")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Dexmedetomidine Attenuates Cellular Injury and Apoptosis in H9c2 Cardiomyocytes by Regulating p-38MAPK and Endoplasmic Reticulum Stress
Authors Zhu Z , Ling X, Zhou H, Zhang C, Yan W
Received 3 June 2020
Accepted for publication 19 September 2020
Published 12 October 2020 Volume 2020:14 Pages 4231—4243
DOI https://doi.org/10.2147/DDDT.S265970
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Zhipeng Zhu,1,* Xiaoyan Ling,2,* Hongmei Zhou,1 Caijun Zhang,1 Weiwei Yan1
1Department of Anesthesiology, The Second Affiliated Hospital of Jiaxing University, Jiaxing City, Zhejiang Province 314000, People’s Republic of China; 2The Outpatient Nursing Department of the Second Affiliated Hospital of Jiaxing University, Jiaxing City, Zhejiang Province 314000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhipeng Zhu
Department of Anesthesiology and Pain, The Second Affiliated Hospital of Jiaxing University, Jiaxing City, Zhejiang Province, People’s Republic of China
Email [email protected]
Background: Myocardial ischaemia-reperfusion injury (IRI) has been confirmed to induce endoplasmic reticulum stress (ERS) when myocardial cell function continues to deteriorate to a certain degree. The clinical applications of effective tested strategies are sometimes inconsistent with the applications evaluated in experiments, although reasonable mechanisms and diverse signalling pathways have been broadly explored. Dexmedetomidine (DEX) has been shown to attenuate IRI of the heart in animal studies. This study aimed to determine whether DEX can protect injured cardiomyocytes under hypoxia/reoxygenation (H/R) at the cellular level and whether the mechanism is related to ERS and the p38 MAPK pathway.
Methods: H9c2 cells were subjected to H/R or thapsigargin (TG) to build a model. DEX or 4-PBA was added to the medium either 1 h or 24 h before modelling, respectively. Model parameters were determined by assessing cell viability and injury, which were measured by assessing cell counting kit-8 (CCK8), lactate dehydrogenase (LDH) release and flow cytometry results, and the expression of GRP78, CHOP and caspase-12. In addition, the protein expression of p38MAPK and p-p38MAPK was examined, and SB202190, a negative regulator, was also preincubated in medium.
Results: Compared to that of cells in the control group, the activity of cells in the H/R and TG groups was decreased dramatically, and the LDH concentration and proportion of apoptotic cells were increased. DEX could correspondingly reverse the changes induced by H/R or TG. Additionally, DEX effectively attenuated ERS defined as increased expression of GRP78, CHOP and caspase-12. Additionally, DEX could obviously depress the P38 MAPK phosphorylation and high p-p38 MAPK expression in the TG group, indicating DEX has a function similar to that of SB202190.
Conclusion: H/R injury in H9c2 cells can lead to abnormal ERS and apoptosis, as well as activation of the p38MAPK signalling pathway. DEX can protect cardiomyocytes by intervening in ERS, regulating p38MAPK and the downstream apoptotic signalling pathway.
Keywords: dexmedetomidine, hypoxia/reoxygenation injury, endoplasmic reticulum stress signalling pathway, p38 mitogen-activated protein kinase
Introduction
Myocardial ischaemia-reperfusion injury (MIRI), which usually occurs in clinical settings in an inevitable manner, always produces severe outcomes for patients if no effective strategies to induce downstream apoptotic cascades to shift in a favourable direction are employed. Fortunately, numerous animal studies evaluating diverse protective mechanisms have confirmed the efficacy of cardioprotection in ameliorating MIRI.1–6 The actual clinical application of some of these strategies, however, does not match the animal study applications, and it is difficult to predict the consequences in clinical practice from those seen in experimental research.7–9 In this context, the unclear mechanism underlying the development of apoptotic pathways or involving key molecules in MIRI might significantly matter, especially for cell death and even associated signalling pathways during ischaemia-reperfusion, such as ERS-related apoptosis or signalling pathways; a noteworthy report by Davidson in 2019 shared the opinion that multitarget strategies must be adopted to reduce MIRI if appropriate because single approaches have a limited capacity to overcome complicated MIRI situations.10 Based on these unknown factors, we explored the possibility that there is a clinical medication with organ-protecting properties that can be adopted to attenuate IRI and then tried to illuminate the underlying mechanism to shorten the development time needed to successfully treat IRI.
DEX, a highly selective α2 adrenergic receptor agonist, is frequently used in clinical practice, especially to protect the heart and other organs during surgery.11–14 At this point, the most likely function of DEX in this application could be related to the anti-inflammatory response and inhibition of ischaemia-reperfusion injury. Regarding ERS, the effects of DEX on ERS and subsequent apoptosis have not been thoroughly illustrated. Judging from existing studies, DEX performs a protective role in inhibiting IRI in the heart of diabetic mice by interfering with ERS or autophagy;6,15 however, these results were partly attributed to the diabetes context. Furthermore, researchers have focused on studying other non-cardiac cells, such as endothelial cells, under IRI or H/R conditions16,17 and examining several crucial ERS chaperones, proteins and apoptosis indicators produced by organs other than the heart under IRI or H/R conditions,6,18–22 finding that DEX can effectively regulate the function of non-cardiac cells and interfere in the endoplasmic reticulum stress signalling pathway under certain circumstances. Few studies have explored the function of DEX in H9c2 cardiomyocytes under H/R conditions;23,24 however, the exact regulatory effect of DEX on ERS remains unknown. In recent years, an increasing number of basic research studies have focused on the p38MAPK signalling pathway; p38MAPK belongs to the family of MAPK-activated protein kinases and participates in many cellular processes, such as cell proliferation, survival and apoptosis. Additionally, it has been shown that stress is the leading cause of p38 MAPK activation. Accumulating evidence has shown that DEX has the capacity to alleviate inflammation and IRI by regulating p38 MAPK signalling.25–27 Wang et al found that DEX could attenuate mouse Neuro 2A neuroblastoma cell apoptosis under H/R conditions by targeting p38 MAPK signalling,28 which confirmed that DEX might positively affect cells under H/R conditions.
In this study, we hypothesized that DEX could prevent cardiac cell injury under ERS conditions, which would verify its involvement in ERS. Moreover, we assume that the cardioprotective function of DEX could be fulfilled by regulating the p38MAPK and ERS signalling pathways.
Methods
Cell Culture
H9c2 cardiomyocytes, a rat embryonic myocardial cell line, were purchased from Process Life and Technology Corporation Limited (Wuhan, China). Cells were cultured in 45 g/L glucose DMEM (Corning, USA) supplemented with 10% foetal bovine serum (FBS, Invitrogen, USA), 100 g/mL streptomycin and 100 U/mL penicillin (Solarbio, China). All the experimental cells were grown in a 95% air plus 5% CO2 incubator. The medium was replaced as needed, according to cell growth, approximately every 1–3 days. When cells reached 70–80% confluence, the cells were used in experiments.
H/R Injury Model
According to our previous study illustrating the optimal in vitro H/R model, H9c2 cells were processed into a single-cell suspension, seeded in advance in 96-well plates at approximately 5×103 cells/mL, and cultivated in two plates (a control plate and an experimental plate). The control plate was incubated in a 37 °C 5% CO2 cell incubator, and the medium was changed at the same time point for both plates. All the groups in the experimental plates underwent a three-hour anoxia period with all the cells cultured at 37 °C in a hypoxia chamber filled with 5% CO2 and 95% N2, and the medium was changed to serum-free DMEM when the anoxia procedure began. According to the different protocols adopted in the study (see below), the experimental groups were subjected to normoxic culture when the reoxygenation procedure started at the end of the previous step. For the second part of the study, an ERS model was induced with thapsigargin (TG) (the exact protocol is explained below). At the end of each procedure, cell viability, the injury rate, apoptosis and several ERS molecular chaperone markers, such as GRP78, CHOP and caspase-12, were detected and examined according to corresponding procedures (see below).
Experimental Protocols
To verify that DEX was an effective protector and attenuated H/R injury in H9c2 cells, and to determine whether DEX is involved in the signalling of ERS, cultured cells in 96-well or 6-well plates were divided into a control group, an H/R group, an H/R+DEX group and an H/R+DEX+4-PBA (4-phenylbutyrate, P-21,005, Sigma, USA) group. 4-PBA, which is a classic chemical chaperone used to reduce protein misfolding and ameliorate ERS, was dissolved in a DMSO solution in a sterile, dark environment and was added to the medium 24 h prior to H/R at a dilution of 1:1000. In addition to cell viability and injury assays, flow cytometry was used to evaluate apoptosis in these groups. In addition, the mRNA and protein expression of GRP78, CHOP and caspase-12 was also detected by RT-PCR and Western blot analysis, respectively (the specific methods are described below).
To test whether p38MAPK is targeted by DEX in the experimental system to attenuate ERS, TG, which is a specific ER stress inducer, was used to induce ERS at the recommended dose of 0.5 µM 10 h before the start of procedures. The other pre-treatments were similar to the above treatment protocol. First, the p38MAPK (dilution ratio 1:1000) and P-P38MAPK (dilution ratio 1:1000) expression of cells in the control group, TG group and DEX + TG group were detected at the end of the experiment by Western blotting, and β-actin was used as the internal control for sample loading. Second, SB202190, a p38MAPK-specific inhibitor, was selected to further study the possible signalling pathway involved in this experiment and was added to the medium at a dose of 5 µM 1 h before the beginning of the experiment. The incubated cells were randomly divided into control, TG, DEX + TG, DEX + TG + 4-PBA, SB202190 + TG, DEX + TG + SB202190, and DEX + TG + 4-PBA + SB202190 groups, and each group followed the appropriate specific procedure. Every group was analysed with cell viability assays, flow cytometry, and detection of the protein and mRNA expression of GRP78, CHOP and caspase-12.
Cell Viability and Injury Assays
According to the instructions of a CCK-8 kit (Dojindo, Japan), H9c2 cells (approximately 5×103 cells/well) were seeded in a 96-well plate and used in the subsequent protocols. When required for viability testing, the cells in each well were covered with 10 μL of Beyotime and incubated for at least 1 h at 37 °C in the incubator. Then, the OD value at 450 nm was measured with a microplate reader (Molecular Devices, Sunnyvale, CA, USA). Cell viability (%) was then calculated. In addition, cell injury was assessed based on the amount of LDH released into the supernatant with an LDH kit (Beyotime Biotechnology, China). According to the instructions of the LDH assay kit, all the cell groups were plated in 96-well plates, with 3 replicates per group, and outcomes were measured with a microplate reader at 450 nm. The LDH concentration and cell injury rate were then calculated.
Apoptosis Assay
The apoptosis rate of the H9c2 cells in each group was examined by flow cytometry with an Annexin V-FITC/PI (propidium iodide) kit (Sigma, USA). Briefly, after digestion and centrifugation, cells were washed with PBS twice, resuspended in binding buffer, and then incubated with 10 μL of Annexin V-FITC and 5 μL of PI for 20 min. Apoptotic cells were detected using a flow cytometer (BD, San Jose, CA), and the apoptosis rate was calculated.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
H9c2 cells were prepared in advance for mRNA analysis. The primers for GRP78, caspase12, CHOP, and β-actin were synthesized by Invitrogen, and the primer sequences are shown in Table 1. Total RNA was extracted from H9c2 cells with TRIzol, and the purity of the RNA from each group of cells was determined. With the Reverse Transcription Kit (Takara, China), 500 ng of RNA from each sample was reverse transcribed into cDNA according to the manufacturer’s instructions. Subsequently, polymerase chain reaction was conducted with 2 µL of cDNA and other necessary reagents according to the instructions of a Premix Ex Taq kit (Takara, China), and the final 25-µL reaction was run on the ABI Prism 7500 System (Bedford, USA). The amplification process was initiated at 95 °C for 3 min, followed by 40 cycles of amplification including denaturation at 95 °C for 30 s, annealing at 55 °C for 20 s, and elongation at 72 °C for 20 s. mRNA expression was calculated as the relative ratio of the target gene level to the β-actin level by using the 2 −ΔΔCT method.
 |
Table 1 Primers Sequences |
Western Blots
Following the treatments described in previous procedures, H9c2 cells were washed well with an ice-cold PBS solution, and then, we added a RIPA solution (Beyotime, China) to the wells and incubated the cells for 30 min on ice. Subsequently, the supernatants were collected after centrifugation of the lysates. The BCA method was used to measure the protein concentration. The proteins were separated by SDS-PAGE and transferred to PVDF membranes at 4 °C and 200 mA for 2 h. Then, the membranes were blocked in a TBST solution for 2 h at room temperature and incubated at 4 °C overnight with the following primary antibodies: anti-dual phospho-p38 MAPK (Thr180 and Tyr182) (Sigma, 1:1000), anti-p38 MAPK (Sigma, 1:1000), anti-CHOP (Affinity, 1:1000), anti-GRP78 (Affinity. 1:1000), anti-caspase12 (Affinity, 1:1000) and mouse anti-β-actin (Affinity, 1:1000). A secondary antibody conjugated to horseradish peroxidase (Jackson, 1:2000) was added and incubated for 2 h at room temperature. Finally, the membranes were washed in TBST, and the signals were visualized with an enhanced chemiluminescence detection kit (ECL, Beyotime, China). The density of the protein bands was quantified by IQuantTL (GE Healthcare, USA). Relative protein expression was calculated with normalization to β-actin expression.
Statistical Analysis
All the data are expressed as the mean ± standard deviation (SD), and one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test was performed to examine the significance of differences among multiple groups with SPSS 19.0 software (SPSS, Chicago, USA). GraphPad Prism (San Diego, USA) was used to construct the graphics. P<0.05 was considered to indicate a statistically significant difference.
Results
DEX Protects H9c2 Cells from H/R Injury
Based on the successful H/R model established in our previous study, the cell viability and LDH release of incubated cells in randomized groups given different treatments were detected (Figure 1). Compared to the control group, the DEX pretreatment group under normal conditions and the 4-PBA pretreatment group under normal conditions showed similar and comparable changes in terms of the two abovementioned variables (P>0.05). However, it was observed that the H/R group showed dramatically decreased cell viability and increased LDH release compared with the control group (P<0.05), which corresponded well with previous results. Compared with the H/R group, the DEX group showed reduced cellular injury and LDH release, as did the 4-PBA group. In addition, 4-PBA successfully reversed the protective effect of DEX on H/R in terms of cell viability and LDH release (Figure 1).
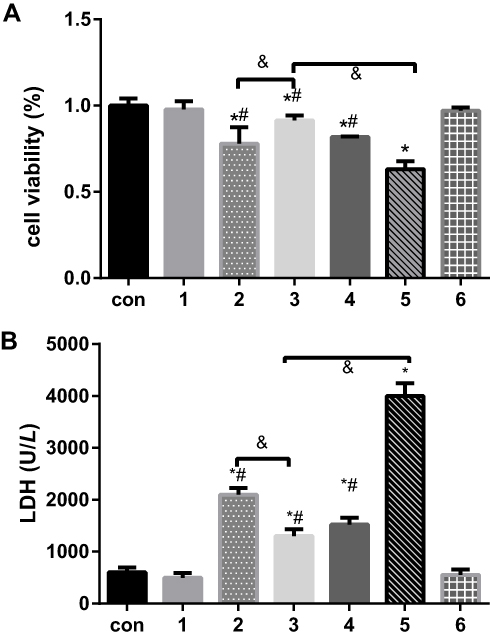 |
Figure 1 The effects of dexmedetomidine on the levels of myocardial cell injury due to H/R under different conditions were determined by measuring (A) the proportions of cell viability and (B) LDH release. The results are expressed as the mean ± SD. *p < 0.05 vs the control group; #P<0.05 vs the H/R group; &P<0.05 vs the DEX+H/R group. Con, control group; 1, normoxic incubation with dexmedetomidine; 2, hypoxic/reoxygenation incubation with dexmedetomidine and 4-phenyl butyric acid; 3, hypoxic/reoxygenation incubation with dexmedetomidine; 4. hypoxic/reoxygenation incubation with 4-phenyl butyric acid; 5. hypoxic/reoxygenation incubation; 6. normoxic incubation with 4-phenyl butyric acid. |
In a cell apoptosis assay, there was an average 10% increase in the H/R group compared with the control group (Figure 2). However, the DEX+ H/R and 4-PBA+ H/R pretreatment groups exhibited reduced cell apoptosis, and apoptosis in these 2 groups was significantly attenuated by 7% or 6%, respectively, compared with that in the H/R group (P<0.05). Additionally, 4-PBA reversed the anti-apoptotic effect of DEX and resulted in an obvious increase of approximately 4% (Figure 2).
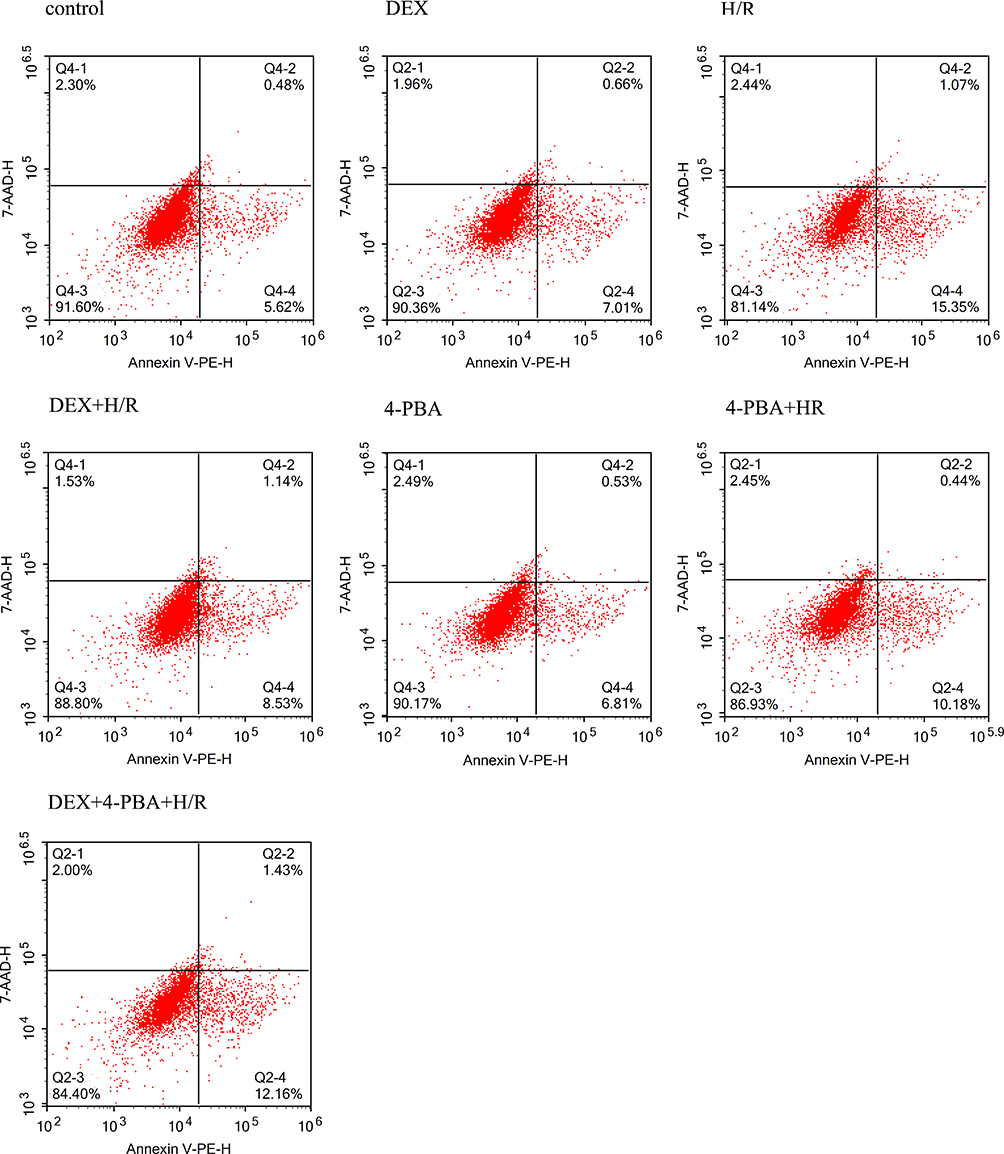 |
Figure 2 The percentages of apoptotic H9C2 cardiomyocytes were examined by flow cytometry under different conditions. |
DEX Reduced the Expression of GRP78, CHOP and Caspase12 in H9c2 Cells After H/R
With the goal of determining the role of DEX, three molecules involved in ERS and related signalling pathways were examined (Figure 3). As illustrated in Figure 3, consistent results were observed between mRNA expression and protein expression. Compared to the control group, the H/R group exhibited obvious upregulation of the indicators of ERS and apoptosis. DEX or 4-PBA pretreatment significantly alleviated the expression of GRP78, CHOP and Caspase-12 induced by H/R (P<0.05). Moreover, the alleviation of ERS and apoptosis in the DEX+H/R group were dramatically reversed by 4-PBA, as was indicated by the results for the DEX+H/R+4-PBA group.
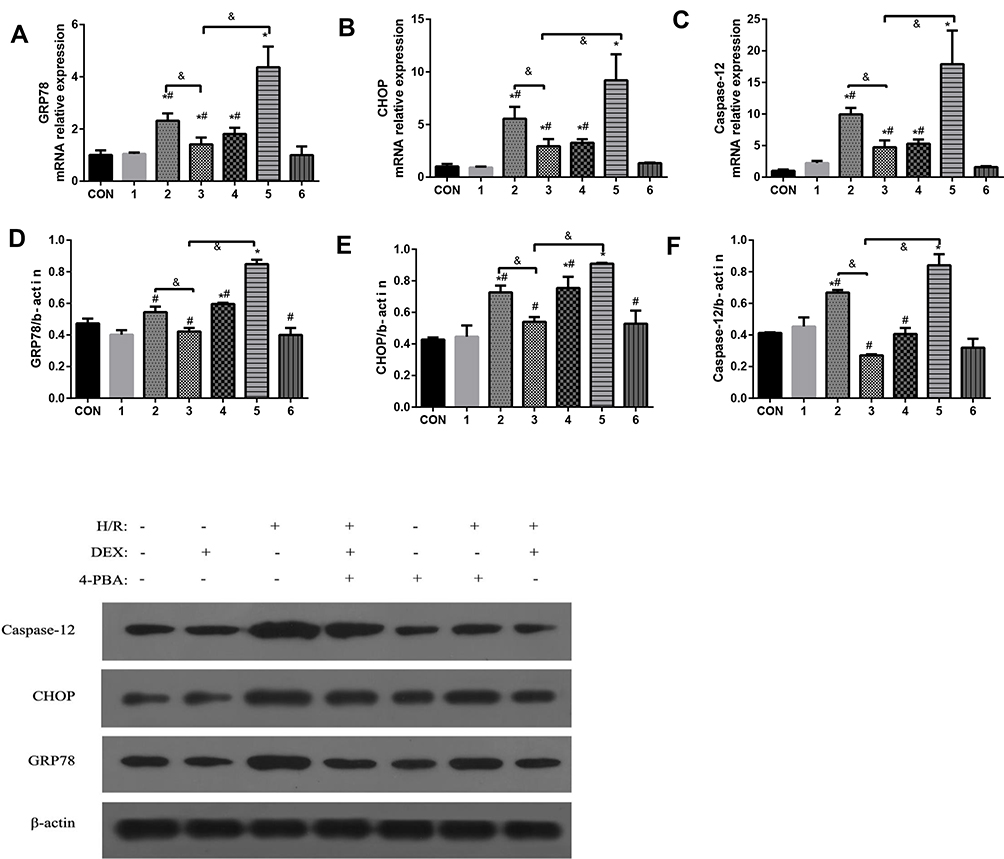 |
Figure 3 The effects of dexmedetomidine on the expression of GRP78, CHOP and caspase-12 in myocardial cells treated under different conditions were examined. (A–C) mRNA expression of GRP78, CHOP and caspase-12; (D–F) Protein expression of GRP78, CHOP and caspase-12. The results are expressed as the mean±SD. *P<0.05 vs the control group; #P<0.05 vs the H/R group; &P<0.05 vs the DEX+H/R group. Con, control group; 1, normoxic incubation with dexmedetomidine; 2, hypoxic/reoxygenation incubation with dexmedetomidine and 4-phenyl butyric acid; 3, hypoxic/reoxygenation incubation with dexmedetomidine; 4. hypoxic/reoxygenation incubation with 4-phenyl butyric acid; 5. hypoxic/reoxygenation incubation; 6. normoxic incubation with 4-phenyl butyric acid. |
DEX Attenuated TG-Induced H9c2 Cell Injury
Previously, ERS in H9c2 cells was shown to be induced by TG, and cell viability, LDH release and the apoptotic proportion were evaluated (Figures 4 and 5). Figure 4 shows that the cell viability in the TG group was significantly decreased compared with that in the normal group, and the LDH concentration was increased dramatically. The cell viability and LDH concentration in the DEX + TG group were increased and decreased, respectively, due to DEX intervention (P < 0.05). Like LDH release, apoptosis showed similar contrasting trends among the different groups (Figure 5). Compared with the DEX + TG group, the sb202190 + TG group had comparable outcomes in terms of cell viability, the LDH concentration and apoptosis (P > 0.05), but those indexes were reversed by sb202190, a p-p38MAPK inhibitor, and showed opposite changes in the DEX + sb202190+TG group (P < 0.05).
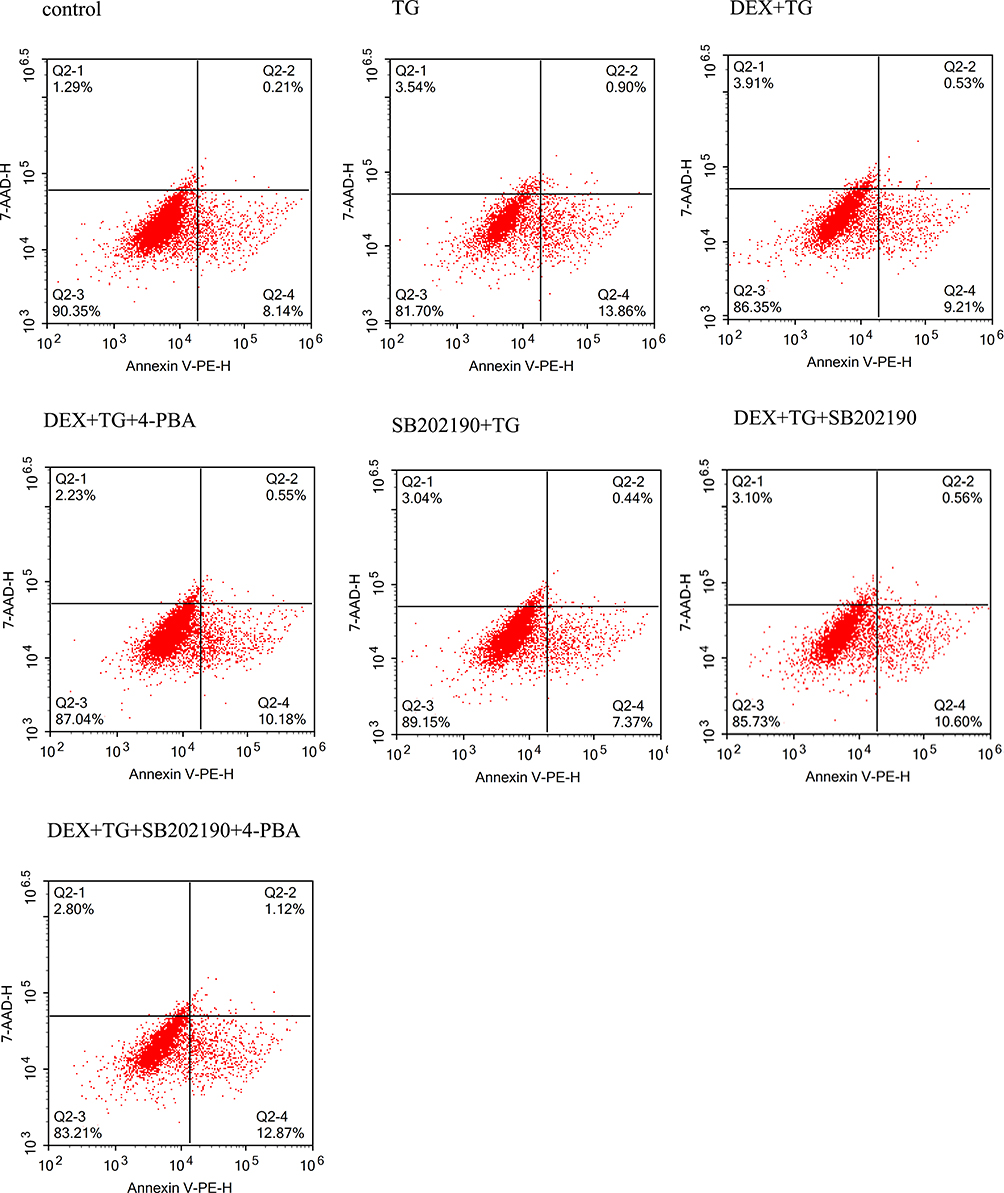 |
Figure 4 The percentages of apoptotic H9C2 cardiomyocytes were examined by flow cytometry under different conditions. |
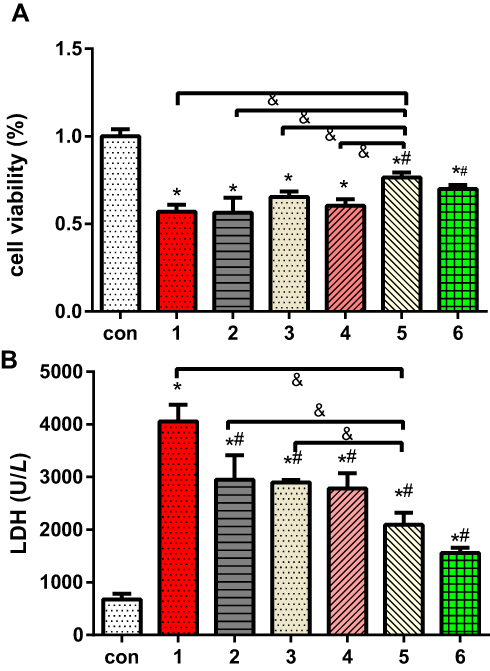 |
Figure 5 The effects of dexmedetomidine on the levels of myocardial cell injury induced by TG and different conditions were determined by measuring (A) the proportions of cell viability and (B) LDH release. The results are expressed as the mean ± SD. *p < 0.05 vs the control group; #P<0.05 vs the TG group; &P<0.05 vs the DEX+TG group. Con, control group; 1, normoxic incubation with TG; 2, incubation with a combination of SB202190, 4-phenyl butyric acid and DEX on TG model; 3, incubation with DEX and 4-phenyl butyric acid on TG model; 4. incubation with SB202190 and DEX on TG model; 5. incubation with DEX on TG model; 6. incubation with a combination of TG and SB202190. |
DEX Inactivated the Phosphorylation of P38 MAPK
As shown in Figure 6, Western blot analysis of P38MAPK and P-P38MAPK was performed for groups with or without DEX pretreatment. In all groups, the overall expression of p38-MAPK remained stable, and no obvious difference was found, even in the TG group (P > 0.05). Compared with that of the control group, the protein expression level of P-P38MAPK in the TG group was almost 3-fold increased (P < 0.05). Compared with the TG group, however, the DEX + TG group had lower protein expression of P-P38MAPK, which is normally accompanied by stress (P < 0.05) (Figure 6). These above results indicate that ERS induced by TG can cause p38MAPK phosphorylation and activate the p38MAPK signalling pathway, which directly affects cell viability, LDH release and apoptosis. DEX has the potential to attenuate cell injury, which may be involved in the activation of p38MAPK.
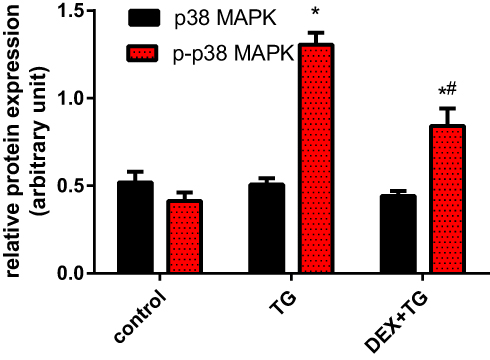 |
Figure 6 The protein expression of p38MAPK and p-p38MAPK in 3 separate groups was detected by Western blotting and is illustrated as the mean ± SD. *p < 0.05 vs the control group; #P<0.05 vs the TG group. DEX, dexmedetomidine; TG, thapsigargin; p38MAPK, p38 mitogen-activated protein kinase; p-p38MAPK, phosphorylated p38MAPK. |
Effects of P38MAPK Inactivation by DEX on the Expression of Markers of the ERS Signalling Pathway
To further verify whether p38MAPK is involved in the process by which DEX attenuates the H9c2 cell damage induced by TG and elucidate the possible mechanism linking DEX pretreatment and the ERS-apoptosis signalling pathway, the expression of GRP78, CHOP and caspase-12 was further evaluated. The results showed that there was no significant difference in GRP78, CHOP and caspase-12 gene or protein expression between the DEX + TG group and SB202190 + TG group (P > 0.05); compared with that in the TG group, the expression of GRP78, CHOP and caspase-12 in the 3 intervention groups including the DEX+ TG group, SB20219+ TG group and 4-PBA+ TG group was decreased (P < 0.05). However, in contrast to that in the DEX + TG group, the expression of those indexes in groups with SB20219 and 4-PBA intervention was significantly increased (P < 0.05). Furthermore, the DEX + TG + sb20219 + 4-PBA group had the highest increase (Figure 7). In conclusion, the SB20219 group exhibited an inhibitory effect similar to that in the 4-PBA group to prevent ERS, and SB20219 could inhibit the protective function of DEX under ERS conditions, which significantly reduced the expression of GRP78, CHOP and caspase-12. These three enzymes might be downstream signalling molecules of the p38MAPK-apoptosis signalling pathway (Figure 7).
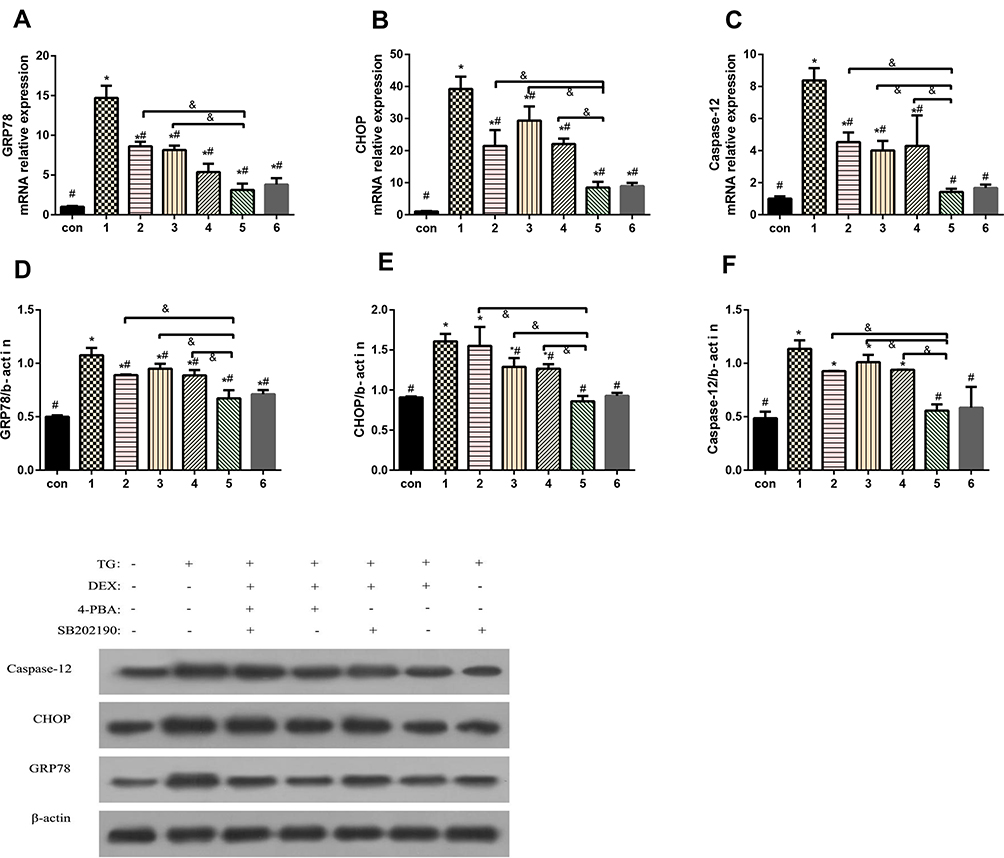 |
Figure 7 The effects of dexmedetomidine on the expression of GRP78, CHOP and caspase-12 in myocardial cells treated under different conditions were evaluated. A-C, mRNA expression of GRP78, CHOP and caspase-12; D-F, protein expression of GRP78, CHOP and caspase-12. The results are expressed as the mean±SD. *P<0.05 vs the con group; #P<0.05 vs the TG group; &P<0.05 vs the DEX+TG group. Con, control group; 1, normoxic incubation with TG; 2, incubation with a combination of SB202190, 4-phenyl butyric acid and DEX on TG model; 3, incubation with DEX and 4-phenyl butyric acid on TG model; 4. incubation with SB202190 and DEX on TG model; 5. incubation with DEX on TG model; 6. incubation with a combination of TG and SB202190. |
Discussion
It is well known that DEX shows a variety of protective properties in clinical use, especially in ischaemic heart disease patients, but the specific mechanism remains unknown. In recent years, a great number of basic research studies have reported that DEX has the capacity to regulate in vivo MIRI and even H/R injury via the regulation of either the inflammatory response pathway or apoptosis,1,12,13,27,29 which are two key downstream branches of the MAPK signalling pathway.30–33 However, few basic research studies have paid attention to the modulation of ERS by DEX in the regulation of IRI outcomes, and the exact mechanism underlying this modulation has not been elucidated. Recently, DEX was reported to alleviate IRI of the intestine or sepsis in the lungs by targeting the p38MAPK signalling pathway.25,26 In our present study, we applied a classic H/R model and TG-induced specific ERS model to examine the effects of DEX on H/R and stress injury based on ERS and further determined if the targeting molecule in this process is p38MAPK. This study confirmed that DEX attenuated the myocardial damage induced by either H/R or TG by attenuating ERS, which subsequently led to the downregulation of the expression of several key molecules related to ERS and apoptosis. Additionally, p38MAPK might be a potential target involved in this process.
It has been well accepted that MIRI can lead to severe ERS, which is usually detected by upregulated GRP78 expression.34,35 If ERS increases, apoptotic cascades would be considered an underlying mechanism of MIRI, and the expression of some specific transcription-related molecules, such as CHOP and caspase-12, would be upregulated in an ERS-dependent manner. Most researchers have confirmed that these molecules can be used to represent the status of ERS and even the developing direction of cell survival as downstream markers of the ERS signalling pathway.36–38 Moreover, ERS in MIRI can be alleviated through the suppression of caspase-12 and CHOP.
In this preclinical study, these 3 molecules were highly expressed under H/R or TG induction but dramatically alleviated by DEX and 4-PBA. This result clearly indicated that DEX is potentially involved in ERS-associated apoptosis and has the capacity to inhibit ERS with a function similar to that of 4-PBA, as reflected by the decreased expression of CHOP, GRP78 and caspase-12 at both the mRNA and protein levels. Similar to studies by Chai and other researchers,19,39,40 which have similar outcomes to ours, DEX could eventually attenuate the apoptotic outcomes induced by CHOP or caspase-12, even though those studies used diverse models or approaches to induce apoptosis, such as IRI, ERS and oxidative stress pathways. In regard to the mechanism at the cellular level, several studies have confirmed the cardioprotective effect of DEX in I/R injury based on H/R models.23,24,41 The consistent conclusions advanced from these studies share similar outcomes with our conclusions, although the H/R models used varied among the studies. The major outcomes evaluated were cell viability, the apoptosis rate and LDH release, and by evaluating the expression of miR-208b,23 intracellular calcium overload24 and TLR4,41 no ERS was found to be involved.
From another aspect, apoptosis in H/R mimic models of IRI can have different causes, and it is extremely difficult to specify the cause and consequence. According to Price and Chaudhari’s research,42,43 hypoxia, to some extent, is considered a beneficial factor in cells that undergo subsequent TG induction, which could affect the efficacy of TG, or hypoxia could be independent of apoptosis, which was unlike the results for the function of TG. Therefore, TG induction was adopted as a specific enhancement model to clarify the inhibitory effect of DEX on the ERS pathway in the present study.
p38 MAPK is a member of the evolutionarily conserved serine/threonine protein kinases that mediate a variety of cellular processes linking cellular signals from outside the cell to intracellular processes.44 It is activated through the phosphorylation of Thr-X-Tyr by mitogen-activated protein kinase (MAPK). Among its family members, p38 MAPK is related to infection and inflammatory stress.45 Then, how is p38 MAPK activated? The first answer is ERS. ERS directly leads to the upregulation of PERK, IRE1 and ATF6 expression, and if ERS is not alleviated, it may lead to an interaction between downstream ASK1 and TRAF2 and even the autophosphorylation of ASK1.32 A study showed that autophosphorylation of ASK1 could activate MKK and p38 MAPK.46 Rasheed et al also noted that ERS can activate multiple signalling pathways, such as the eIF2 α, MAPK and NF-κB pathways, and thus trigger downstream cascade reactions.47 Finally, p38 MAPK phosphorylation is achieved by MAPK3/MAPK6 activation. In addition, infection and other factors can also induce p38 MAPK activation.48 In the present study, consistent results were achieved with TG induction in H9c2 cells, and high expression of p-p38 MAPK was found.
Then, what kinds of downstream changes would be found in the canonical p38 MAPK signalling pathway? It has been reported that p-p38 MAPK can lead to NF-κB-mediated inflammation,31,33 autophagy,49,50 apoptosis30,51 and other processes. Thus, targeting p38 MAPK for the development of novel strategies combating ERS or acute pathologies has been tested for years. For instance, Bonney EA t et al52 reported attenuated ischaemia-reperfusion injury mediated by inhibition of the p38MAPK signalling pathway. Wang et al28 also illustrated the effect of DEX in altering oxygen deprivation/reoxygenation-induced apoptosis via the p38MAPK signalling pathway in N2A cells. In this study, DEX pretreatment was first used to produce a similar experimental result in H9c2 cells, including the high expression of P-P38MAPK produced by ERS and myocardial apoptosis following the activation of the p38MAPK signalling pathway.
Given the importance of p38 MAPK in the ERS-apoptosis pathway, specific p38 MAPK inhibitors are commonly used as experimental tools in pilot studies or the treatment of cancers and inflammatory diseases. SB202190 is an inhibitor that can interact with p38α and p38β via competitive binding to the binding site on p38. In the present study, SB202190 eventually decreased the increases in indexes induced by TG and reversed the decreases in parameters produced by DEX in the DEX+TG group. This conclusion indirectly indicated the roles of DEX in protecting ERS and apoptosis through p38 MAPK intervention.
Several limitations should be taken into consideration. Primarily, human cells or stem cells that more accurately reflect the clinical scenario were not investigated in the present study, so these findings should not be applied to other types of cells, especially human cells. Further in vivo animal studies or clinical trials will be warranted. Likewise, this study was only a pilot study, so no advanced techniques, such as RNAi, were used. Second, the purpose of any in vitro H/R model is to mimic IRI that occurs in a clinical setting so the condition-dependent models might vary. In other words, the outcomes derived from the present study might not apply to other models.
Conclusions
In conclusion, DEX, a promising agent in the clinical setting, has been confirmed to protect H9c2 cells from hypoxia/reoxygenation injury at the cellular level, which occurred via modulation of ERS-induced apoptosis. Furthermore, DEX pretreatment alleviated p-p38 MAPK expression under ERS conditions induced by TG, which significantly reduced the downstream expression of GRP78, CHOP and caspase-12. Additional studies are needed to identify the specific signalling pathway linking p38 MAPK and apoptosis.
Funding
This research was supported by Zhejiang Provincial Natural Science Foundation of China under Grant No.2017C33185, the Project of the Health Commission of Zhejiang Province under Grant No.2019ZD053 and the clinical project of the Zhejiang Medical Association under Grant No.2015ZYC-A70.
Disclosure
The authors report no conflict of interest related to this paper.
References
1. Bunte S, Behmenburg F, Majewski N, et al. Characteristics of dexmedetomidine postconditioning in the field of myocardial ischemia-reperfusion injury. Anesth Analg. 2020;130(1):90–98. doi:10.1213/ANE.0000000000004417
2. Li L, Li X, Zhang Z, et al. Protective mechanism and clinical application of hydrogen in myocardial ischemia-reperfusion injury. Pak J Biol Sci. 2020;23(2):103–112.
3. Xi J, Li Q, Li B, et al. miR‑155 inhibition represents a potential valuable regulator in mitigating myocardial hypoxia/reoxygenation injury through targeting BAG5 and MAPK/JNK signaling. Mol Med Rep. 2020;21(3):1011–1020.
4. Li J, Zhou W, Chen W, et al. Mechanism of the hypoxia inducible factor 1/hypoxic response element pathway in rat myocardial ischemia/diazoxide post‑conditioning. Mol Med Rep. 2020;21(3):1527–1536.
5. Kitazume-Taneike R, Taneike M, Omiya S, et al. Ablation of toll-like receptor 9 attenuates myocardial ischemia/reperfusion injury in mice. Biochem Biophys Res Commun. 2019;515(3):442–447. doi:10.1016/j.bbrc.2019.05.150
6. Li J, Zhao Y, Zhou N, et al. Dexmedetomidine attenuates myocardial ischemia-reperfusion injury in diabetes mellitus by inhibiting endoplasmic reticulum stress. J Diabetes Res. 2019;30::7869318.
7. Heusch G, Gersh BJ. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. 2017;11:774–784.
8. Heusch G. Cardioprotection research must leave its comfort zone. Eur Heart J. 2018;39(36):3393–3395. doi:10.1093/eurheartj/ehy253
9. Heusch G. Critical issues for the translation of cardioprotection. Circ Res. 2017;120(9):1477–1486. doi:10.1161/CIRCRESAHA.117.310820
10. Davidson SM, Ferdinandy P, Andreadou I, et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC Review Topic Of The Week. J Am Coll Cardiol. 2019;73(1):89–99. doi:10.1016/j.jacc.2018.09.086
11. Kong Q, Wu X, Qiu Z, et al. Protective effect of dexmedetomidine on acute lung injury via the upregulation of tumour necrosis factor-α-induced protein-8-like 2 in septic mice. Inflammation. 2020;11:1–14.
12. Zhang Y, Liu M, Yang Y, et al. Dexmedetomidine exerts a protective effect on ischemia-reperfusion injury after hepatectomy: a prospective, randomized, controlled study. J Clin Anesth. 2020;61:109631. doi:10.1016/j.jclinane.2019.109631
13. Xiong J, Quan J, Qin C, et al. Dexmedetomidine exerts brain-protective effects under cardiopulmonary bypass through inhibiting the janus kinase 2/signal transducers and activators of transcription 3 pathway. J Interferon Cytokine Res. 2020;40(2):116–124.
14. Gong J, Zhang R, Shen L, et al. The brain protective effect of dexmedetomidine during surgery for paediatric patients with congenital heart disease. J Int Med Res. 2019;47(4):1677–1684. doi:10.1177/0300060518821272
15. Oh JE, Jun JH, Hwang HJ, et al. Dexmedetomidine restores autophagy and cardiac dysfunction in rats with streptozotocin-induced diabetes mellitus. Acta Diabetol. 2019;1:105–114.
16. He L, Hao S, Wang Y, et al. Dexmedetomidine preconditioning attenuates ischemia/reperfusion injury in isolated rat hearts with endothelial dysfunction. Biomed Pharmacother. 2019;114:108837. doi:10.1016/j.biopha.2019.108837
17. Riquelme JA, Westermeier F, Hall AR, et al. Dexmedetomidine protects the heart against ischemia-reperfusion injury by an endothelial eNOS/NO dependent mechanism. Pharmacol Res. 2016;103:318–327. doi:10.1016/j.phrs.2015.11.004
18. Liu Y, Wang S, Wang Z, et al. Dexmedetomidine alleviated endoplasmic reticulum stress via inducing ER-phagy in the spinal cord of neuropathic pain model. Front Neurosci. 2020;14(14):90. doi:10.3389/fnins.2020.00090
19. Chai Y, Zhu K, Li C, et al. Dexmedetomidine alleviates cisplatin‑induced acute kidney injury by attenuating endoplasmic reticulum stress‑induced apoptosis via the α2AR/PI3K/AKT pathway. Mol Med Rep. 2020;21(3):1597–1605.
20. Sun D, Wang J, Liu X, et al. Dexmedetomidine attenuates endoplasmic reticulum stress-induced apoptosis and improves neuronal function after traumatic brain injury in mice. Brain Res. 2020;1732(1732):146682. doi:10.1016/j.brainres.2020.146682
21. Zhao L, Zhai M, Yang X, et al. Dexmedetomidine attenuates neuronal injury after spinal cord ischaemia-reperfusion injury by targeting the CNPY2-endoplasmic reticulum stress signalling. J Cell Mol Med. 2019;23(12):8173–8183. doi:10.1111/jcmm.14688
22. Liu C, Fu Q, Mu R, et al. Dexmedetomidine alleviates cerebral ischemia-reperfusion injury by inhibiting endoplasmic reticulum stress dependent apoptosis through the PERK-CHOP-Caspase-11 pathway. Brain Res. 2018;1701(1701):246–254. doi:10.1016/j.brainres.2018.09.007
23. Wang Z, Yang Y, Xiong W, et al. Dexmedetomidine protects H9C2 against hypoxia/reoxygenation injury through miR-208b-3p/Med13/Wnt signaling pathway axis. Biomed Pharmacother. 2020;125:110001. doi:10.1016/j.biopha.2020.110001
24. Yuan M, Meng XW, Ma J, et al. Dexmedetomidine protects H9c2 cardiomyocytes against oxygen-glucose deprivation/reoxygenation-induced intracellular calcium overload and apoptosis through regulating FKBP12.6/RyR2 signaling. Drug Des Devel Ther. 2019;2(13):3137–3149. doi:10.2147/DDDT.S219533
25. Shao Y, Chen X, Liu Y, et al. Dexmedetomidine alleviates lung injury in sepsis mice through regulating P38 MAPK signaling pathway. Panminerva Med. 2020.
26. Liu X, Chen QH, Hu Q, et al. Dexmedetomidine protects intestinal ischemia-reperfusion injury via inhibiting p38 MAPK cascades. Exp Mol Pathol. 2020;23(115):104444. doi:10.1016/j.yexmp.2020.104444
27. Qiu Z, Lu P, Wang K, et al. Dexmedetomidine inhibits neuroinflammation by altering microglial M1/M2 polarization through MAPK/ERK pathway. Neurochem Res. 2019;45(2):345–353. doi:10.1007/s11064-019-02922-1
28. Wang K, Zhu Y. Dexmedetomidine protects against oxygen-glucose deprivation/reoxygenation injury-induced apoptosis via the p38 MAPK/ERK signalling pathway. J Int Med Res. 2018;46(2):675–686. doi:10.1177/0300060517734460
29. Zhang J, Xia F, Zhao H, et al. Dexmedetomidine-induced cardioprotection is mediated by inhibition of high mobility group box-1 and the cholinergic anti-inflammatory pathway in myocardial ischemia-reperfusion injury. PLoS One. 2019;7:e0218726. doi:10.1371/journal.pone.0218726
30. Zhang Y, Monomeric HC. C-reactive protein affects cell injury and apoptosis through activation of p38 MAPK in human coronary artery endothelial cells. Bosn J Basic Med Sci. 2020;30.
31. Li X, Zhang Z, Liang W, et al. Data on Tougu Xiaotong capsules may inhibit p38 MAPK pathway-mediated inflammation in vitro. Data Brief. 2020;19(28):105023. doi:10.1016/j.dib.2019.105023
32. Martinez-Limon A, Joaquin M, Caballero M, et al. The p38 pathway: from biology to cancer therapy. Int J Mol Sci. 2020;21(6):1913. doi:10.3390/ijms21061913
33. Zhao J, Cao J, Yu L, et al. Dehydroepiandrosterone resisted E. Coli O157: H7-induced inflammation via blocking the activation of p38 MAPK and NF-κB pathways in mice. Cytokine. 2020;127:154955. doi:10.1016/j.cyto.2019.154955
34. Wang R, Yang M, Wang M, et al. Total saponins of aralia elata (Miq) seem alleviate calcium homeostasis imbalance and endoplasmic reticulum stress-related apoptosis induced by myocardial ischemia/reperfusion injury. Cell Physiol Biochem. 2018;1:28–40. doi:10.1159/000493954
35. Bi X, Zhang G, Wang X, et al. Endoplasmic reticulum chaperone GRP78 protects heart from ischemia/reperfusion injury through Akt activation. Circ Res. 2018;11:1545–1554. doi:10.1161/CIRCRESAHA.117.312641
36. Li H, Chen H, Li R, et al. Cucurbitacin I induces cancer cell death through the endoplasmic reticulum stress pathway. J Cell Biochem. 2018;11.
37. Huang ZH, Zhang SX, Wang C, et al. Downregulated long non-coding RNA FOXD3-AS1 promotes endoplasmic reticulum stress-induced apoptosis by inhibiting RCN1 via let-7e-5p in nasopharyngeal carcinoma. American journal of physiology. Cell Physiol. 2020.
38. Chen J, Chen J, Cheng Y, et al. Mesenchymal stem cell-derived exosomes protect beta cells against hypoxia-induced apoptosis via miR-21 by alleviating ER stress and inhibiting p38 MAPK phosphorylation. Stem Cell Res Ther. 2020;1:97. doi:10.1186/s13287-020-01610-0
39. Liu XR, Li T, Cao L, et al. Dexmedetomidine attenuates H2O2-induced neonatal rat cardiomyocytes apoptosis through mitochondria- and ER-medicated oxidative stress pathways. Mol Med Rep. 2018;5:7258–7264.
40. Kim SH, Jun JH, Oh JE, et al. Renoprotective effects of dexmedetomidine against ischemia-reperfusion injury in streptozotocin-induced diabetic rats. PLoS One. 2018;8:e0198307. doi:10.1371/journal.pone.0198307
41. Gao JM, Meng XW, Zhang J, et al. Dexmedetomidine protects cardiomyocytes against hypoxia/reoxygenation injury by suppressing TLR4-MyD88-NF- κ B signaling. Biomed Res Int. 2017;1674613.
42. Chaudhari AA, Seol J-W, Lee Y-J, et al. Hypoxia protects articular chondrocytes from thapsigargin-induced apoptosis. Biochem Biophys Res Commun. 2009;381(4):513–517. doi:10.1016/j.bbrc.2009.02.073
43. Price BD, Mannheim-Rodman LA, Brefeldin SKC. A, thapsigargin, and AIF4- stimulate the accumulation of GRP78 mRNA in a cycloheximide dependent manner, whilst induction by hypoxia is independent of protein synthesis. J Cell Physiol. 1992;152(3):545–552. doi:10.1002/jcp.1041520314
44. Kyriakis JM, Avruch J. Mammalian mapk signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi:10.1152/physrev.00028.2011
45. Yan BC, Adachi T, Tsubata T. ER stress is involved in B cell antigen receptor ligation-induced apoptosis. Biochem Biophys Res Commun. 2008;365(1):143–148. doi:10.1016/j.bbrc.2007.10.137
46. Rasheed Z, Haqqi TM. Endoplasmic reticulum stress induces the expression of COX-2 through activation of eIF2α, p38-MAPK and NF-κB in advanced glycation end products stimulated human chondrocytes. Biochim Biophys Acta. 2012;12:2179–2189. doi:10.1016/j.bbamcr.2012.08.021
47. Li YX, Ren YL, Fu HJ, et al. Hepatitis B virus middle protein enhances IL-6 production via p38 MAPK/NF-κB pathways in an ER stress-dependent manner. PLoS One. 2016;7:e0159089. doi:10.1371/journal.pone.0159089
48. Li L, Zhang J, Zhang Q, et al. High glucose suppresses keratinocyte migration through the inhibition of p38 MAPK/autophagy pathway. Front Physiol. 2019;28(10):24. doi:10.3389/fphys.2019.00024
49. Ning J, Zhao C, Chen JX, et al. Oleate inhibits hepatic autophagy through p38 mitogen-activated protein kinase (MAPK). Biochem Biophys Res Commun. 2019;18(514):92–97. doi:10.1016/j.bbrc.2019.04.073
50. Qian Z, Chang J, Jiang F, et al. Excess administration of miR-340-5p ameliorates spinal cord injury-induced neuroinflammation and apoptosis by modulating the P38-MAPK signaling pathway. Brain Behav Immun. 2020;31:S0889–S1591.
51. Kumphune S, Surinkaew S, Chattipakorn SC, et al. Inhibition of p38 MAPK activation protects cardiac mitochondria from ischemia/reperfusion injury. Pharm Biol. 2015;53(12):1831–1841. doi:10.3109/13880209.2015.1014569
52. Bonney EA. Mapping out p38MAPK. Am J Reprod Immunol. 2017;77(5):e12652. doi:10.1111/aji.12652
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.