")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Dexmedetomidine reduces the neuronal apoptosis related to cardiopulmonary bypass by inhibiting activation of the JAK2–STAT3 pathway
Authors Chen Y, Zhang X, Zhang B, He G, Zhou L, Xie Y
Received 28 April 2017
Accepted for publication 21 August 2017
Published 26 September 2017 Volume 2017:11 Pages 2787—2799
DOI https://doi.org/10.2147/DDDT.S140644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Yanhua Chen,1,* Xu Zhang,2,* Bingdong Zhang,1 Guodong He,2 Lifang Zhou,2 Yubo Xie2
1Department of Anesthesiology, Cardiovascular Institute, 2Department of Anesthesiology, the First Affiliated Hospital of Guangxi Medical University, Nanning, China
*These authors contributed equally to this work
Abstract: Cardiopulmonary bypass (CPB) constitutes one of the primary methodologies pertaining to cardiac surgery. However, this form of surgery can cause damage to the body. Many studies have reported that dexmedetomidine confers cerebral protection. In this study, we aimed to investigate the effect and mechanism of dexmedetomidine on neuronal apoptosis caused by CPB. Here, rats were treated with different doses of dexmedetomidine by intravenous infusion 2 hours after CPB. We observed that dexmedetomidine treatment to rats reduces the S100ß, NSE levels in plasma, and neuronal apoptosis following CPB in a dose-dependent manner. Furthermore, we observed that the beneficial effect of dexmedetomidine treatment following CPB was associated with a reduction in IL6, an inflammatory cytokine in plasma and cortex. Our results suggest that dexmedetomidine provides neuroprotective effects by inhibiting inflammation and reducing neuronal apoptosis. There was a correlation between the protective effect on the brain and the dose of dexmedetomidine. In addition, dexmedetomidine administration inhibits phosphorylation of JAK2 and STAT3 proteins in the hippocampus of rats 2 hours after CPB. Therefore, we speculate that the JAK2–STAT3 pathway plays an important role in the neuroprotective effects of dexmedetomidine following brain injury induced by CPB.
Keywords: apoptosis, cardiopulmonary bypass, dexmedetomidine, neuroprotective effect, JAK2, STAT3
Introduction
Cardiopulmonary bypass (CPB) is the predominant technique associated with cardiovascular surgery, and this form of surgery is known to improve the curative rate pertaining to numerous cardiac diseases. However, CPB can induce an abnormal physiological status, which can cause damage to the body.1 Following blood contact with the inner surfaces of the CPB apparatus, the complement system is activated and in turn stimulates release of proinflammatory cytokines, thereby influencing inflammatory response.2 Consequently, nervous system complications have become a concern after CPB.3 Many studies have been performed to elucidate the mechanisms that mediate the occurrence of brain injury following CPB,4,5 and an increasing number of treatments are now used to prevent brain injury caused by CPB.6 Several studies have shown that CPB-mediated brain injury is associated with embolism, hypoperfusion, arrhythmias, rapid rewarming, and inflammation.3,4,7 Moreover, a substantial amount of research has been conducted into brain injury associated with CPB in relation to different cell factors and signal pathways.5,8
Dexmedetomidine is an α2-adrenergic receptor agonist, and is known to exhibit sedative, analgesic, and anti-sympathetic nerve activities. It has been widely used in clinical anesthesia and sedation in intensive care units. Dexmedetomidine can significantly reduce cerebral injury induced by transient global cerebral ischemia–reperfusion in diabetic rats, mainly through altering the oxidative stress markers and inflammatory cytokines.9 Recent studies have shown that dexmedetomidine exhibits renoprotective effects following renal ischemia–reperfusion injury by inhibiting JAK–STAT signaling pathway. Investigators have also found similar renoprotective effects upon AG490 treatment, an inhibitor of JAK2.10,11
The JAK–STAT signaling pathway is not only involved in renal protection but also involved in brain protection.12 AG490 reduces brain injury and inflammatory responses in rats following heatstroke by inhibiting the activation of the JAK2–STAT3 pathway.13 The STAT3 signaling pathway is activated following traumatic brain injury, and may be involved in the recovery of neurological function.14,15 We investigated the effect of different doses of dexmedetomidine on cerebral injury related to CPB. Furthermore, the study aimed to characterize whether JAK2–STAT3 signaling was involved in dexmedetomidine-induced neuroprotection following CPB.
Materials and methods
Animals and grouping
A total of 96 adult male Sprague Dawley rats weighing 300–350 g were provided by the Guangxi Medical University Laboratory Animal Center. Animal experiments and protocols were approved by the medical ethics committee of the First Affiliated Hospital of Guangxi Medical University (2016[KY-E-002]).
Rats were randomly divided into six experimental groups of 16 rats in each group. In group 1, rats did not undergo surgical operation for CPB, and hence served as sham controls (group Sham). In group 2, all 16 rats underwent surgery to develop CPB as a model system. Rats in group 2 did not receive any treatment, and hence served as CPB control (group CPB). Rats in groups 3 and 4 received 1 μg/mL dexmedetomidine (Jiangsu Hengrui Medicine, Lianyungang, China), which was diluted with 0.9% saline. In addition, rats in groups 3 and 4 received loading doses of dexmedetomidine 2.5 μg/kg (low dose [group L]) and 5 μg/kg (high dose [group H]) intravenously using a microinfusion pump 15 minutes before CPB. Then, maintenance doses of dexmedetomidine – 2.5 μg/kg/h (group L) or 5 μg/kg/h (group H) – were administered intravenously during the CPB procedure after the loading the dose. In group 5, rats received AG490 (Selleck Chemicals, Dallas, TX, USA), a JAK2 inhibitor, at a dose of 10 mg/kg intraperitoneally, and served as the AG490 group (10 mg AG490 diluted with 0.6796 mL dimethyl sulfoxide [DMSO] and then diluted with physiological saline).11 In group 6, rats received DMSO at a dose of 0.6796 mL/kg, intraperitoneally (0.6796 mL DMSO in physiological saline as vehicle dose), and served as the DMSO group.
CPB-model preparation
Rats were anesthetized with pentobarbital sodium (65 mg/kg intraperitoneally) and mechanically ventilated to maintain end-tidal carbon dioxide between 35 and 45 mmHg. The protocol followed for the development of this CPB model has been described previously.16 To sustain mean arterial pressure of 70–85 mmHg and hematocrit of approximately 20%−25%, each rat was covered with a heat lamp to maintain the body central temperature at 36.5°C±1°C throughout the experiment. The CPB procedure continued for 2 hours.
Specimen collection and processing
Venous blood samples (n=8) were collected prior to CPB (t0), 1 hour after CPB (t1), and 2 hours after CPB (t2). Blood samples were centrifuged at 1,000 g for 15 minutes. Plasma supernatants were harvested and stored at −20°C for enzyme-linked immunosorbent assay (ELISA).
Rats were killed and fresh brain-tissue samples collected 2 hours after CPB (n=8). Hippocampal tissue samples were harvested to determine JAK2, pJAK2, STAT3, pSTAT3, and cleaved caspase-3 protein levels. Tissue samples were frozen and stored at −80°C prior to Western blot analysis. Parietal cortices were weighed according to the proportion of 100 mg hippocampal tissue homogenized in 1 mL PBS, and stored overnight at −20°C. After two freeze–thaw cycles had been performed to break the cell membranes, homogenates were centrifuged for 5 minutes at 5,000 g, 2°C−8°C. The supernatant was collected and stored at −20°C for analysis of IL6 and IL10 using ELISA. The remaining fresh brain tissue was used for determination of brain water content.
Other rats (n=7 for groups CPB and DMSO, n=8 for other groups) were subjected to cerebral perfusion for TUNEL assay and immunohistochemistry. Transcardial perfusions were performed after blocking the abdominal aorta (100 mL 0.9% saline, followed by 200 mL refrigerated 4% paraformaldehyde solution). Brains were removed, cut open along the coronal incision at 4 mm after the optic chiasma, and 2 mm-thick hippocampal tissue was excised and stored in refrigerated paraformaldehyde (<24 hours). Specimens were then dehydrated by being passed through graded series of ethanol, cleared in xylene, and embedded in paraffin wax. The paraffin-embedded hippocampal tissues were cut into 4 μm sections.
Enzyme-linked immunosorbent assay
All biomarkers were analyzed using commercially available ELISA kits. Plasma samples were analyzed for IL6 (Cusabio Biotech, Wuhan, China), IL10 (Thermo Fisher Scientific, Waltham, MA, USA), S100β (Cusabio Biotech), and NSE (Cusabio Biotech). Cortex samples were analyzed using ELISA according to the ELISA-kit manufacturer’s instructions for IL6 and IL10.
Western blot analysis for JAK2, pJAK2, STAT3, pSTAT3, and cleaved caspase 3
For Western blot analysis, proteins from different groups were extracted and protein concentration quantified. Then, samples were used for routine Western blot analysis; the detailed protocol has been described previously.17 Antibodies were replaced by the primary antibodies JAK2 (1:1,000, AB_2128522; Cell Signaling Technology, Danvers, MA, USA), phospho-JAK2 (phospho-Y1007 + Y1008, 1:1,000, AB_775808; Abcam, Cambridge, UK), STAT3 (1:1,000, AB_331269; Cell Signaling Technology), phospho-STAT3 (Tyr705, 1:1,000, AB_1658549; Abcam), cleaved caspase 3 (1:1,000, AB_2070042; Cell Signaling Technology), and β-actin (1:3,000; Bioworld Technology, Minneapolis, MN, USA). The secondary antibody was horseradish peroxidase conjugated to goat antirabbit/mouse IgG (1:10,000; Cell Signaling Technology). The membranes were developed using an Odyssey two-color infrared scanner (LI-Cor Biosciences, Lincoln, NE, USA).
Brain water content
Fresh brain tissue was weighed promptly after removal of the parietal cortex and hippocampus. Brain water content was measured using the standard wet/dry weight method.18
Detection of apoptosis of hippocampal neurons and cortex by TUNEL assay
Apoptosis of hippocampal neurons in the CA1 region and the parietal cortex adjacent to the CA1 region was monitored using a TUNEL-assay kit according to the manufacturer’s instructions and finally observed under microscopy. All TUNEL-positive cells were counted, and numbers of apoptotic cells were calculated for each area.9
Immunohistochemistry
The immunohistochemistry protocol followed in this study was as previously described.19,20 After being blocked in 3% H2O2 and 3% normal goat serum, tissue sections were incubated with anti-pJAK2 antibody (dilution 1:200) or anti-pSTAT3 antibody (dilution 1:100) rabbit monoclonal antibodies in PBS overnight.
Statistical analysis
Data are presented as means ± SEM. All statistical analyses were performed using SPSS 16.0 (SPSS Inc, Chicago, IL, USA). Multiple comparisons were performed with one-way analysis of variance, followed by Tukey’s post hoc test. P-values <0.05 were considered significant.
Results
A total of 96 rats were used. Two rats (one in group CPB and one in group DMSO) were excluded, due to survival failure. The remaining 94 rats were used for analysis of various parameters. Venous blood samples and cortex samples were collected for ELISA (n=8). Hippocampal tissue samples were harvested for Western blot analysis (n=8). The remaining fresh brain-tissue samples were used for determination of brain water content (n=8). Other rats (n=7 for group CPB and group DMSO, n=8 for other groups) were subjected to cerebral perfusion for TUNEL assay and immunohistochemistry.
Dexmedetomidine reduced brain water content in rats following CPB
The brain water content in group CPB was significantly increased compared with group Sham (P<0.05). The brain water content in group AG490, group L, and group H was significantly decreased compared with group CPB (P<0.05). However, there was no significant difference between group L and group H in brain water content (P>0.05, Figure 1). Not surprisingly, DMSO-administered rats did not shown significant decreases in brain water level, which indicates the vehicle dose of DMSO used in this study did not play any pharmacological role.
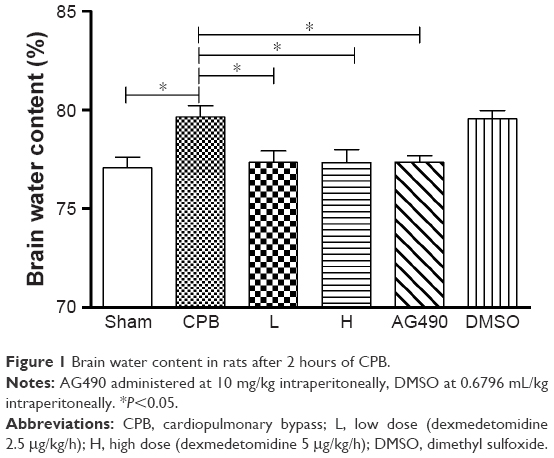 | Figure 1 Brain water content in rats after 2 hours of CPB. |
Dexmedetomidine decreased S100β and NSE plasma levels in CPB rat model
S100β and NSE are major biomarkers of brain injury and mainly used in brain trauma, cerebral stroke and hypoxic ischemia encephalopathy. We measured levels of these markers by ELISA. Remarkably, ELISA indicated levels of S100β and NSE in plasma were significantly increased in group CPB compared with group Sham at t1 and t2, respectively (P<0.05). Compared with group CPB, a decrease in S100β and NSE plasma levels was observed in group L at t2 (P<0.05). Compared with group CPB, a decrease in S100β and NSE plasma levels was also observed in group AG490 and group H at the t1 and t2 time points (P<0.05), whereas group L showed a significant effect at t2. S100β and NSE plasma levels in group H were significantly decreased compared with group L at t1 and t2 (P<0.05, Figure 2).
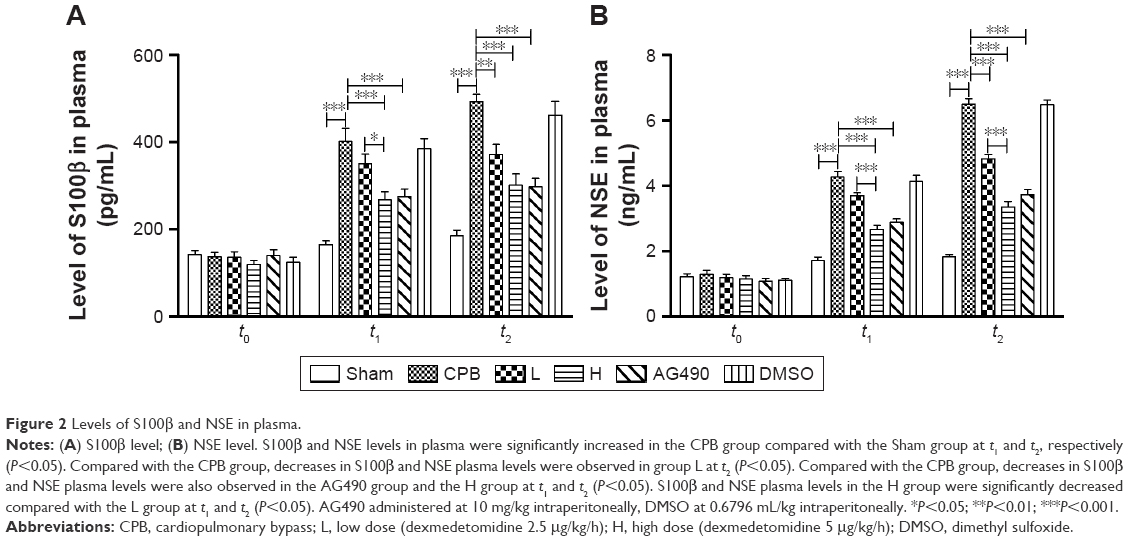 | Figure 2 Levels of S100β and NSE in plasma. |
Dexmedetomidine reduced apoptosis of hippocampus and cortex
To evaluate apoptosis of hippocampal neurons and cortex region induced by CPB, a TUNEL assay was performed. Interestingly, we observed marked TUNEL-positive cells indicated apoptosis of hippocampal neurons in the CA1 region of CPB-group rats, which was significantly comparable to that of group Sham (P<0.05). Apoptosis of hippocampal neurons in the CA1 region in group AG490, group L, and group H was significantly decreased compared with group CPB (P<0.05). Apoptosis of hippocampal neurons in the CA1 region in group H was significantly decreased compared with group L (P<0.05, Figure 3).
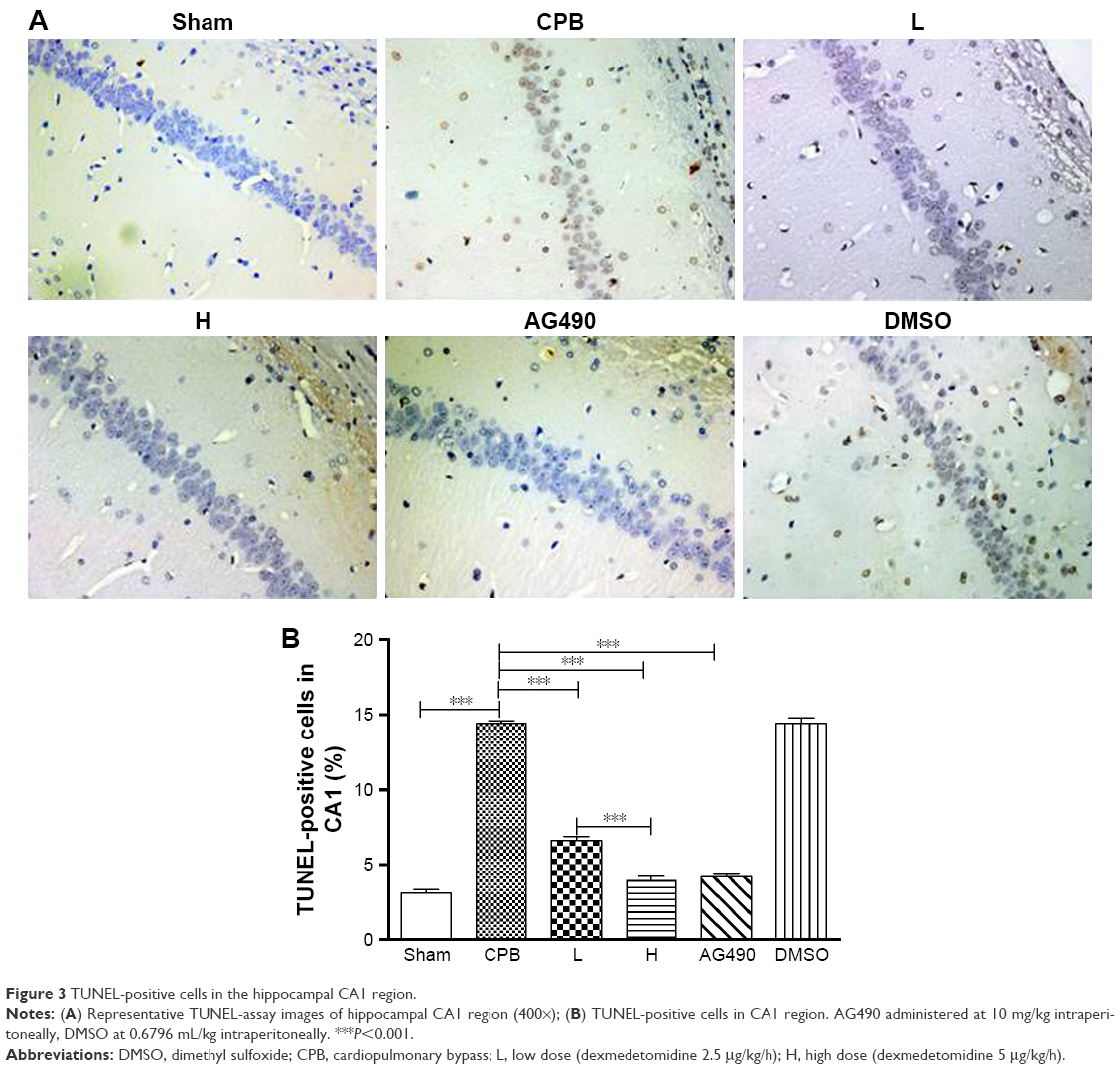 | Figure 3 TUNEL-positive cells in the hippocampal CA1 region. |
As expected, apoptosis of cortex in group CPB was significantly increased compared with group Sham (P<0.05). Apoptosis of cortex in group AG490, group L, and group H was significantly decreased compared with group CPB (P<0.05). Apoptosis of cortex in group H was significantly decreased compared with group L (P<0.05, Figure 4).
 | Figure 4 TUNEL-positive cells in cortex. |
Dexmedetomidine reduced IL6 levels in plasma and cortex
Levels of IL6 and IL10 in plasma were significantly increased in group CPB compared with group Sham at t1 and t2 (P<0.05). Compared with group CPB, decreased IL6 was observed in group AG490, group L, and group H at t2 (P<0.05). Compared with CPB group, no significant difference in IL10 was observed in group DMSO, group AG490, group L, or group H at t1 or t2 (P>0.05). There was no significant difference between group L and group H in plasma levels of IL6 or IL10 (P>0.05, Figure 5).
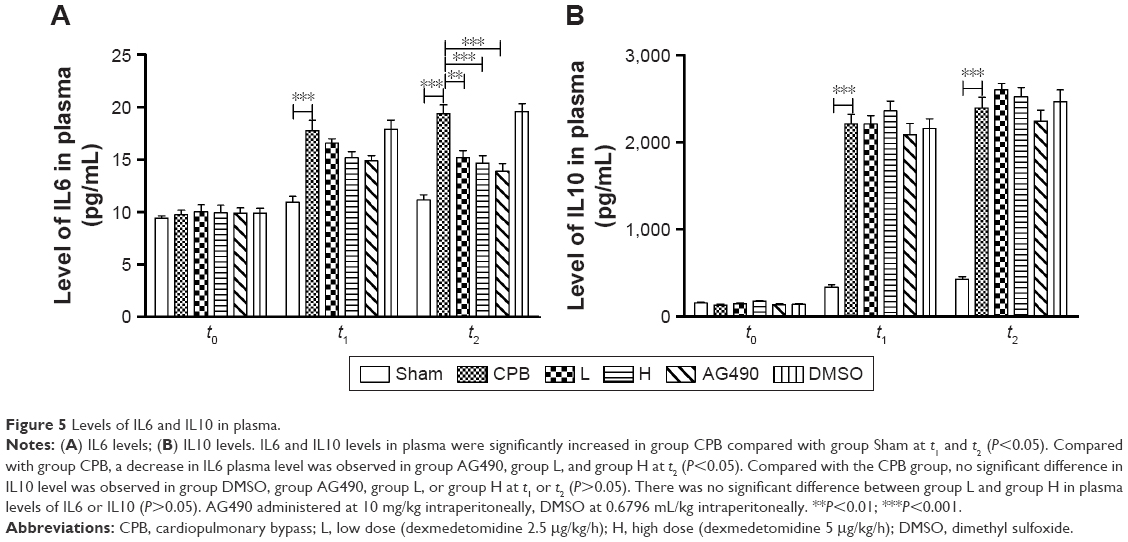 | Figure 5 Levels of IL6 and IL10 in plasma. |
Levels of IL6 and IL10 in cortex in group CPB were significantly increased compared with group Sham (P<0.05). Compared with group CPB, a decrease in cortex IL6 was observed in group AG490, group L, and group H (P<0.05). Compared with the CPB group, no significant difference in cortex IL10 was observed in group AG490, group L, or group H (P>0.05). There was no significant difference between group L and group H in IL6 or IL10 cortex levels (P>0.05, Figure 6).
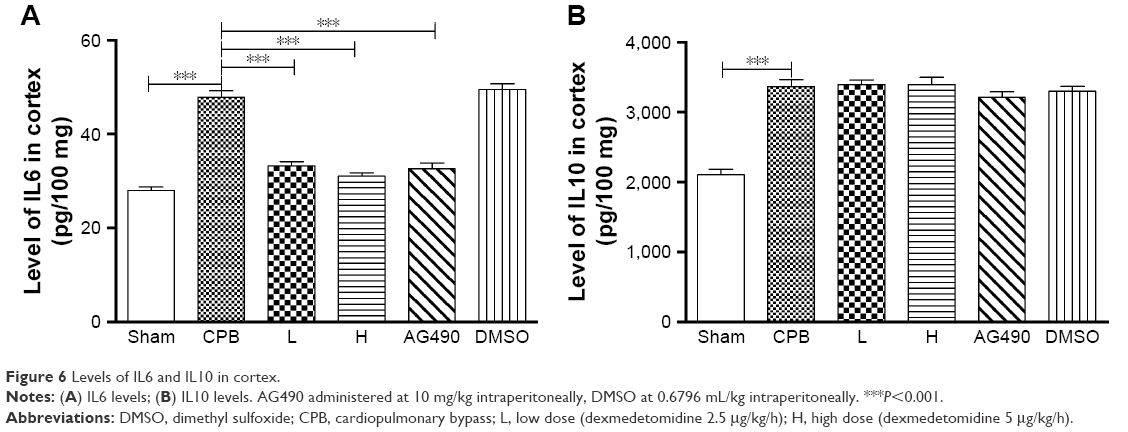 | Figure 6 Levels of IL6 and IL10 in cortex. |
Dexmedetomidine decreased expression of cleaved caspase-3 protein in hippocampus
To confirm TUNEL positivity for apoptotic cell death in hippocampal neurons and the cortex region, we assessed cleaved caspase-3 protein expression, which is the most important protein in intrinsic apoptotic signals. Western blot was performed, and showed significantly increased expression of cleaved caspase 3 in group CPB rat hippocampi when compared with group Sham (P<0.05). However, dramatically decreased expression of cleaved caspase 3 was seen in group AG490, group L, and group H compared with group CPB (P<0.05). These data revealed that dexmedetomidine can potentially protect cells from CPB-mediated apoptotic cell death. However, there was no significant difference between group L and group H in cleaved caspase-3 protein levels (P>0.05, Figure 7), which indicated a more pronounced effect of dexmedetomidine.
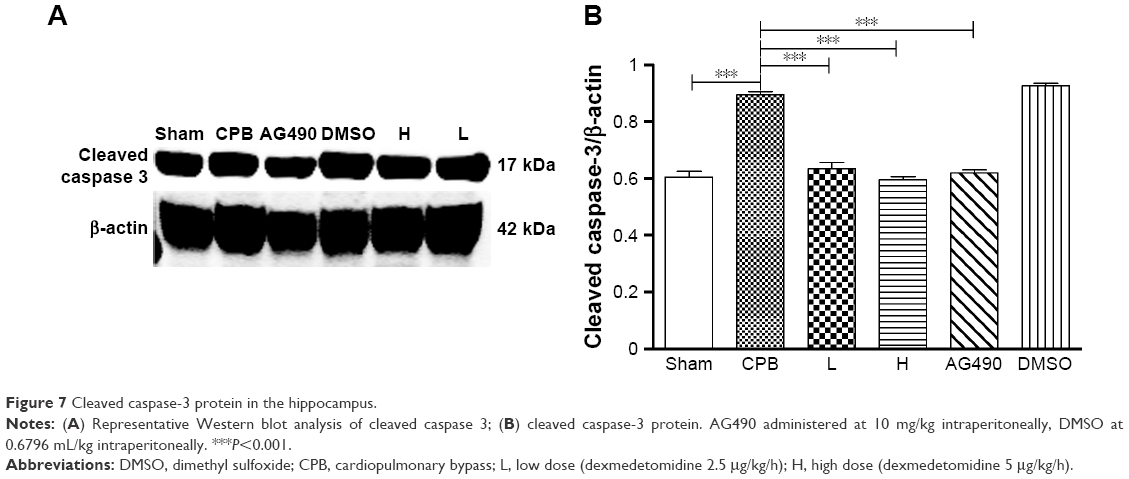 | Figure 7 Cleaved caspase-3 protein in the hippocampus. |
Dexmedetomidine decreased expression of pJAK2 and pSTAT3 proteins in hippocampus
Next, we investigated the underlying mechanisms of the neuroprotective effect of dexmedetomidine. Western blot analysis and immunohistochemistry data revealed that expression of pJAK2 and pSTAT3 proteins in group CPB was significantly increased compared with group Sham (P<0.05). Furthermore, pJAK2 and pSTAT3 expression in group AG490, group L, and group H was significantly decreased compared with group CPB (P<0.05), whereas there were no significant changes in total JAK2 or STAT3 proteins. However, there was no significant difference between group L and group H in the expression of pJAK2 or pSTAT3 protein (P>0.05, Figures 8–11). This significant inhibition of JAK2 and STAT3 phosphorylation by dexmedetomidine treatment shows promising anti-inflammatory effects.
 | Figure 8 JAK2 and pJAK2 protein levels in the hippocampus. |
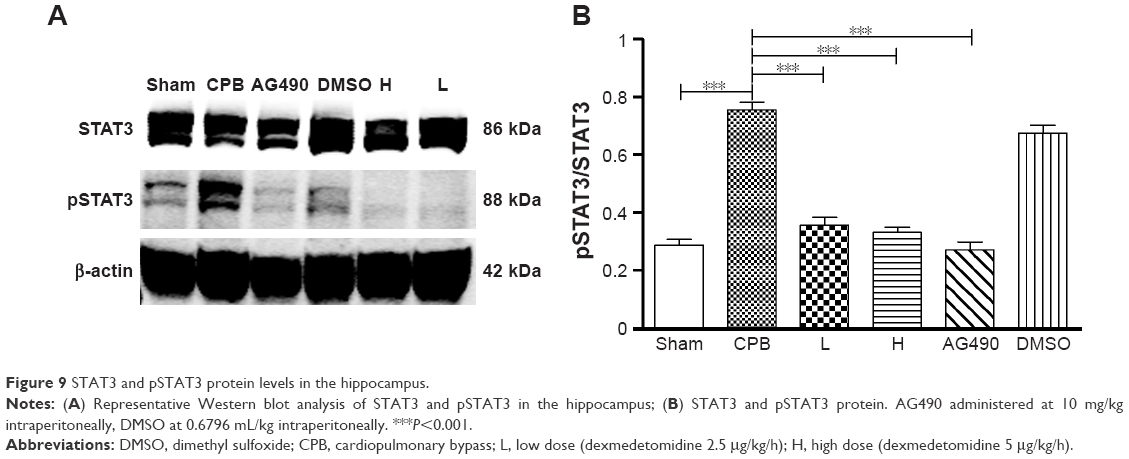 | Figure 9 STAT3 and pSTAT3 protein levels in the hippocampus. |
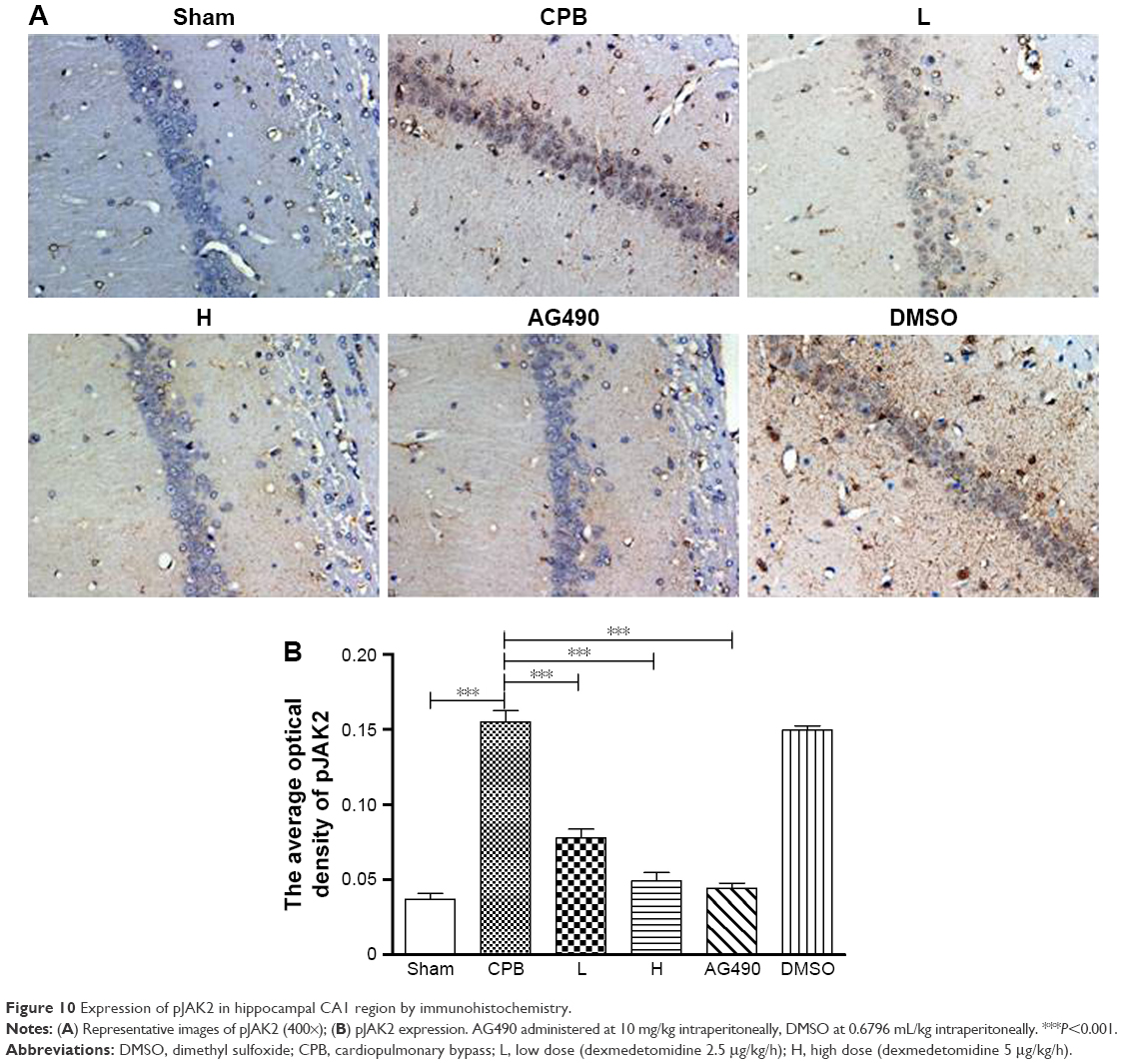 | Figure 10 Expression of pJAK2 in hippocampal CA1 region by immunohistochemistry. |
 | Figure 11 Expression of pSTAT3 in hippocampal CA1 region by immunohistochemistry. |
AG490 attenuated neuroapoptosis related to CPB and decreased pJAK2 and pSTAT3
To elucidate the role of JAK2–STAT3 signaling in dexmedetomidine-attenuated CPB-related neuroapoptosis further, we administered a JAK2 inhibitor (AG490), which resulted in a significant decrease in the apoptosis of hippocampus and cortex (Figures 3 and 4). pJAK2 and pSTAT3 protein expression decreased significantly in group AG490 compared with group CPB on both Western blot analysis and immunohistochemistry positivity (Figures 8–11). The effect of dexmedetomidine in CPB rats was similar to that of AG490. This observation suggested that the JAK2–STAT3 signaling pathway played a significant role in dexmedetomidine-mediated neuroprotection.
Dexmedetomidine attenuated brain injury related to CPB in a dose-dependent manner
To determine the protective effect of different doses of dexmedetomidine on brain injury in rats undergoing CPB, a maintenance dose of 2.5 μg/kg/h was administered in group L and a maintenance dose of 5 μg/kg/h administered in group H. Dexmedetomidine administration to CPB rats resulted in significant decreases in brain-injury biomarkers, such as S100β and NSE, in plasma levels at t1 and t2, and S100β and NSE plasma levels in group H were more pronounced when compared with group L at t1 and t2 (Figure 2). Meanwhile, dexmedetomidine significantly reduced apoptosis of hippocampal neurons and the cortex region, and apoptosis of hippocampus and cortex in group H was significantly decreased compared with group L (Figures 3 and 4). These results suggest that dexmedetomidine attenuated brain injury related to CPB in a dose-dependent manner.
Discussion
In this study, we investigated the neuroprotective effects of dexmedetomidine using a rat model of CPB. We observed that dexmedetomidine resulted in a significant decrease in S100β and NSE plasma levels at t1 and t2 and reduced neuronal apoptosis following CPB in a dose-dependent manner. Furthermore, we found that the beneficial effects of dexmedetomidine treatment were associated with reduced plasma and cortex levels of the inflammatory cytokine IL6. In addition, dexmedetomidine treatment resulted in a decrease in pJAK2 and pSTAT3 protein levels in the hippocampi of rats that underwent CPB for 2 hours. These effects of dexmedetomidine were similar to those of AG490.
According to clinical data from several medical institutions, the average duration of CPB, included valve, valve and coronary artery-bypass graft, coronary artery-bypass graft, and others (aortic root, ascending aneurysm), is close to 120 minutes.21,22 Extracorporeal circulation flow time that was employed in this study was 2 hours. Previous reports have shown that dexmedetomidine exhibits neuroprotective, myocardial-protective and renal-protective functions in animal experiments, clinical anesthesia, and intensive care units.23–26 The mechanisms that underpin these phenomena may be related to reductions in inflammation and oxidative stress markers.9 A previous report observed that treatment with 5 μg/kg/h dexmedetomidine can alleviate cerebral damage in diabetic rats,9 while treatment with 10 or 20 μg/kg/h dexmedetomidine has also been shown to reduce renal dysfunction.27 According to dose-conversion coefficients following different animal conversions,28 rat dosages of 2.5 μg/kg/h and 5 μg/kg/h dexmedetomidine are near clinical dosages of 0.3 μg/kg/h and 0.7 μg/kg/h, respectively, both which are commonly used in clinical procedures.29 However, single doses of 1–160 μg/kg have been employed for a variety of brain injuries in several animal experiments.30–32 We prefer continuous infusion with a micropump.
S100β and NSE are specific and sensitive biochemical markers for central nervous system damage. Elevations in these markers can be reflective of brain injury.33 In this study, S100β and NSE plasma levels were increased after 2 hours of CPB, while dexmedetomidine reduced plasma levels of S100β and NSE. At the same time, dexmedetomidine 5 μg/kg/h significantly decreased plasma levels of S100β and NSE compared with dexmedetomidine 2.5 μg/kg/h. These results revealed that dexmedetomidine attenuated brain injury related to CPB in a dose-dependent manner. Several reports have suggested that an increase in the level of cleaved caspase 3 results in extensive apoptosis.34,35 In this study, increased cleaved caspase-3 levels were observed in group CPB, while dexmedetomidine administration resulted in a decrease in cleaved caspase 3. These results are in agreement with the observation that dexmedetomidine reduces brain water content and apoptosis of neurons following CPB. Therefore, dexmedetomidine administration confers protection against brain injury induced by CPB.
Systemic inflammatory responses can be induced by CPB. This results in a concomitant increase in inflammatory cytokines, a phenomenon that can result in injury to various organs of the body.5,36,37 Several researchers have reported that IL6 and IL10 levels are increased after CPB, and the associated increase in cytokines activates the inflammatory response.36,38 Our results were in accordance with previous studies. In this study, dexmedetomidine administration resulted in a reduction in IL6 levels in plasma and cortex, while also inhibiting systemic inflammatory reactions that are normally associated with CPB. These results were also in accordance with a previous study that reported that dexmedetomidine reduced the inflammatory response to coronary artery-bypass graft surgery under mini-CPB.39
The protein that encodes for IL6 is a major proinflammatory cytokine involved in the inflammatory response associated with CPB.40 An increase in IL6 levels can activate intracellular signaling.41 de Jong et al demonstrated that STAT3 signaling plays a crucial role in the course of systemic inflammation caused by CPB.42 The JAK–STAT pathway is an important intracellular signal-transduction pathway that relays extracellular signals to nuclei. Activated STAT3 (pSTAT3) is involved in the transcription of genes that mediate inflammation, apoptosis, endothelial cell differentiation, and angiogenesis.43
Previous studies have reported that activation of the JAK–STAT signaling pathway may play an important role in organ protection and cell survival during ischemia–reperfusion injury.20,44 Kim et al45 demonstrated that sevoflurane postconditioning can reduce neuronal apoptosis by increasing pJAK2 and pSTAT3 expression after transient global ischemia in rats. Zhao et al15 showed that activation of the JAK2–STAT pathway may be involved in neurological function recovery after traumatic brain injury.
However, some studies have shown that inhibition of activation of the JAK2–STAT3 pathway has organ protection. Hristova et al46 demonstrated that inhibition of STAT3 phosphorylation can reduce neonatal hypoxic–ischemic brain injury. Si et al11 showed that administration of dexmedetomidine protects against renal ischemia and reperfusion injury by inhibiting JAK–STAT signaling activation. Jia et al47 demonstrated that propofol postconditioning attenuates hippocampus ischemia–reperfusion injury by reducing the expression of JAK2 and STAT3 in rats after autogenous orthotropic liver transplantation.
These different results may be related to the experimental model, experimental object, experimental conditions, and degree of injury. We hypothesize that when the body is injured, the body can play a protective role by activating the JAK2–STAT3 pathway. However, if the JAK2–STAT3 pathway is activated excessively after the body is injured, the use of drugs and other methods to protect the body can appear as inhibition of activation of the JAK2–STAT3 pathway, such that it can play an organ-protective effect.
In this study, we observed that dexmedetomidine caused a decrease in pJAK2 and pSTAT3 protein levels in the hippocampi of rats that underwent CPB. AG490, an inhibitor of JAK2, was used to verify that dexmedetomidine exerted its neuroprotective effect via inhibiting JAK2–STAT3 signaling. All dexmedetomidine effects were similar to those in group AG490. These results suggest that dexmedetomidine plays a role in reducing the expression of pJAK2 and pSTAT3 in the protective effect of brain injury following CPB. These findings further validate the importance of the JAK–STAT signaling pathway in injury amelioration induced by dexmedetomidine.
Conclusion
In conclusion, dexmedetomidine can reduce inflammatory reactions and neuronal apoptosis related to CPB. The JAK2–STAT3 pathway plays an important role in the neuroprotective effects of dexmedetomidine. Dexmedetomidine provides neuroprotection during CPB in a dose-dependent manner.
Acknowledgments
This work was supported by the Natural Science Foundation of China (81373498), Science Study and Technology Development Program of Guangxi (1355005-4-2), Natural Science Foundation of Guangxi (2015GXNSFBA139139), Youth Science Foundation of Guangxi Medical University (GXMUYSF2014041) and the Department of Education Scientific Research Subject of Guangxi (KY2016YB079), China.
Author contributions
YHC and YBX conceived and designed the experiments, YHC, XZ, and LFZ performed the experiments, YHC, XZ, BDZ, and GDH analyzed the data, YHC and GDH wrote the paper, YBX and YHC were in charge of administration, and BDZ and YBX were in charge of project supervision. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Caputo M, Mokhtari A, Miceli A, et al. Controlled reoxygenation during cardiopulmonary bypass decreases markers of organ damage, inflammation, and oxidative stress in single-ventricle patients undergoing pediatric heart surgery. J Thorac Cardiovasc Surg. 2014;148(3):792–801.e8. | ||
Homi HM, Jones WL, de Lange F, Mackensen GB, Grocott HP. Exacerbation of systemic inflammation and increased cerebral infarct volume with cardiopulmonary bypass after focal cerebral ischemia in the rat. J Thorac Cardiovasc Surg. 2010;140(3):660–666.e1. | ||
McDonagh DL, Berger M, Mathew JP, Graffagnino C, Milano CA, Newman MF. Neurological complications of cardiac surgery. Lancet Neurol. 2014;13(5):490–502. | ||
Patel N, Minhas JS, Chung EM. Risk factors associated with cognitive decline after cardiac surgery: a systematic review. Cardiovasc Psychiatry Neurol. 2015;2015:370612. | ||
Howell KW, Cleveland JC Jr, Meng X, et al. Interleukin 6 production during cardiac surgery correlates with increasing age. J Surg Res. 2016;201(1):76–81. | ||
Ouk T, Amr G, Azzaoui R, et al. Lipid-lowering drugs prevent neurovascular and cognitive consequences of cardiopulmonary bypass. Vascul Pharmacol. 2016;80:59–66. | ||
Zhang W, Weng G, Li M, et al. Original Research: Establishment of an early embolus-related cerebral injury model after cardiopulmonary bypass in miniature pigs. Exp Biol Med (Maywood). 2016;241(16):1819–1824. | ||
Liao Z, Cao D, Han X, et al. Both JNK and P38 MAPK pathways participate in the protection by dexmedetomidine against isoflurane-induced neuroapoptosis in the hippocampus of neonatal rats. Brain Res Bull. 2014;107:69–78. | ||
Zeng X, Wang H, Xing X, Wang Q, Li W. Dexmedetomidine protects against transient global cerebral ischemia/reperfusion induced oxidative stress and inflammation in diabetic rats. PLoS One. 2016;11(3):e0151620. | ||
Si YN, Bao HG, Xu L, et al. Dexmedetomidine protects against ischemia/reperfusion injury in rat kidney. Eur Rev Med Pharmacol Sci. 2014;18(13):1843–1851. | ||
Si Y, Bao H, Han L, et al. Dexmedetomidine protects against renal ischemia and reperfusion injury by inhibiting the JAK/STAT signaling activation. J Transl Med. 2013;11:141. | ||
Zhao J, Li G, Zhang Y, Su X, Hang C. The potential role of JAK2/STAT3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhEPO) after experimental traumatic brain injury of rats. Cytokine. 2011;56(2):343–350. | ||
Tao Z, Cheng M, Wang SC, et al. JAK2/STAT3 pathway mediating inflammatory responses in heatstroke-induced rats. Int J Clin Exp Pathol. 2015;8(6):6732–6739. | ||
Oliva AA Jr, Kang Y, Sanchez-Molano J, Furones C, Atkins CM. STAT3 signaling after traumatic brain injury. J Neurochem. 2012;120(5):710–720. | ||
Zhao JB, Zhang Y, Li GZ, Su XF, Hang CH. Activation of JAK2/STAT pathway in cerebral cortex after experimental traumatic brain injury of rats. Neurosci Lett. 2011;498(2):147–152. | ||
Kim J, Yin T, Yin M, et al. Examination of physiological function and biochemical disorders in a rat model of prolonged asphyxia-induced cardiac arrest followed by cardio pulmonary bypass resuscitation. PLoS One. 2014;9(11):e112012. | ||
Lv J, Wei Y, Chen Y, et al. Dexmedetomidine attenuates propofol-induce neuroapoptosis partly via the activation of the PI3k/Akt/GSK3β pathway in the hippocampus of neonatal rats. Environ Toxicol Pharmacol. 2017;52:121–128. | ||
Hatashita S, Hoff JT, Salamat SM. Ischemic brain edema and the osmotic gradient between blood and brain. J Cereb Blood Flow Metab. 1988;8(4):552–559. | ||
Komine-Kobayashi M, Zhang N, Liu M, et al. Neuroprotective effect of recombinant human granulocyte colony-stimulating factor in transient focal ischemia of mice. J Cereb Blood Flow Metab. 2006;26(3):402–413. | ||
Liu X, Zhang X, Zhang J, et al. Diosmin protects against cerebral ischemia/reperfusion injury through activating JAK2/STAT3 signal pathway in mice. Neuroscience. 2014;268:318–327. | ||
Hori D, Hogue CW Jr, Shah A, et al. Cerebral autoregulation monitoring with ultrasound-tagged near-infrared spectroscopy in cardiac surgery patients. Anesth Analg. 2015;121(5):1187–1193. | ||
Matata BM, Scawn N, Morgan M, et al. A single-center randomized trial of intraoperative zero-balanced ultrafiltration during cardiopulmonary bypass for patients with impaired kidney function undergoing cardiac surgery. J Cardiothorac Vasc Anesth. 2015;29(5):1236–1247. | ||
Zhang XK, Zhou XP, Zhang Q, Zhu F. The preventive effects of dexmedetomidine against intestinal ischemia-reperfusion injury in Wistar rats. Iran J Basic Med Sci. 2015;18(6):604–609. | ||
Wang Y, Wu C, Han B, et al. Dexmedetomidine attenuates repeated propofol exposure-induced hippocampal apoptosis, PI3K/Akt/Gsk-3β signaling disruption, and juvenile cognitive deficits in neonatal rats. Mol Med Rep. 2016;14(1):769–775. | ||
Hwang L, Choi IY, Kim SE, et al. Dexmedetomidine ameliorates intracerebral hemorrhage-induced memory impairment by inhibiting apoptosis and enhancing brain-derived neurotrophic factor expression in the rat hippocampus. Int J Mol Med. 2013;31(5):1047–1056. | ||
Cheng XY, Gu XY, Gao Q, Zong QF, Li XH, Zhang Y. Effects of dexmedetomidine postconditioning on myocardial ischemia and the role of the PI3K/Akt-dependent signaling pathway in reperfusion injury. Mol Med Rep. 2016;14(1):797–803. | ||
Sugita S, Okabe T, Sakamoto A. Continuous infusion of dexmedetomidine improves renal ischemia-reperfusion injury in rat kidney. J Nippon Med Sch. 2013;80(2):131–139. | ||
Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659–661. | ||
Kunisawa T, Ueno M, Kurosawa A, et al. Dexmedetomidine can stabilize hemodynamics and spare anesthetics before cardiopulmonary bypass. J Anesth. 2011;25(6):818–822. | ||
Zhu YJ, Peng K, Meng XW, Ji FH. Attenuation of neuroinflammation by dexmedetomidine is associated with activation of a cholinergic anti-inflammatory pathway in a rat tibial fracture model. Brain Res. 2016;1644:1–8. | ||
Sifringer M, von Haefen C, Krain M, et al. Neuroprotective effect of dexmedetomidine on hyperoxia-induced toxicity in the neonatal rat brain. Oxid Med Cell Longev. 2015;2015:530371. | ||
Pan W, Lin L, Zhang N, et al. Neuroprotective effects of dexmedetomidine against hypoxia-induced nervous system injury are related to inhibition of NF-κB/COX-2 pathways. Cell Mol Neurobiol. 2016;36(7):1179–1188. | ||
Luo X, Zheng X, Huang H. Protective effects of dexmedetomidine on brain function of glioma patients undergoing craniotomy resection and its underlying mechanism. Clin Neurol Neurosurg. 2016;146:105–108. | ||
Deng H, Zuo X, Zhang J, et al. α-Lipoic acid protects against cerebral ischemia/reperfusion-induced injury in rats. Mol Med Rep. 2015;11(5):3659–3665. | ||
Kim YR, Kim HN, Jang JY, et al. Effects of electroacupuncture on apoptotic pathways in a rat model of focal cerebral ischemia. Int J Mol Med. 2013;32(6):1303–1310. | ||
Fujii Y, Shirai M, Inamori S, et al. Insufflation of hydrogen gas restrains the inflammatory response of cardiopulmonary bypass in a rat model. Artif Organs. 2013;37(2):136–141. | ||
Trop S, Marshall JC, Mazer CD, et al. Perioperative cardiovascular system failure in South Asians undergoing cardiopulmonary bypass is associated with prolonged inflammation and increased Toll-like receptor signaling in inflammatory monocytes. J Surg Res. 2014;187(1):43–52. | ||
Jongman RM, Zijlstra JG, Kok WF, et al. Off-pump CABG surgery reduces systemic inflammation compared with on-pump surgery but does not change systemic endothelial responses: a prospective randomized study. Shock. 2014;42(2):121–128. | ||
Bulow NM, Colpo E, Pereira RP, et al. Dexmedetomidine decreases the inflammatory response to myocardial surgery under mini-cardiopulmonary bypass. Braz J Med Biol Res. 2016;49(4):e4646. | ||
Podgoreanu MV, White WD, Morris RW, et al. Inflammatory gene polymorphisms and risk of postoperative myocardial infarction after cardiac surgery. Circulation. 2006;114(1 Suppl):I275–I281. | ||
Steinbrenner H, Bilgic E, Pinto A, et al. Selenium pretreatment for mitigation of ischemia/reperfusion injury in cardiovascular surgery: influence on acute organ damage and inflammatory response. Inflammation. 2016;39(4):1363–1376. | ||
de Jong PR, Schadenberg AW, van den Broek T, et al. STAT3 regulates monocyte TNF-alpha production in systemic inflammation caused by cardiac surgery with cardiopulmonary bypass. PLoS One. 2012;7(4):e35070. | ||
Owais K, Huang T, Mahmood F, et al. Cardiopulmonary bypass decreases activation of the signal transducer and activator of transcription 3 (STAT3) pathway in diabetic human myocardium. Ann Thorac Surg. 2015;100(5):1636–1645. | ||
Li L, Li H, Li M. Curcumin protects against cerebral ischemia-reperfusion injury by activating JAK2/STAT3 signaling pathway in rats. Int J Clin Exp Med. 2015;8(9):14985–14991. | ||
Kim HC, Kim E, Bae JI, et al. Sevoflurane postconditioning reduces apoptosis by activating the JAK-STAT pathway after transient global cerebral ischemia in rats. J Neurosurg Anesthesiol. 2017;29(1):37–45. | ||
Hristova M, Rocha-Ferreira E, Fontana X, et al. Inhibition of signal transducer and activator of transcription 3 (STAT3) reduces neonatal hypoxic-ischaemic brain damage. J Neurochem. 2016;136(5):981–994. | ||
Jia L, Wang F, Gu X, et al. Propofol postconditioning attenuates hippocampus ischemia-reperfusion injury via modulating JAK2/STAT3 pathway in rats after autogenous orthotropic liver transplantation. Brain Res. 2017;1657:202–207. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.