")
Back to Journals » Journal of Inflammation Research » Volume 15
Dexmedetomidine Activates Akt, STAT6 and IRF4 Modulating Cytoprotection and Macrophage Anti-Inflammatory Phenotype Against Acute Lung Injury in vivo and in vitro
Authors Chen Q , Qin Z, Sun Y, Liu X, Pac Soo A , Chang E, Sun Q, Yi B, Wang DX , Zhao H, Ma D , Gu J
Received 4 January 2022
Accepted for publication 13 April 2022
Published 26 April 2022 Volume 2022:15 Pages 2707—2720
DOI https://doi.org/10.2147/JIR.S357012
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Qian Chen,1,2,* Zhigang Qin,1,* Yibing Sun,3 Xiangfeng Liu,1 Aurelie Pac Soo,2 Enqiang Chang,2 Qizhe Sun,2 Bin Yi,1 Dong-Xin Wang,3 Hailin Zhao,2 Daqing Ma,2 Jianteng Gu1
1Department of Anaesthesiology, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, People’s Republic of China; 2Division of Anaesthetics, Pain Medicine and Intensive Care, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, Chelsea & Westminster Hospital, London, UK; 3Department of Anaesthesiology and Critical Care Medicine, Peking University First Hospital, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianteng Gu, Department of Anaesthesiology, Southwest Hospital, Third Military Medical University, 30 Gaotanyan Road, Chongqing, People’s Republic of China, Tel +86 23 68765366, Fax +86 2365463270, Email [email protected] Daqing Ma, Division of Anaesthetics, Pain Medicine and Intensive Care, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, Chelsea and Westminster Hospital, London, UK, Tel +44 020 3315 8495, Fax +44 020 3315 5109, Email [email protected]
Purpose: This study aims to investigate the cytoprotective and anti-inflammatory effects of an α2-adrenoreceptor (α2-AR) agonist, dexmedetomidine (Dex), on lipopolysaccharides (LPS)-induced acute lung injury and underlying mechanisms with focus on alveolar macrophage polarization modulation.
Methods: C57BL/6 mice were intraperitoneally injected LPS (10 mg/kg) with or without Dex (25 μg/kg) and/or α2-AR antagonist atipamezole (Atip, 500 μg/kg). Lung tissues were then analysed to determine injuries. In vitro, human pulmonary epithelial cells (A549) and mice alveolar macrophages (MH-S) were exposed to LPS (10 ng/mL) with or without different concentrations of Dex (0.1– 100 nM). Alveolar macrophage polarization, NLRP3 inflammasome activation and inflammatory responses were determined. PTEN/Akt signaling and its downstream transcriptional factors as targets for macrophage polarization were assessed.
Results: Dex treatment significantly reduced pro-inflammatory M1 macrophage polarization and NLRP3 inflammasome activation in the lungs relative to the mice treated with LPS. The similar pattern reduction of NLRP3 inflammasome activation by Dex was also found in A549 cells. Atip partly reversed the anti-inflammatory effects of Dex. In cultured alveolar macrophages, Dex reduced LPS-mediated expression of IL-1, − 6 and TNF-α receptors while promoting alveolar macrophages differentiation towards a M2 anti-inflammatory phenotype. Additionally, LPS increased Akt signaling activation in a time-dependent manner, which was further activated by Dex via inhibiting phosphatase and tensin homolog (PTEN). The action of Dex on Akt signaling shifted alveolar macrophages from M1 to M2 phenotype through increasing STAT6 and IRF4 transcriptional factors.
Conclusion: Dex protected against LPS-induced lung injury and suppressed LPS-induced pulmonary inflammatory responses by attenuating the NLRP3 inflammasome activation and promoting anti-inflammatory M2 macrophage polarization.
Keywords: sepsis, acute lung injury, macrophage polarization, dexmedetomidine, Akt signaling
Introduction
Sepsis is characterized by an aberrant and excessive inflammatory immune response that causes life-threatening organ dysfunction.1 The incidence and mortality of sepsis impose great challenges on global healthcare services, as indicated by an annual global incidence of 31.5 million cases and 5.3 million deaths.2 The lungs are the most affected organ during sepsis, and sepsis-induced acute lung injury (ALI) may progress to acute respiratory distress syndrome (ARDS), resulting in significant mortality.3–5 Dexmedetomidine (Dex) is a highly selective α2-adrenoreceptor (α2-AR) agonist that has potent organoprotective and anti-inflammatory effects that would be favorable in the context of ALI.6,7 We previously reported that Dex reduced pulmonary endothelial apoptosis and decreased endothelium barrier hyper-permeability via α2-AR activation during ALI.8,9 However, the cellular and molecular mechanisms regarding the immunomodulation of Dex affording cytoprotection and anti-inflammation in the lungs remain unknown.
Monocyte/macrophage phenotype changes determine the severity of organ injury during sepsis.10,11 At the early stage of sepsis-related ALI, alveolar macrophages are activated and then polarize to be pro-inflammatory M1 phenotype. M1 macrophages-induced lung tissue damage directly through excessive release of toxic species12 and also indirectly exacerbates tissue inflammation through the recruitment of neutrophils and TH 17 cells to the lungs.13 At the end stage of inflammation, the overall macrophage population transitions to M2 anti-inflammatory phenotype that participates in tissue repair and cell proliferation through destroying pathogens, removing cellular debris and forming blood vessels.14,15 Evidence has suggested that macrophages represent a spectrum of activated states rather than stable phenotypes, and the original polarization is easily reversible upon environmental changes.16,17 Therefore, the development of adequate anti-inflammatory strategies, such as increasing the M2/M1 ratio, is necessary to suppress lung inflammation and to promote survival of patients with sepsis. This study, therefore, aims to determine whether Dex provides protection to the lungs by modulating alveolar macrophage polarization in response to lipopolysaccharides (LPS) challenge in vivo and in vitro.
Materials and Methods
Animals and Drug Administration
Inbred C57BL/6 adult male mice (20–25 g) were purchased from the Experimental Animal Centre of Peking University (Beijing, China). All animal experimental procedures were approved by the Peking University experimental animal welfare subcommittee (Approval No. J201807) and were performed in adherence to the International Guidelines for Animal Experiment18 and followed the ARRIVE guidelines.19 The sepsis model was established accordingly.20 Briefly, mice were intraperitoneally injected with 10 mg/kg LPS (L2630, Sigma-Aldrich). And, 25 µg/kg Dex (SML0956, Sigma-Aldrich) with/without 500 µg/kg Atip (α2-adrenergic antagonist, A9611, Sigma-Aldrich) were then intraperitoneally injected every 2 hrs for 3 times immediately after LPS administration. Controls received comparable volume injections of saline. At 24 hrs after the treatment, animals were sacrificed with a lethal dose of pentobarbitone (P0500000, Sigma-Aldrich).
Haematoxylin and Eosin (HE) Staining
Lung sections (5 μm) were stained with hematoxylin/eosin, and 10 fields/animal were randomly chosen to assess lung morphology under an Olympus (Watford, UK) BX4 microscope. The severity of lung injury was blindly assessed with a semi-quantitative scale system from 0 to 3 grades: nil (0), mild (1), moderate (2), or severe (3) injury based on the presence of exudates, hyperemia or congestion, infiltration of neutrophils, alveolar hemorrhage, presence of debris, and cellular hyperplasia.21,22
In vitro Cell Culture and Treatment
Human pulmonary epithelial cells (A549 cell line) and mice alveolar macrophages (MH-S cell line) were purchased from the European Collection of Authenticated Cell Culture (ECACC, Salisbury, UK). The cells were incubated with 1 µM Atip for 3 hrs, then exposed simultaneously to 10 ng/mL LPS or/and 0.1–100 nM Dex for another 24 hrs.7 The activation of NLRP3 inflammasome complex and endoplasmic reticulum (ER) stress in A549 cells were assessed by immunofluorescence; macrophage polarization and inflammatory responses were assessed by light microscope,23 Western blot, immunofluorescence, or flow cytometry. To determine the activation of PTEN/Akt pathway, macrophages were separately stimulated with 10 ng/mL LPS or 100 nM Dex for 3 to 60 min, then assessed by Western Blot. Additionally, 100 nM bpV (SML0885, Sigma-Aldrich) was given to macrophages for 1 hr prior to Dex or LPS administration, macrophage polarization and its associated transcription factors, transcription factor signal transducer and activator of transcription 6 (STAT6) and interferon regulatory factor 4 (IRF4), were detected after 24 hrs using Western Blot.
Intracellular Staining and Flow Cytometry
After being washed with PBS, the cells were fixed with fixation buffer and incubated with permeabilization buffer (BD Bioscience) and stained with the following antibodies: anti-HMGB1 and anti-TNF-α antibodies. For cell surface receptor experiments, fixed MH-S cells were directly incubated with the following antibodies: anti-CD206, anti-CD86, anti-CD121a, anti-CD126, and anti-CD54 antibodies (BD Life Sciences) for 30 min at 37°C. Flow Cytometry was performed on a FACS Calibur (BD Life Sciences, UK).
Immunofluorescence
For in vivo double fluorescent staining, 25-µm paraffin-embedded lung sections were first dewaxed and then subjected to heat-mediated antigen retrieval. The sections were incubated in 4% normal donkey serum in PBS-T for 1 hr, then incubated with the mouse anti-iNOS (1:200, Abcam), and rabbit anti-caspase-1 (1:200, Abcam) antibodies at 4°C overnight. Followed by the second primary goat anti-NLRP3 (1:200, Invitrogen) and goat anti-ASC (apoptosis-associated speck-like protein containing a CARD, 1:200, Abcam) antibodies at 4°C overnight. For in vitro fluorescent staining, cells were incubated with mouse anti-GRP78 (1:100, Santa Cruz) or rabbit anti-PTEN (1:100, Abcam) antibodies at 4°C overnight. For in vitro double-labelled immunofluorescence, cell samples were incubated with the first primary mouse anti-iNOS (1:200), goat anti-NLRP3 (1:200), and goat anti-ASC (1:200) antibodies at 4°C overnight, and then the second primary rabbit anti-Arg1 (1:200) and anti-caspase-1 (1:100) antibodies. All tissue slices or cells were then incubated with the second secondary antibodies for another 1 hr, then were counterstained with nuclear dye DAPI and mounted with VECTASHIELD Mounting Medium (Vector lab, USA), and then examined using a Zeiss Axio Observer 7 microscope (Zeiss, UK) under constant exposure level. Immunofluorescence staining was quantified using ImageJ (National Institutes of Health). Ten representative regions per section (in vivo) or field (in vitro) were randomly selected by an assessor blinded to the treatment groups. Values were then calculated as percentages of the mean value for naive controls and expressed as the percentage of fluorescence intensity.
Western Blot Analysis
The treated cells were mechanically homogenized and centrifuged at 3000 g for 30 min at 4°C to collect supernatant. The protein extracts were denatured and were heated for 10 min at 98°C. The samples (40 μg/sample) were then loaded on a NuPAGE 4–12% Bis-Tris gel (Invitrogen) for electrophoresis and transferred to a PVDF membrane. The membrane was incubated with blocking solution for 1 hr and probed with the following primary antibodies: anti-α2-AR (Antibodies.com), anti-iNOS, anti-Arg1, anti-Ym1 (Abcam), anti-PPAR (this antibody and below were all from Cell Signaling Technology), anti-Akt, anti-p-Akt (Ser473), anti-p-PTEN (Ser380), anti-PTEN, anti-p-GSK-3β (Ser9), anti-p-PDK (Ser241), anti-STAT6, and anti-IRF-4 in 1:1000 dilution overnight at 4°C, followed by HRP-conjugated secondary antibody for 1 hr. The loading control was probed with anti-GADPH antibody (1:10,000, Sigma-Aldrich). The membrane was washed with TBST and visualized with an enhanced chemiluminescence (ECL) system (Santa Cruz, UK). The protein bands were captured with an image processor (GeneSnap; SYNGENE®, Cambridge, UK).
Statistical Analysis
Data were expressed as mean ± standard deviation (SD) and presented as a histogram or dot plot. All numerical data were analysed with analysis of variance (ANOVA) followed by Kruskal–Wallis non-parametric test (lung injury scoring and macrophage numbers) or Bonferroni’s post hoc test (measurement) for comparisons where appropriate. The activation of PTEN/Akt signaling was analysed with two-way ANOVA followed by Bonferroni’s multiple comparison test. All statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, US). A p value less than 0.05 was considered to be of statistical significance.
Results
Dex Treatment Reduced ALI and NLRP3 Inflammasome in the Lungs After LPS Challenge
Intraperitoneal injection with LPS resulted in severe lung injury (Figure 1A), as evidenced by lung alveolar damage, massive leukocyte infiltration, and an increase in lung injury score (3.14 ± 0.36, p < 0.0001; Figure 1B) compared with the control (0.67 ± 0.47). Dex treatment significantly reduced LPS-induced leukocyte infiltration and lung injury score (2.67 ± 0.66, p < 0.05), while Atip reversed the protective effect of Dex (3.19 ± 0.68, p < 0.05). To evaluate the activation of NLRP3 inflammasome complex (Figure 1C), the data showed that LPS increased the expression of ASC (p < 0.0001; Figure 1D), caspase-1 (p < 0.0001; Figure 1E), and NLRP3 (p < 0.001; Figure 1F) in the lungs. Dex treatment significantly downregulated the expression of ASC, caspase-1, and NLRP3, relative to control LPS treatment (p < 0.05, respectively). Atip reversed the effect of Dex by increasing the expression of ASC and NLRP3 (p < 0.05, respectively), but failed to reverse the caspase-1 reduction. Additionally, iNOS staining was used to evaluate the number of M1 macrophages (Figure 1G). The result showed that LPS administration promoted macrophage polarized to M1 phenotype by increasing the number of inducible nitric oxide synthase (iNOS) positive macrophages (4.50 ± 1.84/field, p < 0.001) compared with the control (0.50 ± 0.23/field). Dex treatment significantly reduced the number of iNOS positive macrophages (2.60 ± 1.35/field, p < 0.05), while Atip failed to reverse the effect of Dex (Figure 1H).
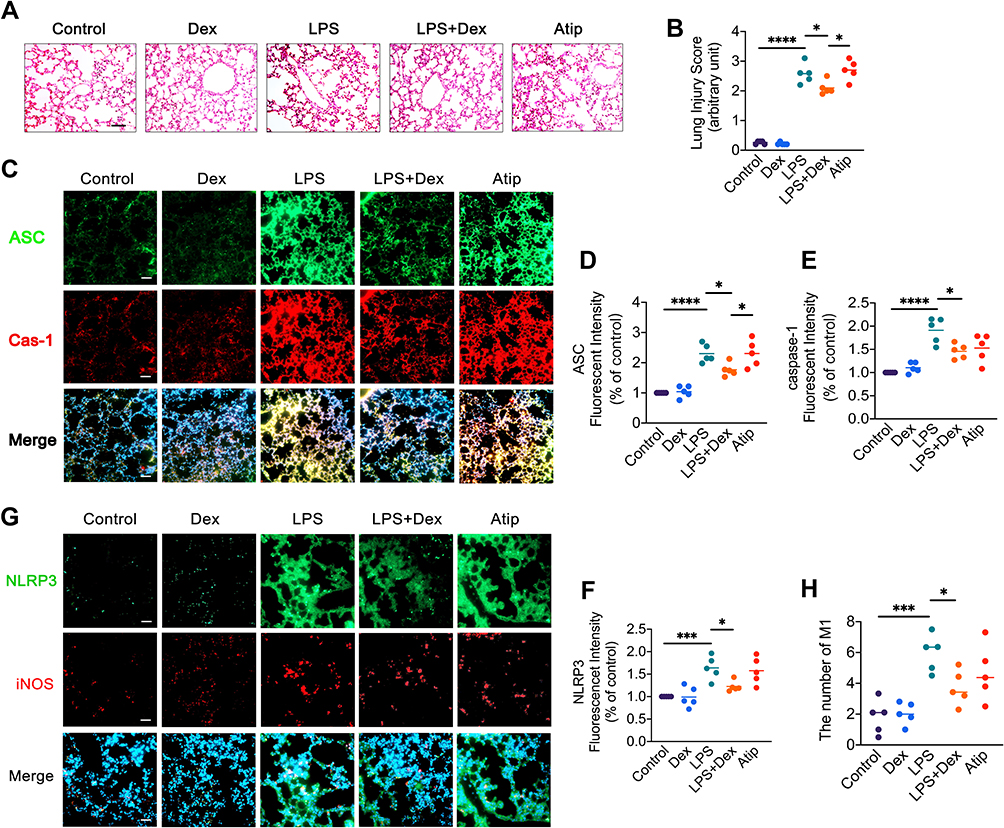 |
Figure 1 Dex treatment reduced ALI and NLRP3 inflammasome in the lungs after LPS challenge. C57BL/6 adult male mice were injected intraperitoneally with 10 mg/kg LPS, followed 25 µg/kg Dex with or without 500 µg/kg Atip every 2 hrs for 3 times. Lung injury was evaluated after 24 hrs of the treatment. Normal lung tissues served as the control. (A) Histology (H&E staining) of the lung tissues; (B) Lung morphology was evaluated by a lung injury scoring system; (C) NLRP3 inflammasome activation was detected by immunofluorescence; Fluorescent intensity of (D) ASC, (E) caspase-1, and (F) NLRP3 in lung tissue; (G) NLRP3 expression and iNOS intracellular staining of M1 macrophages were detected by immunofluorescence; (H) Number of M1 macrophages. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Scale bar: 50 μm. Data were presented as scatter plot with mean (n = 5); *p < 0.05, ***p < 0.001 and ****p < 0.0001. |
Dex Treatment Suppressed the Formation of NLRP3 Inflammasome and ER Stress Induced by LPS in A549 Cells
LPS stimuli promoted the formation of NLRP3 inflammasome complex (Figure 2A) in A549 cells by significantly increasing the expression of ASC (Figure 2B), caspase-1 (Figure 2C), and NLRP3 (Figure 2D) (p < 0.001, respectively) compared with those in the control group. Dex treatment down-regulated the expression of ASC (p < 0.05), caspase-1 (p < 0.05), and NLRP3 (p < 0.01) relative to LPS stimuli. Atip partially reversed this protective effect of Dex by increasing the expression of NLRP3 and ASC (p < 0.05, respectively). To further explore the protective effects of Dex in lung epithelial cells, an ER intraluminal protein (Grp78) was stained to evaluate ER stress (Figure 2E). The results (Figure 2F) demonstrated that compared with the control group, Grp78 expression significantly increased after LPS stimuli (p < 0.01), while Dex blocked the increasing of Grp78 (p < 0.05), and Atip reversed the effect of Dex (p < 0.05).
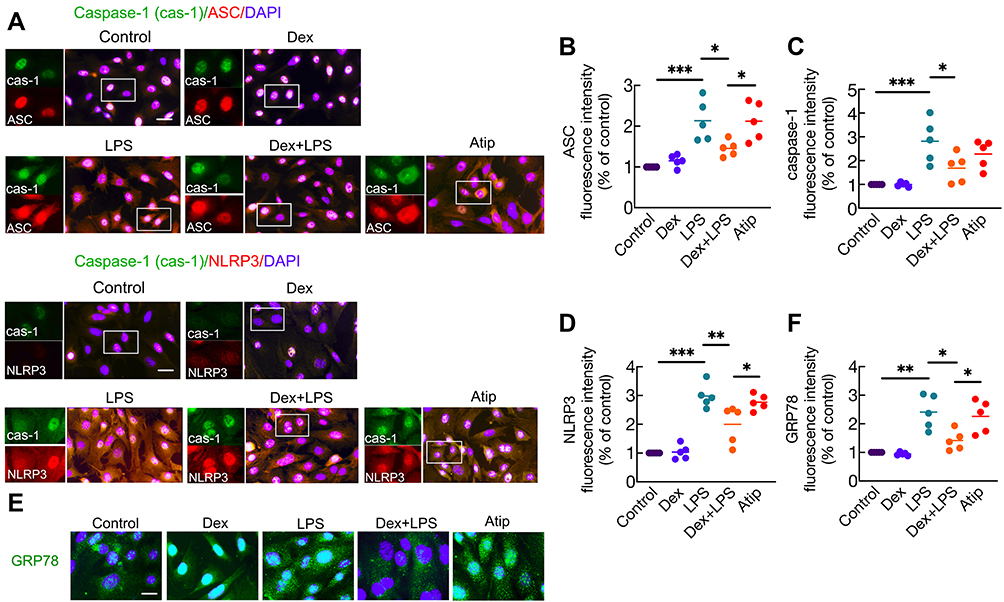 |
Figure 2 Dex treatment suppressed the formation of NLRP3 inflammasome and ER stress induced by LPS in A549 cells. Human pulmonary epithelial cells (A549 cell line) were incubated with 1 µM Atip for 3 hrs, then exposed simultaneously to 10 ng/mL LPS or/and 100 nM Dex for 24 hrs. Normal A549 cells served as the control. (A) inflammasome complex – NLRP3, ASC, and caspase-1 were assessed by immunofluorescence. Fluorescent intensity of (B) ASC (C) caspase-1 and (D) NLRP3 in A549 cells. (E) Endoplasmic reticulum (ER) stress was evaluated by labelling cells with Grp78. (F) Fluorescent intensity of Grp78. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Scale bar: 10 μm. Data were presented as scatter plot with mean (n = 5), *p < 0.05, **p < 0.01 and ***p < 0.001. |
Dex Treatment Suppressed Inflammatory Responses Induced by LPS in Alveolar Macrophages
Flow cytometry was used to assess LPS-induced inflammatory responses and the anti-inflammatory property of Dex (Figure 3). The results showed that LPS stimuli significantly increased the expression of inflammatory receptors CD121a (IL-1R) (p < 0.001, Figure 3A) and CD126 (IL-6R) (p < 0.001, Figure 3B) compared with the control group. Dex treatment reduced the expression of CD121a and CD126 (p < 0.05, respectively), while Atip reversed the effect of Dex by increasing the expression of CD126 (p < 0.01), but failed to reverse the CD121a reduction. In addition, LPS stimuli also increased the content of intracellular TNF-α (p < 0.001; Figure 3C), CD54 (ICAM1) (p < 0.01; Figure 3D), and HMGB1 (p < 0.001; Figure 3E), respectively. Dex treatment reduced the increase of TNF-α and CD54 (p < 0.05, respectively) but failed to block the increase of HMGB1. Atip reversed the effect of Dex by increasing intracellular CD54 (p < 0.05).
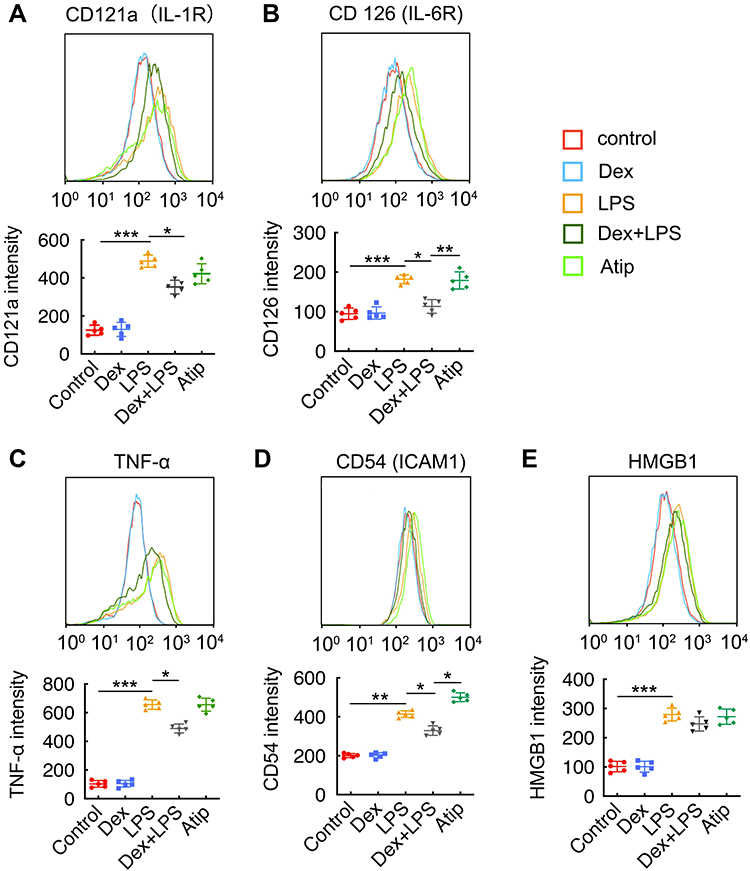 |
Figure 3 Dex treatment suppressed inflammatory response induced by LPS in alveolar macrophages. Mice alveolar macrophage cells (MH-S cell line) were incubated with 1 µM Atip for 3 hrs, then exposed simultaneously to 10 ng/mL LPS or/and 100 nM Dex for 24 hrs. Flow cytometry analysis of (A) CD121 (IL-1R), (B) CD126 (IL-6R), (C) TNF-α, (D) CD54 (ICAM1), (E) HMGB1. Data were presented as scatter plot with mean ± SD (n = 5). *p < 0.05; **p < 0.01; ***p < 0.01. |
Dex Promoted Macrophages Polarized to M2 Phenotype Under LPS Stimuli
Western Blot was used to determine the expression of α2-AR on MH-S cells following incubation with 10 ng/mL LPS or 100 nM Dex for 24 hrs (Figure 4A). The expression of α2-AR was not significantly different between the control group and the LPS or Dex group (Figure 4B). The morphology of the alveolar macrophages was assessed by light microscope. More round and small M1 polarized macrophages were noticed in the LPS group, while more adherent spindle-shaped M2 polarized macrophages were found in the Dex group (Figure 4C). Meanwhile, flow cytometry was used to determine the effect of different concentrations of LPS and Dex on macrophage polarization (Figure 4D). M1 macrophages were identified as CD206−CD86+ and M2 macrophages as CD206+CD86−. The results showed that, after 24 hrs, the percentage of M2 macrophages increased from 17.8 ± 5.54% to 31.2 ± 4.97% (p < 0.01) when treated with 100 nM Dex alone, whereas this percentage increased from 8.92 ± 3.72% to 22.8 ± 3.11% (p < 0.05) when treated with 100 nM Dex combined with 10 ng/mL LPS (Figure 4E). Moreover, 100 nM Dex also significantly decreased the percentage of M1 macrophages from 52.2 ± 4.67% to 32.2 ± 3.98 (p < 0.01) in the presence of 10 ng/mL LPS (Figure 4F). In addition, several M2 macrophage biomarkers were used to further evaluate the effect of Dex on macrophage polarization (Figure 4G). The results demonstrated that Dex treatment promoted macrophages polarized to M2 phenotype by increasing the expression of Ym1 (p < 0.05 at 1 and 10 nM, p < 0.001 at 100 nM; Figure 4H), Arg1 (p < 0.01 at 10 and 100 nM; Figure 4I), and PPARγ (p < 0.01 at 10 nM, p < 0.001 at 100 nM; Figure 4J) under LPS stimuli.
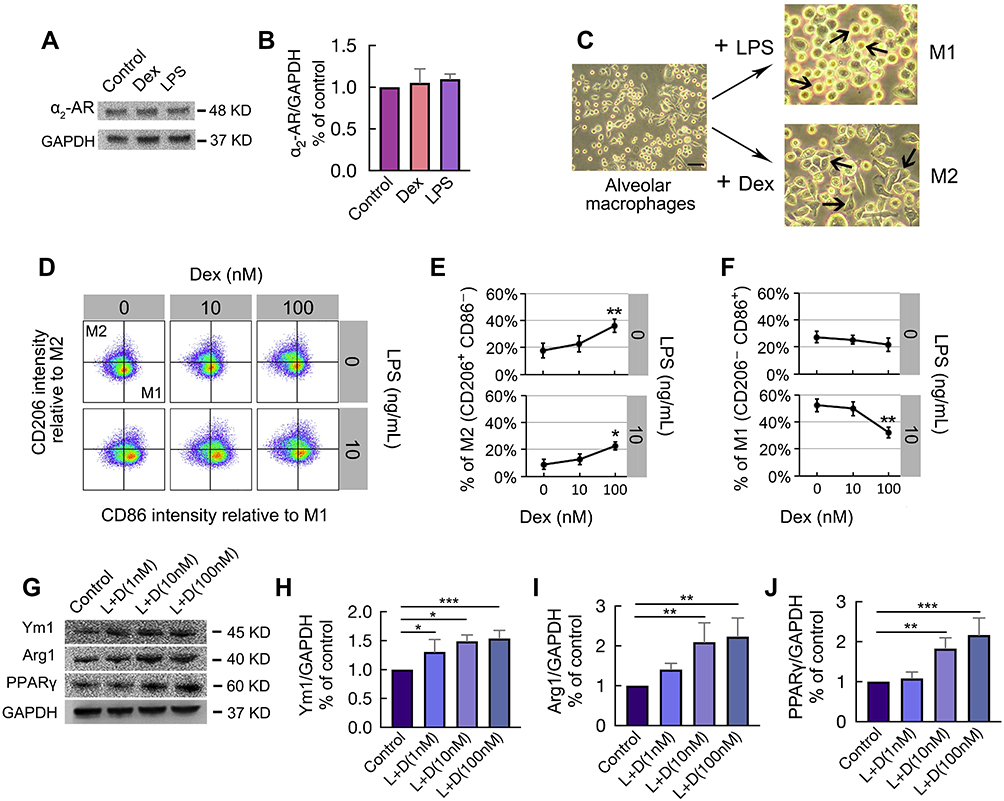 |
Figure 4 Dex promoted macrophages polarized to M2 phenotype under LPS stimuli. MH-S cells were stimulated with 10 ng/mL LPS or 100 nM Dex for 24 hrs. The expression of (A) α2-AR was assessed by Western Blot, GAPDH as a loading control; Western Blot analysis showing the expression of (B) α2-AR. (n = 3); (C) Microscope observation on MH-S cells polarization. Populations of globular M1 polarized macrophages (M1) and adherent spindle-shaped M2 polarized macrophages (M2) were obtained. (D) Pseudocolor of normalized CD86 (M1 marker) versus CD206 (M2 marker) staining intensity of MH-S cells subjected 10 ng/mL LPS with/without different concentrations of Dex (10, 100 nM) for 24 hrs, assessed by flow cytometry. (E) Percentage of M2 subtype (CD206+CD86−) and (F) Percentage of M1 subtype (CD206−CD86+) (n = 5). (G) The expression of M2 markers Ym1, Arg1, and PPARγ were assessed by Western Blot after being stimulated with 10 ng/mL LPS in combination with different concentrations of Dex (1, 10, 100 nM) for 24 hrs. Western Blot analysis of (H) Ym1, (I) Arg1, and (J) PPARγ (n = 3). Scale bar: 50 μm. Data were presented as mean ± SD. *p < 0.05; **p < 0.01, ***p < 0.001. |
Dex Promoted M2 Polarization Through Activating α2-AR
M1 and M2 macrophages were determined by the presence of their respective markers iNOS and Arginase-1 (Arg-1) following stimulation with LPS with or without Dex and Atip (Figure 5A). The results demonstrated that LPS significantly promoted M1 macrophage polarization by increasing the expression of iNOS (p < 0.001, Figure 5B) and decreasing the expression of Arg-1 (p < 0.001, Figure 5C) when compared to the control group. Dex treatment down-regulated M1 polarization as evidenced by decreasing iNOS expression and increasing Arg-1 expression (p < 0.05, respectively). Atip significantly blocked the effect of Dex. Immunofluorescence staining of MH-S for iNOS and Arg-1 was also conducted to assess the presence of M1 and M2 (Figure 5D). The red arrows point towards round shaped M1 macrophages and the green arrows indicate M2 macrophages with a more elongated features.
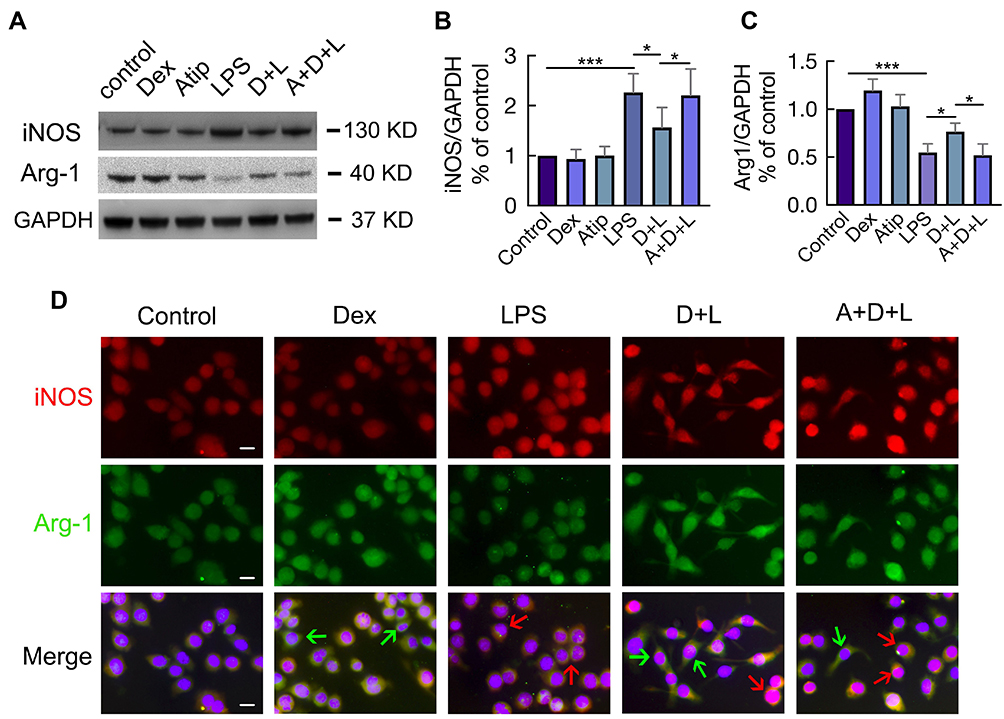 |
Figure 5 Dex promoted M2 polarization through activating α2-AR. MH-S cells incubated with 1 µM Atip for 3 hrs, then exposed simultaneously to 10 ng/mL LPS or/and 100 nM Dex for another 24 hrs. (A) Western Blot was used to detect the expression of M1 marker iNOS and M2 marker Arg-1, GAPDH as a loading control. Western Blot analysis of the expression of (B) iNOS and (C) Arg1. Data were presented as mean ± SD (n = 4). *p < 0.05, ***p < 0.001. (D) Immunofluorescence shows the expression of both iNOS (red) and Arg1 (green) in individual cell. Red arrows indicate enhanced iNOS expression representing M1 macrophages, while green arrows indicate enhanced Arg1 expression representing M2 macrophages. Scale bar: 10 μm. |
Dex Enhanced the Activation of Akt Signaling by Inhibiting PTEN Phosphorylation
Following stimulation with LPS or Dex, MH-S cells were analysed for the activation of PTEN and Akt signaling using Western Blot (Figure 6A). We found that LPS significantly up-regulated PTEN phosphorylation in a time-dependent manner with a peak between 9 and 15 min; on the other hand, PTEN phosphorylation was inhibited by the Dex treatment when compared with the LPS group (Figure 6B). Unexpectedly, both LPS and Dex upregulated the activation of Akt signaling by increasing the expression of p-Akt (Figure 6C), p-GSK3β (Figure 6D), and p-PDK1 (Figure 6E) over time and peak at 12–30 min, respectively. Of note, the activation of Akt signaling in macrophages treated with Dex was much higher than that of cells treated with LPS alone. Therefore, we assessed whether PTEN is an upstream regulator in modulating the activation of Akt signaling (Figure 6F and G). We found that PTEN inhibitor bpV blocked PTEN phosphorylation induced by LPS stimuli (p < 0.0001, Figure 6H); meanwhile, an increase in Akt phosphorylation was observed after PTEN phosphorylation was inhibited (p < 0.01, Figure 6I), demonstrating PTEN is a negative regulator of Akt signal transduction that Dex may improve the level of Akt signaling activation through inhibiting PTEN.
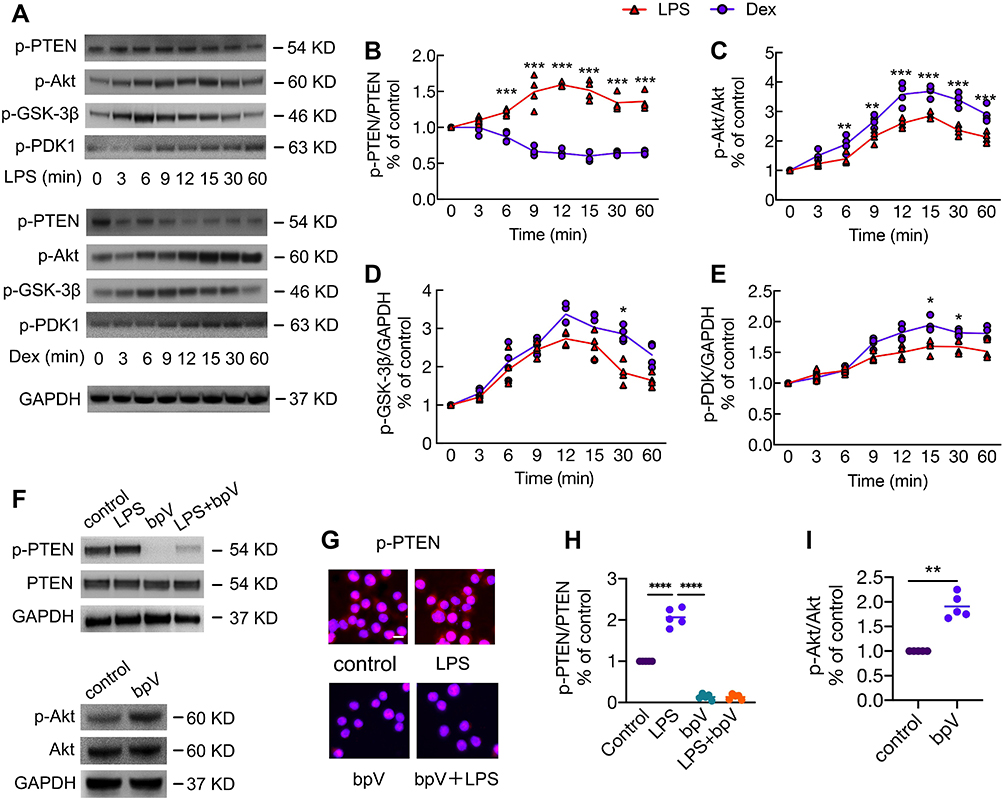 |
Figure 6 Dex enhanced the activation of Akt signaling by inhibiting PTEN phosphorylation. MH-S cells were stimulated with 10 ng/mL LPS or 100 nM Dex for 3–60 min. (A) Western Blot was performed to detect the phosphorylation of PTEN, Akt, GSK-3β, and PDK1. GAPDH as a loading control. Western Blot analysis of the expression of (B) p-PTEN, (C) p-Akt, (D) p-GSK-3β, and (E) p-PDK1. (n = 3). MH-S cells were cultured with or without the PTEN inhibitor bpV (Pic) 100 nM for 1 hr, followed by the stimulation of LPS 10 ng/mL for another 10 min; or cells were stimulated with bpV (Pic) 100 nM alone for 1 hr. (F) Western Blot and (G) Immunofluorescence was used to analyse the phosphorylation of PTEN and Akt. Western Blot analysis of the expression of (H) p-PTEN and (I) p-Akt. Scale bar: 10 μm. Data were presented as scatter plot with mean (n = 5). *p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Dex Promoted M2 Polarization via Inhibiting PTEN Phosphorylation
We then proceed to determine whether PTEN participates in macrophage polarization via modulating Akt signalling. We found that LPS significantly promoted M1 polarization by increasing the expression of iNOS (p < 0.001; Figure 7A and B) and decreasing the expression of Arg-1 (p < 0.001; Figure 7A and C). The inhibition of PTEN down-regulated M1 polarization, as evidenced by the fact that bpV treatment decreased iNOS expression (bpV vs control, p < 0.01; bpV+LPS vs LPS, p < 0.0001) and increased Arg-1 expression (bpV vs control, p < 0.01; bpV+LPS vs LPS, p < 0.0001) with or without LPS stimulation. We then analysed MH-S cells for the expression of transcriptional proteins STAT6 and IRF-4 (Figure 7D), both of which are involved in the pathways leading to M2 polarization. The results (Figure 7E) showed that STAT6 and IRF-4 were significantly down-regulated after LPS stimuli (p < 0.05 and p < 0.01, respectively), but significantly up-regulated after treatment with bpV (p < 0.05 and p < 0.0001, respectively) or bpV combined with LPS (p < 0.05 and p < 0.0001, respectively). Meanwhile, Dex treatment also significantly increased the expression of M2 transcriptional factors STAT6 and IRF-4 (p < 0.01 and p < 0.0001, respectively; Figure 7D and F).
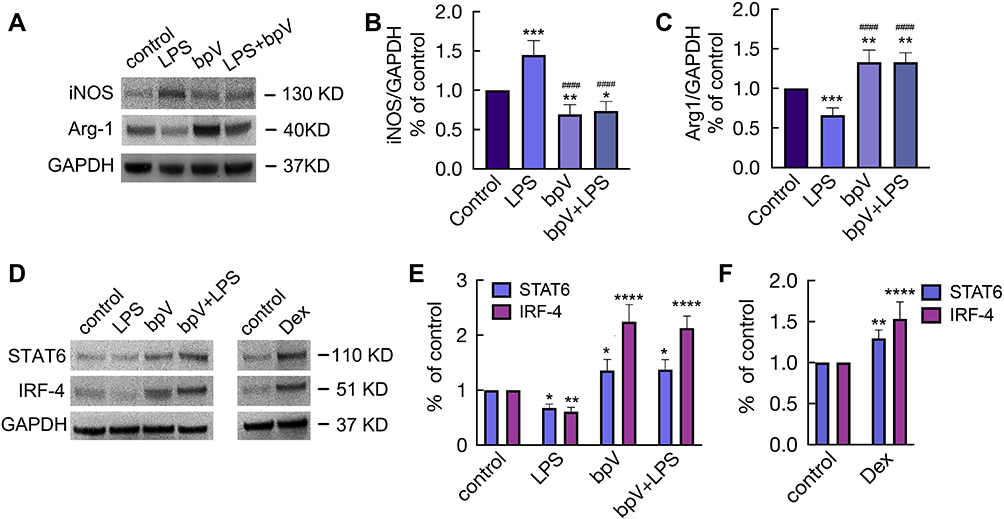 |
Figure 7 Dex promoted M2 polarization via inhibiting PTEN phosphorylation. MH-S cells were cultured with LPS 10 ng/mL or bpV (Pic) 100 nM for 24 hrs or stimulated with bpV (pic) 100 nM for 1 hr, followed by the stimulation of LPS 10 ng/mL for another 24 hrs. (A) Western Blot was used to detect the expression of iNOS and Arg1, GAPDH as a loading control. Western Blot analysis of the expression of (B) iNOS and (C) Arg1. (D) Western Blot was used to detect the expression of STAT6 and IRF4 after the cells were cultured with (E) 10 ng/mL LPS or/and 100 nM bpV(pic), or (F) cultured with 100 nM Dex alone for 24 hrs, GAPDH as a loading control. Data were presented as mean ± SD (n = 4). *p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001 vs control. ####p < 0.0001 vs LPS group. |
Discussion
Our data showed that LPS increased the activation of NLRP3 inflammasome in pulmonary epithelium, and promoted polarization of alveolar macrophages to M1 pro-inflammatory phenotype. Dex treatment reduced LPS-mediated NLRP3 inflammasome activation, and promoted the alternative activation of alveolar macrophages polarized to M2 anti-inflammatory phenotype, therefore suppressing inflammatory responses in the lungs as well as protecting against lung injury. Furthermore, we identified PTEN as a potent counter-regulator of macrophage polarization through modulating the level of Akt activation, as evidenced by the fact that inhibiting PTEN enhanced the activation of Akt signaling subsequently promoting M2 polarization. Dex treatment may be a promising lung protectant, which is able to alleviate acute lung injury induced by endotoxin (Figure 8).
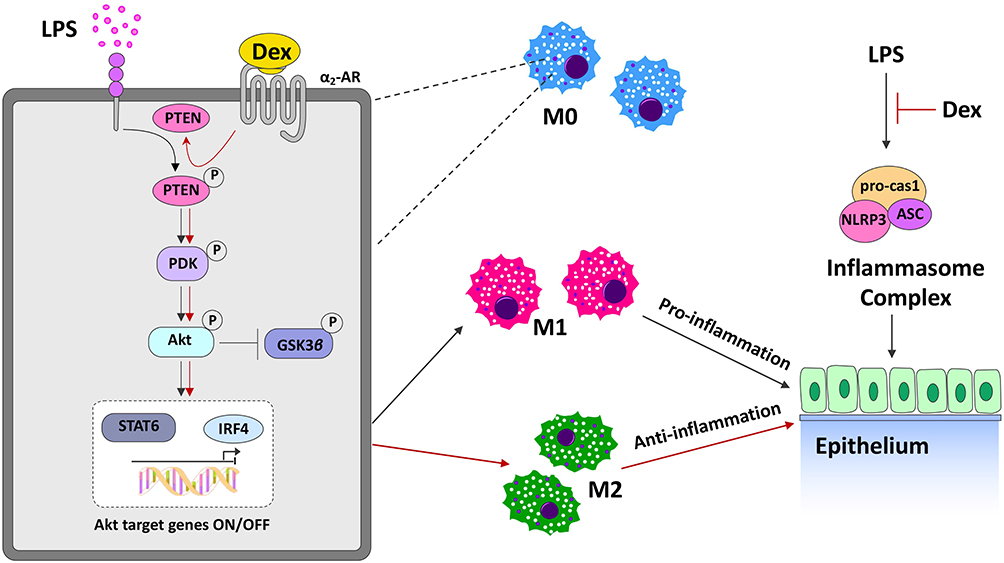 |
Figure 8 The putative molecular mechanisms of Dex mediated protection against LPS-induced lung injury. LPS stimulation enhances the NLRP3, ASC, and caspase-1 inflammasome complex activation in epithelium, and promotes alveolar macrophages polarize to M1 pro-inflammatory phenotype, leading to acute lung injury. Dex treatment ameliorates LPS-mediated NLRP3 inflammasome activation and promotes the differentiation of alveolar macrophages towards M2 anti-inflammatory phenotype, resulting in the reduction of pulmonary inflammation. Alveolar macrophages respond to LPS with pro-inflammatory changes that increase the activation of Akt signaling by promoting the phosphorylation of PTEN, PDK1 and Akt. α2-AR activation induced by Dex further activating Akt signaling by inhibiting PTEN phosphorylation, as PTEN is a negative regulator of Akt signal transduction. High levels of activated Akt shift alveolar macrophages from the inflammatory M1 phenotype toward the anti-inflammatory M2 phenotype through increasing M2 transcriptional factors STAT6 and ARF4, and thereby suppressing the pulmonary inflammatory response. All of these protective effects could ameliorate lung damage associated with LPS stimulation. |
M1 macrophages have a significant role in sustaining acute inflammation, partially due to secretion of pro-inflammatory cytokines that accelerate the acute phase pathology of infection characterized by leukocytosis, vascular hyperpermeability, and pulmonary edema.24–26 On the other hand, M2 macrophages play an important role in resolving inflammation and tissue repair in late stage ALI by enhancing pulmonary microvascular endothelial cell (PMVEC) regeneration, decreasing lung microvascular hyper-permeability, and reducing lung edema after LPS stimulation.27 Here, we demonstrated that Dex treatment alone or LPS and Dex combination decreased M1 macrophages and increased M2 macrophages, suggesting that Dex promoted macrophages polarized to M2 phenotype both under the physiological and pathological conditions. Furthermore, in line with previous studies,28,29 our data also suggested that Dex promoted macrophage M2 phenotype changes through the transcription of STAT6 and IRF4, which may subsequently reduce the release of pro-inflammatory factors. In fact, a previous study similarly suggested that Dex regulates 6-hydroxydopamine-induced microglial polarization by promoting the M2 phenotype and subsequently interrupting the process of microgliosis.30 Therefore, it may be concluded that Dex promotes polarization towards the M2 phenotype and this may be a key mechanism for its anti-inflammatory properties in addition to its direct cytoprotection8,9 to protect lung against LPS-induced injury per se.
PTEN mediates inflammatory responses via intracellular regulation of Akt signaling;31 however, the role of PTEN/Akt signaling in macrophage polarization remains controversial. Previous studies indicated that Akt and its upstream regulator phosphatidylinositol-3-kinase (PI3K) positively regulated pro-inflammatory M1 polarization, while others have reported that Akt appears to promote anti-inflammatory M2 polarization, since inhibiting Akt activation attenuated the induction of M2 genes.32,33 In our study, LPS administration not only activated Akt signaling but also led alveolar macrophages polarized to M1 phenotype; unexpectedly, cells exposed to mixed conditions with LPS and Dex further activated Akt signaling with higher levels of M2 markers when compared to cells treated with LPS alone, suggesting that macrophage reprogramme to a contrasting phenotype may be dependent on the levels of Akt activation. Inhibition of PTEN using a PTEN inhibitor bpV resulted in elevated levels of Akt activation and increased the expression of IRF4 and Stat6 for transcriptional promoting M2 polarization. This suggests that the regulatory effect of PTEN on Akt signaling is a central mediator for controlling macrophage polarization. It is also possible that PTEN modulates the activation of Akt by acting on different PI3K subunits, as evidenced that membrane targeted PI3K (myrp110) seems to have lower capability to activate Akt.34
In addition to its direct cytoprotection, Dex leads to phenotypic change in lung alveolar macrophages from pro-inflammatory M1 to anti-inflammatory M2 in LPS-induced sepsis models. This likely indicates the high therapeutic value of Dex for sepsis patients who need to be intubated and ventilated under sedation. Indeed, Dex used in the ICU setting has shown a favourable effectiveness as reported previously.35,36 Thus, the data reported here may call more clinical studies of Dex for the best interests of sepsis patients. However, it is worth pointing out that the inhibition of PTEN in the macrophages of tumor micro-environment initiates tumor cell migration by increasing M2 macrophage polarization.37,38 Therefore, the risk of Dex in promoting cancer proliferation and metastasis due to its direct cellular effects as shown in our previous studies,39 together with its action of M2 macrophage polarization found in the current study cannot be ignored when used in cancer patients with sepsis.
In our study, it has been noted that α2 adrenergic receptor antagonist atipamezole failed to counteract Dex-mediated expression changes of caspase-1, CD121a, and HMGB1 release. These indicated that Dex might potentially act via non-α2 receptor-mediated pathways. Indeed, although Dex has high affinity to α2 receptor compared with α1 receptor (α2/α1 affinity ratio is 1600/1),40 the effects of α1 receptor activation and its downstream pathways have not be excluded. In addition, affinities of Dex to the various subtypes (a, b, and c) of α2 receptor differ and whether these subtypes may be involved in triggering other pathways remains unknown. Furthermore, a previous study has shown that Dex conferred cyto-protection against oxygen-glucose deprivation-induced injury through the I2 imidazoline receptor-PI3K/Akt pathway in rat C6 glioma cells.41 How these receptors are potentially involved in inducing the effects of Dex found in the current study warrants further study.
Our study is not without limitations. Firstly, immortalized epithelial and macrophage cells were used. Primary cultured cells should be considered in future studies to strengthen our conclusions. Secondly, three different experimental settings, ie, mice in vivo, mouse-immortalized macrophages and human epithelial cells, were used. Given well-known different interspecies in immune responses, the data reported herein are complicated and this certainly warrants further investigation in future. However, the therapeutic effects of Dex found in our study in a unified manner in different model settings may, arguably, consolidate our conclusions per se. NLRP3 might have different functions in cytoplasm or in nucleus. The cytosolic NLRP3 is activated in response to DAMPs and initiates an inflammatory response, while NLRP3 might play a transcriptional regulatory role in the nucleus.42 Based on this, we have carried out assessment of NLRP3 protein intensity between nucleus and cytoplasm, but we have found no significant difference. The regulatory role of NLRP3 in nucleus in addition to its function as inflammasome could not be simply excluded. This certainly needs to be explored in further studies.
In summary, we have demonstrated that Dex treatment reduces the activation of NLRP3 inflammasome in pulmonary epithelial cells and promotes the differentiation of alveolar macrophages towards the M2 phenotype, resulting in significant reduction of lung inflammation. All of which together with the direct lung cellular protection of Dex significantly attenuated ALI induced by LPS.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81772050, 81801900), The Basic and Frontier Research Fund of Chongqing, China (No. cstc2018jcyjAX0007, cstc2018jcyjAX0577), Excellent Talents Foundation of Army Medical University (XZ2019-505-028), Chongqing, China, The British Medical Research Council, The Chelsea-Westminster Hospital Joint Research Committee Grant and BJA/RCoA project grant, London, UK.
Disclosure
The authors report no conflicts of interest for this work.
References
1. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17(7):407–420. doi:10.1038/nri.2017.36
2. Fleischmann C, Scherag A, Adhikari NK, et al. Assessment of global incidence and mortality of hospital-treated sepsis. current estimates and limitations. Am J Respir Crit Care Med. 2016;193(3):259–272. doi:10.1164/rccm.201504-0781OC
3. Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800. doi:10.1001/jama.2016.0291
4. Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34(2):344–353. doi:10.1097/01.CCM.0000194725.48928.3A
5. Islam MN, Das SR, Emin MT, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18(5):759–765. doi:10.1038/nm.2736
6. Zhang Q, Wu D, Yang Y, Liu T, Liu H. Dexmedetomidine alleviates hyperoxia-induced acute lung injury via inhibiting NLRP3 inflammasome activation. Cell Physiol Biochem. 2017;42(5):1907–1919. doi:10.1159/000479609
7. Feng X, Guan W, Zhao Y, et al. Dexmedetomidine ameliorates lipopolysaccharide-induced acute kidney injury in rats by inhibiting inflammation and oxidative stress via the GSK-3beta/Nrf2 signaling pathway. J Cell Physiol. 2019;234(10):18994–19009. doi:10.1002/jcp.28539
8. Chen Q, Yi B, Ma J, et al. alpha2-adrenoreceptor modulated FAK pathway induced by dexmedetomidine attenuates pulmonary microvascular hyper-permeability following kidney injury. Oncotarget. 2016;7(35):55990–56001.
9. Li J, Chen Q, He X, et al. Dexmedetomidine attenuates lung apoptosis induced by renal ischemia-reperfusion injury through alpha2AR/PI3K/Akt pathway. J Transl Med. 2018;16(1):78. doi:10.1186/s12967-018-1455-1
10. Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol. 2012;47(4):417–426. doi:10.1165/rcmb.2012-0090OC
11. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404. doi:10.1038/nri3671
12. Farley KS, Wang LF, Razavi HM, et al. Effects of macrophage inducible nitric oxide synthase in murine septic lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;290(6):L1164–1172. doi:10.1152/ajplung.00248.2005
13. Sakaguchi R, Chikuma S, Shichita T, et al. Innate-like function of memory Th17 cells for enhancing endotoxin-induced acute lung inflammation through IL-22. Int Immunol. 2016;28(5):233–243. doi:10.1093/intimm/dxv070
14. Allard B, Panariti A, Martin JG. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front Immunol. 2018;9:1777. doi:10.3389/fimmu.2018.01777
15. Garlet GP, Giannobile WV. Macrophages: the bridge between inflammation resolution and tissue repair? J Dent Res. 2018;97(10):1079–1081. doi:10.1177/0022034518785857
16. Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci. 2018;19(6):1801. doi:10.3390/ijms19061801
17. Smith TD, Tse MJ, Read EL, Liu WF. Regulation of macrophage polarization and plasticity by complex activation signals. Integr Biol. 2016;8(9):946–955. doi:10.1039/c6ib00105j
18. Demers G, Griffin G, De Vroey G, Haywood JR, Zurlo J, Bedard M. Animal research. Harmonization of animal care and use guidance. Science. 2006;312(5774):700–701. doi:10.1126/science.1124036
19. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412. doi:10.1371/journal.pbio.1000412
20. Sun YB, Zhao H, Mu DL, et al. Dexmedetomidine inhibits astrocyte pyroptosis and subsequently protects the brain in in vitro and in vivo models of sepsis. Cell Death Dis. 2019;10(3):167. doi:10.1038/s41419-019-1416-5
21. Hirano Y, Aziz M, Yang WL, et al. Neutralization of osteopontin attenuates neutrophil migration in sepsis-induced acute lung injury. Crit Care. 2015;19:53. doi:10.1186/s13054-015-0782-3
22. Aziz M, Ode Y, Zhou M, et al. B-1a cells protect mice from sepsis-induced acute lung injury. Mol Med. 2018;24(1):26. doi:10.1186/s10020-018-0029-2
23. McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A. 2013;110(43):17253–17258. doi:10.1073/pnas.1308887110
24. Lavin Y, Winter D, Blecher-Gonen R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–1326. doi:10.1016/j.cell.2014.11.018
25. Hou L, Yang Z, Wang Z, et al. NLRP3/ASC-mediated alveolar macrophage pyroptosis enhances HMGB1 secretion in acute lung injury induced by cardiopulmonary bypass. Lab Invest. 2018;98(8):1052–1064. doi:10.1038/s41374-018-0073-0
26. Vernooy JH, Reynaert N, Wolfs TG, et al. Rapid pulmonary expression of acute-phase reactants after local lipopolysaccharide exposure in mice is followed by an interleukin-6 mediated systemic acute-phase response. Exp Lung Res. 2005;31(9–10):855–871. doi:10.1080/01902140600611645
27. Shen Y, Song J, Wang Y, et al. M2 macrophages promote pulmonary endothelial cells regeneration in sepsis-induced acute lung injury. Ann Transl Med. 2019;7(7):142. doi:10.21037/atm.2019.02.47
28. Zhu W, Lonnblom E, Forster M, et al. Natural polymorphism of Ym1 regulates pneumonitis through alternative activation of macrophages. Sci Adv. 2020;6(43). doi:10.1126/sciadv.aba9337
29. Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFkappaB in inflammation. Front Immunol. 2018;9:2542. doi:10.3389/fimmu.2018.02542
30. Zhang P, Li Y, Han X, Xing Q, Zhao L. Dexmedetomidine regulates 6-hydroxydopamine-induced microglial polarization. Neurochem Res. 2017;42(5):1524–1532. doi:10.1007/s11064-017-2209-9
31. Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28(7):1379–1386. doi:10.1093/carcin/bgm052
32. Perdiguero E, Kharraz Y, Serrano AL, Munoz-Canoves P. MKP-1 coordinates ordered macrophage-phenotype transitions essential for stem cell-dependent tissue repair. Cell Cycle. 2012;11(5):877–886. doi:10.4161/cc.11.5.19374
33. Wang G, Shi Y, Jiang X, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A. 2015;112(9):2853–2858. doi:10.1073/pnas.1501441112
34. Liu Z, Roberts TM. Human tumor mutants in the p110alpha subunit of PI3K. Cell Cycle. 2006;5(7):675–677. doi:10.4161/cc.5.7.2605
35. Scheer CS, Kuhn SO, Rehberg S. Dexmedetomidine in patients with sepsis requiring mechanical ventilation. JAMA. 2017;318(5):479–480. doi:10.1001/jama.2017.7853
36. Kawazoe Y, Miyamoto K, Morimoto T, et al. Effect of dexmedetomidine on mortality and ventilator-free days in patients requiring mechanical ventilation with sepsis: a randomized clinical trial. JAMA. 2017;317(13):1321–1328. doi:10.1001/jama.2017.2088
37. Li N, Qin J, Lan L, et al. PTEN inhibits macrophage polarization from M1 to M2 through CCL2 and VEGF-A reduction and NHERF-1 synergism. Cancer Biol Ther. 2015;16(2):297–306. doi:10.1080/15384047.2014.1002353
38. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi:10.1016/j.cell.2010.03.014
39. Wang C, Datoo T, Zhao H, et al. Midazolam and dexmedetomidine affect neuroglioma and lung carcinoma cell biology in vitro and in vivo. Anesthesiology. 2018;129(5):1000–1014. doi:10.1097/ALN.0000000000002401
40. Weerink MAS, Struys M, Hannivoort LN, Barends CRM, Absalom AR, Colin P. Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin Pharmacokinet. 2017;56(8):893–913. doi:10.1007/s40262-017-0507-7
41. Zhang F, Ding T, Yu L, Zhong Y, Dai H, Yan M. Dexmedetomidine protects against oxygen-glucose deprivation-induced injury through the I2 imidazoline receptor-PI3K/AKT pathway in rat C6 glioma cells. J Pharm Pharmacol. 2012;64(1):120–127. doi:10.1111/j.2042-7158.2011.01382.x
42. Bruchard M, Rebe C, Derangere V, et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol. 2015;16(8):859–870. doi:10.1038/ni.3202
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.