:S20–7. doi:10.1111/j.1440-1746.2006.04640.x
27. Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem. 2005;280:16456–16460. doi:10.1074/jbc.M501485200
28. Xu F, Gao Z, Zhang J, et al. Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1± mice: a role of lipid mobilization and inflammation. Endocrinology. 2010;151:2504–2514. doi:10.1210/en.2009-1013
29. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci. 2016;61:1294–1303. doi:10.1007/s10620-016-4049-x
30. Song NY, Na HK, Baek JH, Surh YJ. Docosahexaenoic acid inhibits insulin-induced activation of sterol regulatory-element binding protein 1 and cyclooxygenase-2 expression through upregulation of SIRT1 in human colon epithelial cells. Biochem Pharmacol. 2014;92:142–148. doi:10.1016/j.bcp.2014.08.030
31. Inoue T, Tanaka M, Masuda S, et al. Omega-3 polyunsaturated fatty acids suppress the inflammatory responses of lipopolysaccharide-stimulated mouse microglia by activating SIRT1 pathways. Biochim Biophys Acta. 2017;1862:552–560. doi:10.1016/j.bbalip.2017.02.010
32. Zhu X, Liu Q, Wang M, et al. Activation of Sirt1 by resveratrol inhibits TNF-alpha induced inflammation in fibroblasts. PLoS One. 2011;6:e27081. doi:10.1371/journal.pone.0027081
33. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25:1939–1948. doi:10.1016/j.cellsig.2013.06.007
34. Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi:10.1038/sj.emboj.7600244
35. Jhou JP, Chen SJ, Huang HY, Lin WW, Huang DY, Tzeng SJ. Upregulation of FcgammaRIIB by resveratrol via NF-kappaB activation reduces B-cell numbers and ameliorates lupus. Exp Mol Med. 2017;49:e381. doi:10.1038/emm.2017.144
36. Chen LF, Williams SA, Mu Y, et al. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol. 2005;25:7966–7975. doi:10.1128/MCB.25.18.7966-7975.2005
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.