")
Back to Journals » OncoTargets and Therapy » Volume 11
Developments in the management of advanced soft-tissue sarcoma – olaratumab in context
Authors Moroncini G, Maccaroni E , Fiordoliva I , Pellei C, Gabrielli A, Berardi R
Received 24 October 2017
Accepted for publication 17 January 2018
Published 16 February 2018 Volume 2018:11 Pages 833—842
DOI https://doi.org/10.2147/OTT.S127609
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yao Dai
Gianluca Moroncini,1,* Elena Maccaroni,2,* Ilaria Fiordoliva,2 Chiara Pellei,2 Armando Gabrielli,1 Rossana Berardi2
1Department of Clinical and Molecular Sciences, Università Politecnica delle Marche, Ancona, Italy; 2Medical Oncology Unit, Università Politecnica delle Marche, Azienda Ospedaliero-Universitaria Ospedali Riuniti Umberto I, GM Lancisi, G Salesi, Ancona, Italy
*These authors contributed equally to this work
Abstract: Lartruvo® (olaratumab) is a fully human immunoglobulin G subclass 1 (IgG1) monoclonal antibody that inhibits platelet-derived growth factor receptor alpha (PDGFRα). The antitumor activity of olaratumab has been tested in vitro and in vivo, and inhibition of tumor growth has been observed in cancer cell lines, including glioblastoma and leiomyosarcoma cells. It represents the first-in-class antibody to be approved by regulatory authorities for the treatment of advanced soft-tissue sarcomas (STSs) in combination with doxorubicin, based on the results of the Phase Ib/II trial by Tap et al. The median progression-free survival (PFS), which was the primary end point of the study, was improved for patients treated with olaratumab plus doxorubicin compared to those treated with doxorubicin monotherapy (6.6 vs 4.1 months, respectively; HR 0.672, 95% CI 0.442–1.021, p=0.0615). Moreover, final analysis of overall survival (OS) showed a median OS of 26.5 months with olaratumab plus doxorubicin vs 14.7 months with doxorubicin, with a gain of 11.8 months (HR 0.46, 95% CI 0.30–0.71, p=0.0003). In October 2016, olaratumab was admitted in the Accelerated Approval Program by the US Food and Drug Administration (FDA) for use in combination with doxorubicin for the treatment of adult patients with STSs. In November 2016, the European Medicines Agency (EMA) granted conditional approval for olaratumab in the same indication under its Accelerated Assessment Program. A double-blind, placebo-controlled, randomized Phase III study (ANNOUNCE trial, NCT02451943) is being performed in order to confirm the survival advantage of olaratumab and to provide definitive drug confirmation by regulators. The study is ongoing, but enrollment is closed. The purpose of this review was to evaluate the rationale of olaratumab in the treatment of advanced STSs and its emerging role in clinical practice.
Keywords: anti-PDGFR antibodies, soft-tissue sarcoma, PDGFRα, doxorubicin, olaratumab
Introduction
Soft-tissue sarcomas (STSs) represent a group of rare mesenchymal tumors derived from connective tissues,1 which can arise from any site within the body, such as extremities, retroperitoneum, uterus, trunk, and head and neck.
STSs are heterogeneous diseases that are classified into more than 50 subtypes with different molecular, histological, and clinical characteristics. The most common subtypes of high-grade STSs include leiomyosarcoma, dedifferentiated sarcoma, undifferentiated pleomorphic sarcoma, synovial sarcoma, and malignant peripheral nerve sheath tumors (MPNSTs). Altogether, STSs account for <1% of adult tumors.2
Management of STSs requires a multidisciplinary approach including surgery, radiotherapy, and chemotherapy.
Radical resection represents the cornerstone treatment for localized disease, in combination with radiotherapy in selected cases, while systemic treatment, mostly chemotherapy, still remains the main option for advanced or metastatic disease.3
To date, anthracyclines (doxorubicin and epirubicin) remain the standard of care for first-line therapy of high-grade advanced STSs, regardless of histological subtype, clinical presentation, and patients’ characteristics.4
For advanced or metastatic disease, palliative chemotherapy determines an advantage in overall survival (OS) with median OS of 12–18 months for patients treated with first-line anthracycline-based regimes.5–8
Several clinical trials compared doxorubicin monotherapy and combinations of doxorubicin and other drugs, such as ifosfamide, showing a significant increase in terms of response rates (RRs) and progression-free survival (PFS) but failing to show an improvement in OS.9,10
Moreover, newer studies such as the PICASSO III and the TH CR-406/SARC021 trials have compared doxorubicin alone to doxorubicin in combination with recently developed fosfamides, palifosfamide and evofosfamide, respectively. No benefit in terms of OS and PFS was found in favor of the combinations, but the median OS of patients treated with doxorubicin in monotherapy has improved from 12 months to ~16 months in PICASSO III and 19.0 months in TH CR-406/SARC021.11,12
Targeted therapies play an emerging role in STS treatment, following discovery of molecular patterns involved in tumor biology. For example, upon detection of somatostatin receptor subtypes13 in intermediate and malignant soft tissue tumors,14 initial cases of patients with multiple drug-resistant, metastatic STSs successfully treated with radiolabeled somatostatin analogs have been recently reported.15 However, these agents are not approved.
In addition, recent studies highlighted the potential therapeutic value of blocking platelet-derived growth factor receptors (PDGFRs) as a viable antitumor approach. PDGFR, in particular the alpha (α) isoform, drives tumor growth and metastatic spread in several types of cancer, including ovarian cancer, prostate cancer, lung cancer, and STS.16,17
Overexpression or aberrant activation of this receptor has been demonstrated on both tumor cells and stroma.18 Several preclinical and clinical data suggest that PDGFR–ligand binding could play a significant role in stemness, senescence, and apoptosis in sarcoma cells and is also associated with metastatic progression.19 PDGFR amplification and activating mutations have also been found in gastrointestinal stromal tumor (GIST).20
Several tyrosine kinase inhibitors, blocking also platelet-derived growth factor receptor alpha (PDGFRα), such as pazopanib, regorafenib, sunitinib, and sorafenib, have been evaluated in advanced STSs; however, in all these studies, the median PFS ranged from 1.8 to 3.2 months, which did not differ from PFS associated with cytotoxic monotherapy.21–23
To date, pazopanib, a multikinase tyrosine kinase inhibitor that targets multiple receptors, including vascular endothelial growth factor (VEGF), PDGFR, and c-KIT, is the only US Food and Drug Administration (FDA)- and European Medicines Agency (EMA)-approved oral agent for high-grade STSs, on the basis of the results of the multi-institutional, Phase III PALETTE trial, that randomized 369 pretreated, metastatic STS (excluding liposarcoma) patients 2:1 to pazopanib vs placebo. Pazopanib improved median PFS (4.6 vs 1.6 months, HR 0.31, 95% CI 0.24–0.40, p<0.001) compared with placebo, even if no differences in terms of OS were observed.24
An in vitro study also showed that PDGFR overexpression represents a negative prognostic marker for survival in STS patients: the median OS in patients with a high vs low expression of PDGFRα was 27 vs 91 months, respectively.25
Lartruvo® (olaratumab) is a PDGFRα antagonist. It is a first-in-class recombinant human immunoglobulin G subclass 1 (IgG1) monoclonal antibody that binds to PDGFRα, blocking PDGF-AA, PDGF-BB, and PDGF-CC receptor binding and activation.
It was studied in a Phase Ib and in a randomized Phase II study (randomization between doxorubicin alone and in combination with olaratumab plus doxorubicin) in previously anthracycline-naive STS patients, showing both a statistically significant improvement in PFS and an improvement in OS to 26.9 months. This is the first randomized trial to show a significant improvement in OS compared to doxorubicin alone.26
This review aims at providing a comprehensive overview on this innovative drug that represents an important therapeutic novelty in the treatment of advanced STSs, in particular focusing on its clinical development and its application in clinical practice.
Platelet-derived growth factor axis
The PDGFR ligands, ie, PDGF-A, PDGF-B, PDGF-C, and PDGF-D, are a family of disulfide-bound polypeptide chains that form hetero- and homodimers before binding and activating two structurally related tyrosine kinase receptors, PDGFRα and PDGFRβ.27,28 These receptors are transmembrane proteins containing an extracellular ligand-binding domain, a transmembrane domain, and a cytoplasmic tyrosine kinase domain. After binding to the receptor, PDGF induces homodimerization or heterodimerization, resulting in the formation of PDGFRαα, PDGFRαβ, or PDGFRββ.29 After dimerization, the receptors phosphorylate each other on tyrosine residues: this process increases the kinase activity of the receptor and creates a high-affinity docking site for Src homology 2 (SH2) domain-containing adapter molecules (such as growth factor receptor-bound protein 2 [Grb2], Grb7, Crk, Nck, and Shc) that work as a link to other downstream effectors. Thus, phosphorylation of the receptors activates signaling pathways that promote growth and regulate stromal-derived fibroblasts30–32 and angiogenesis.33,34 Moreover, PDGFRα regulates stromal fibroblasts’ production of VEGF, an essential growth factor for angiogenesis and tumorigenesis.35
If unbalanced, this pathway could play a role in influencing tumor microenvironment in promoting diffusion and growth of cancer cells, hence further suggesting the need for development of drugs targeting the PDGF/PDGFRα axis as means of antitumor development.36
Preclinical evidence for olaratumab
Monoclonal antibodies directed against the ligand-binding site of PDGFRα have been demonstrated to be potent antagonists against receptor function. Neutralizing antibodies to PDGFRα have been reported previously.37,38
Olaratumab (LY3012207; formerly IMC-3G3; Eli Lilly and Company, Indianapolis, IN, USA) is an IgG1 monoclonal antibody identified as the most potent PDGFRα-neutralizing antibody (Figure 1). Olaratumab binds to the extracellular domain of PDGFRα with a high affinity (Kd 0.04 nM) and specificity, as it does not cross-react with PDGFR-β isoform.39 High-affinity binding of olaratumab to PDGFRα blocks ligand (PDGF-AA, PDGF-BB, PDGF-CC) binding in a dose-dependent manner with a 50% inhibitory concentration (IC50) of 0.24–0.58 nM. By blocking ligand binding, olaratumab inhibits ligand-induced receptor autophosphorylation and subsequent cellular events such as phosphorylation of the downstream signaling molecules (Akt and mitogen-activated protein kinase) and cell mitogenesis.40 In addition, olaratumab induces internalization of surface PDGFR into intracellular space and down modulation of PDGF/PDGFR signaling.39
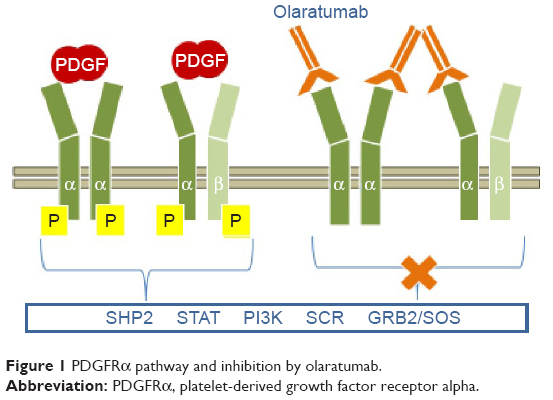 | Figure 1 PDGFRα pathway and inhibition by olaratumab. |
It is interesting to note that the potent anti-PDGFRα neutralizing activity of olaratumab is likely due to the affinity for receptor lying in the picomolar range, higher than the PDGF itself (Kd 0.2 nM), and the receptor binding site, since autoantibodies binding specific PDGFRα epitopes with nanomolar affinity (Kd 71–17 nM) have been cloned from memory B cells of patients affected by systemic sclerosis and shown to be potent receptor agonists in vitro and in vivo.41–44
The first evidence of antimitogenic activity of olaratumab in cancer cell lines was provided by the significant inhibition of growth of human leiomyosarcoma (SKLMS-1) xenografts in mice (p<0.0001) compared to the control group.39 Later, it was also shown that olaratumab enhances the antitumor activity of doxorubicin against the same human soft-tissue sarcoma xenograft model.45
The study by Loizos et al39 established that plasma olaratumab concentrations should be maintained in the 155–258 μg/mL range to obtain antitumor activity; tested doses of 6, 20, and 60 mg/kg administered twice per week resulted in statistically significant tumor growth inhibition. Monoclonal antibodies are not metabolized by cytochrome P450 enzymes; thus, co-administered drug do not affect the pharmacokinetic properties of olaratumab.46
These data represent the preclinical rationale for the dose cohorts in Phase I studies.
Clinical development
Phase I studies
Based on the favorable preclinical data, olaratumab monotherapy has been studied in two Phase I trials (the first one in predominantly Caucasian and the second one in Asian patients’ population) in order to assess its pharmacokinetic properties.47,48
The first trial47 was an open-label, dose-escalation study conducted in Caucasian and Black patients with advanced solid tumors. The aim of this study was to assess the safety, maximum tolerated dose (MTD), recommended Phase II dose (RP2D), pharmacokinetics, and preliminary antitumor activity of olaratumab. Patients were enrolled into five dose-escalating cohorts of three to six patients each. Olaratumab was administered intravenously weekly at 4, 8, or 16 mg/kg (cohorts 1–3) or once every other week at 15 or 20 mg/kg (cohorts 4–5), with 4 weeks/cycle. The study was conducted from December 2006 to March 2009, and totally, 19 patients were treated in five cohorts. Among all cohorts, the median number of infusions was 9 (range 1.0–54.0) and the median treatment duration was 12.1 weeks (range 1.0–57.9 weeks). Interestingly, no dose-limiting toxicities (DLTs) were observed during the study, and therefore, the MTD was not identified. The most common adverse events (AEs) considered at least possibly, probably, or definitely related to olaratumab were fatigue and infusion-related reactions (IRRs; 10.5% each). With the exception of one patient (enrolled in the 20 mg/kg cohort) experiencing two grade 3 drug-related AEs (increased alkaline phosphatase) after assessment period of DLTs, all drug-related AEs were of grade 1 or 2. Following multiple doses, the trough concentrations (Cmin) for 16 mg/kg weekly and 20 mg/kg biweekly were >155 μg/mL, which was the efficacious concentration in animal xenograft models. Regarding efficacy measurements, there were no complete responses (CRs) or partial responses (PRs) in this study, but 12 (63.2%) patients had a best response of stable diseases (SDs) with a median duration of 3.9 months (95% CI 2.3–8.7 months). The authors concluded that olaratumab was well tolerated and showed preliminary antitumor activity. Identified RP2Ds were both 16 mg/kg weekly and 20 mg/kg biweekly.
The second Phase I trial48 was a single-center, open-label, dose-escalation study conducted in Japanese patients with advanced/refractory solid malignancies. The primary objective was to establish the safety and pharmacokinetic profile of olaratumab. Three to six patients were enrolled into each of three cohorts: patients received intravenous olaratumab at 10 mg/kg on days 1 and 8 every 3 weeks (cohort 1), 20 mg/kg every 2 weeks (cohort 2), and 15 mg/kg on days 1 and 8 every 3 weeks (cohort 3). Doses were escalated from cohort 1 through cohort 3. A total of 16 patients were treated across three cohorts, and the median duration of treatment was 13.1 weeks (range 7.0–13.6 weeks), 6.0 weeks (range 3.0–13.4 weeks), and 7.0 weeks (range 7.0–25.1 weeks) in cohort 1, cohort 2, and cohort 3, respectively.
Similar to the previous Phase I trial conducted in Caucasian patients, there were no DLTs; thus, the MTD was not reached. The most frequently reported olaratumab-related events across the three cohorts included proteinuria (25.0%) and increased aspartate aminotransferase levels (12.5%), while no IRRs were reported. Based on the pharmacokinetic concentration profile of olaratumab, the trough concentrations following single and multiple doses at 15 mg/kg on days 1 and 8 every 3 weeks (cohort 3) and multiple doses at 20 mg/kg every 2 weeks (cohort 2) were above the 155 μg/mL target. Regarding efficacy, like in the other Phase I trial, the best overall response was SD. The disease control rate (CRs + PRs + SDs) was 66.7% in cohort 1, 42.9% in cohort 2, and 33.3% in cohort 3. The median duration of SD was 2.8 months in cohort 1 and cohort 2 and 4.9 months in cohort 3. Thus, the authors concluded that olaratumab had an acceptable safety profile and was well tolerated, and the aforementioned two doses represent an acceptable schedule for future trials in Japanese patients.
Therefore, olaratumab safety profile emerging from Phase I monotherapy trials was consistent with its toxicology profile and the toxicity profile observed in patients with advanced-stage solid tumors.
Phase Ib/II studies
On the basis of the favorable safety profile from the Phase I studies and preclinical data regarding olaratumab combination with doxorubicin in human sarcoma xenograft models,39,45 Tap et al26 performed a multicentric, open-label Phase Ib and randomized Phase II study of doxorubicin alone or doxorubicin plus olaratumab in patients with unresectable or metastatic STSs, previously untreated with an anthracycline.
The enrollment of Phase Ib–Phase II ran from October 2010 to January 2013.
The primary end point of Phase Ib was safety and that of Phase II was PFS, while OS, RR, safety, and pharmacokinetics were secondary end points.
In the Phase Ib part of the study, patients received olaratumab (15 mg/kg) intravenously on day 1 and day 8 plus doxorubicin (75 mg/m2) on day 1 of each 21-day cycle up to eight cycles. After eight cycles of the combination therapy, if no disease progression or unacceptable toxicities occurred, patients were allowed to receive olaratumab monotherapy until disease progression.
In the Phase II part of the study, patients were randomly assigned in a 1:1 ratio to receive either olaratumab (15 mg/kg) plus doxorubicin (as described in the Phase Ib part) or doxorubicin alone (75 mg/m2) on day 1 of each 21-day cycle for up to eight cycles. After completion of eight cycles of doxorubicin, patients in the combination group could receive olaratumab monotherapy until disease progression, while patients in the doxorubicin-alone group were observed and could receive olaratumab monotherapy after disease progression.
Overall, 15 patients were enrolled and treated in the Phase Ib part of the study. In the Phase II portion, a total of 129 patients (out of 133 randomized patients) received at least one dose of the investigational therapy, including 64 patients who received the combination of olaratumab and doxorubicin (investigational arm) and 65 patients who received doxorubicin alone (control arm). A total of 30 patients treated in the control arm received olaratumab monotherapy after discontinuing doxorubicin, as permitted by the protocol. Baseline characteristics were well balanced in the two groups, except for a slight predominance of women in the investigational arm. Approximately 40% of patients were in the first-line setting; the remaining sample had performed at least one treatment line. The most represented histological subtypes were leiomyosarcoma (36% of patients in the investigational arm and 40% of patients in the control arm), undifferentiated pleomorphic sarcoma, and liposarcoma.
The Phase II primary end point was PFS. It was 6.6 months (95% CI 4.1–8.3 months) for patients treated with olaratumab plus doxorubicin and 4.1 months (95% CI 2.8–5.4 months) for those treated with doxorubicin monotherapy. This PFS improvement in favor of the experimental arm met the protocol-defined significance level of 0.1999 for final PFS (stratified HR 0.672, 95% CI 0.442–1.021, p=0.0615). Moreover, the most relevant finding of this study was the significant improvement in OS, which was a secondary end point, for patients treated in the experimental arm: final analysis of OS, performed after 91 (71%) patients had died in the intention-to-treat population, showed a median OS of 26.5 months (95% CI 20.9–31.7 months) with olaratumab plus doxorubicin vs 14.7 months (95% CI 9.2–17.1 months) with doxorubicin, with a gain of 11.8 months (HR 0.46, 95% CI 0.30–0.71, p=0.0003). This impressive benefit was consistent across the subgroup stratification factors, including histological tumor type (leiomyosarcoma vs non-leiomyosarcoma), number of previous treatments, and PDGFRα expression. PDGFRα expression, assessed by immunohistochemistry, did not show any predictive value on efficacy.
No difference in terms of objective RR was observed, which was 18.2% (9.8%–29.6%) for patients in the experimental arm compared with 11.9% (5.3–22.2) for patients in the control arm (p=0.3421).
AEs were more frequent with combination therapy vs doxorubicin alone and included neutropenia (58% vs 35%, respectively), mucositis (53% vs 35%, respectively), nausea (73% vs 52%, respectively), vomiting (45% vs 18%, respectively), and diarrhea (34% vs 23%, respectively). Grade 3 febrile neutropenia rate was similar in both groups (experimental arm 13% vs control arm 14%). The number of IRRs in the olaratumab plus doxorubicin arm was 13% vs 0% for doxorubicin. Cardiac toxicity is an expected risk of doxorubicin treatment, in particular with increasing cumulative exposure; however, the incidence of cardiac dysfunction in Phase II portion of the study was similar between the two treatment arms.
Ongoing trials
An ongoing double-blind, placebo-controlled, randomized Phase III study (ANNOUNCE trial, NCT02451943)49 was planned in order to confirm the survival advantage of olaratumab and to provide definitive drug confirmation by regulators. The primary end point of the study was OS, with an enrolled population of 460 patients. From August 2015 to June 2017, patients with unresectable or metastatic STSs were randomized to receive olaratumab plus doxorubicin vs placebo plus doxorubicin, with the same schedule used in the Phase II trial. The study is still ongoing, but the enrollment is closed. The first efficacy results are expected in 2019, and the study is expected to be completed in 2020. The Phase III ANNOUNCE study will enroll a higher number of patients compared with the Phase Ib/II trial, which will make the study more powerful and may help to detect tissue biomarkers to predict survival in treated patients. Preliminary data from post hoc biomarker analysis failed to identify a significant association between PDGFRα or PDGFRβ expression and OS or PFS in patients included in Tap et al’s Phase Ib/II study of doxorubicin with or without olaratumab. Low PDGF-BB or CXCR4 expression was associated with improved PFS in the combination arm.50
Recently, Barker et al51 presented results of a subgroup analysis including patients who received olaratumab monotherapy in the Phase II trial of Tap et al.26
The subgroup receiving olaratumab as maintenance of previous doxorubicin plus olaratumab treatment showed a median OS of 31.7 months and a 2-year survival rate of 67.6%. Conversely, the subgroup receiving olaratumab as a switch therapy after disease progression under doxorubicin showed a median OS of 13.5 months and a 2-year survival rate of 28.7%. These data, although not yet published, suggest a clinical benefit from olaratumab monotherapy as a maintenance strategy after a combination first-line treatment.
Several other trials are evaluating efficacy and safety of olaratumab in STS patients and are listed in Table 1.
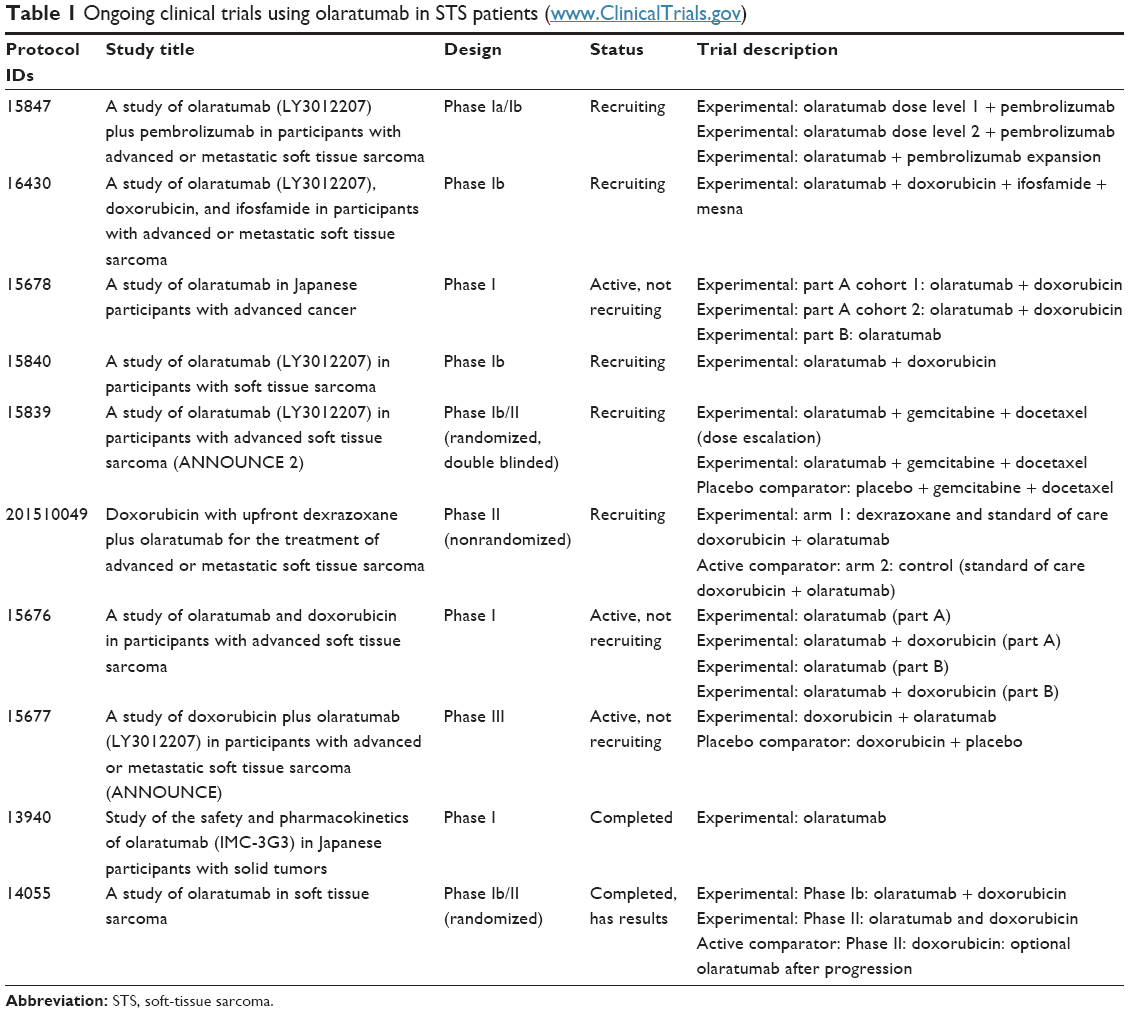 | Table 1 Ongoing clinical trials using olaratumab in STS patients (www.ClinicalTrials.gov) |
Among them, the Phase Ib/II ANNOUNCE-2 trial has been designed in order to assess safety and efficacy of the chemotherapy association of gemcitabine and docetaxel with or without olaratumab in patients with pretreated advanced STSs. OS is the primary end point of the Phase II portion, and the study is still recruiting patients.52
Moreover, this drug is being evaluated in Phase II trials regarding other cancer histotypes, such as advanced non-small cell lung cancer (NSCLC), GIST, and ovarian cancer.53,54
The Phase II study of olaratumab in previously treated patients with metastatic GIST by Wagner et al showed a statistically different disease control rate in two different cohorts including patients with or without PDGFRα mutations. The clinical benefit rate at 12 weeks was 50.0% and 14.3%, respectively.53
Agencies’ approval and regulatory affairs
In October 2014, the FDA granted olaratumab, in combination with doxorubicin, orphan drug status for the treatment of STS patients not amenable to curative treatment with surgery or radiotherapy.55 The same indication was also given by the EMA in February 2015.56
Owing to the impressive results in terms of OS improvement observed in the Phase Ib/II study, in October 2016, the FDA also granted first accelerated approval to use olaratumab in combination with doxorubicin for treatment of adult patients with advanced STS, and the same approval was also given by the EMA in November 2016.
Both regulatory agencies’ approval is contingent upon confirmation of a significant clinical benefit in the ongoing randomized, Phase III ANNOUNCE trial.52
Technical information
Olaratumab has an approximate molecular weight of 154 kDa and is produced in genetically engineered mammalian NS0 cells. Olaratumab is supplied in single-use vials containing 500 mg/50 mL (10 mg/mL) solution, and it is administered at a dose of 15 mg/kg via intravenous infusion over 60 min; for patients requiring higher doses, the duration of infusion should be increased such that the maximum infusion rate of 25 mg/minute is not exceeded. The approved recommended dose and schedule of treatment is 15 mg/kg administered by intravenous infusion over 60 minutes on day 1 and day 8 of each 21-day cycle, in combination with 75 mg/m2 of doxorubicin on day 1 following the olaratumab infusion, for up to eight cycles of treatment or until the patient experiences an unacceptable toxicity or disease progression, followed by olaratumab monotherapy until disease progression or unacceptable toxicity.57,58 Premedication with intravenous diphenhydramine and dexamethasone can be administered on day 1 of the first cycle.
Maximum serum concentrations (Cmax) are reached in a median time of 2 hours after the start of the infusion, and the drug reaches steady state during the third cycle. At the steady state, the volume of distribution (Vd) is 7.7 L and the estimated elimination half-life is ~11 days (range 6–24 days). No differences in pharmacokinetics are noticed in patients treated by a combination of olaratumab and doxorubicin.57,58
A recent review59 described the pharmacokinetic properties of olaratumab using data from four Phase II clinical studies in NSCLC, glioblastoma, STSs, and GIST populations. This study substantially confirmed the data from Tap et al analysis, showing a linear drug disposition due to full target saturation at the dose levels tested. Moreover, tumor burden seemed to be associated with a higher clearance (CL).
The mechanism of action of olaratumab has been well defined, as a specific PDGFRα ligand and receptor dimerization and signaling blocker. Moreover, the internalization of olaratumab-bound PDGFRα dimer may further contribute to drug activity.
As demonstrated by preclinical sarcoma models, the PDGFRα inhibition is necessary but not sufficient for durable control of cell growth. In fact, the antitumor activity of olaratumab is enhanced by co-administration with doxorubicin or cisplatin. Thus, combination with other targeted agents or chemotherapy drugs may be the future scenario of olaratumab clinical application.60,61
An additional possible mechanism under investigation is the increased tumor cell permeability to doxorubicin due to olaratumab inhibition of PDGFRα in stromal cells.62
This pharmacodynamic interaction may reduce the number of doxorubicin plus olaratumab cycles compared to the eight administrations of doxorubicin expected from the trial of Tap et al.26 In fact, this schedule leads to 600 mg doxorubicin/m2 cumulative dose, which is not recommended in clinical practice.
Conclusion
Olaratumab, a first-in-class monoclonal antibody blocking the PDGF/PDGFRα cellular signaling pathway, is the first agent in many years to show a survival advantage for patients with advanced STSs.
In their randomized, double-blind Phase II trial, Tap et al26 showed a clinically meaningful OS improvement in patients treated with olaratumab and doxorubicin combination vs patients receiving doxorubicin alone, with a median survival gain of 11.8 months, with manageable toxicity: although an increased rate of neutropenia, mucositis, vomiting, diarrhea, and musculoskeletal pain was observed, these AEs were primarily of grade 2 and, with the exception of febrile neutropenia, severe events were not observed. By contrast, PFS, the primary end point of the study, was extended by only 2.5 months in the olaratumab arm (6.6 months vs 4.1 months in the control arm). These findings suggest a change to the treatment strategy in patients with high-grade, advanced or metastatic, anthracycline-naive STSs in the first-line setting: this was the first time that a monoclonal antibody in combination with doxorubicin was able to influence OS in soft-tissue sarcomas (perhaps also owing to the survival gain obtained by olaratumab maintenance therapy after primary induction with the combination).
These results appear remarkable also because previous studies evaluating the superiority of combination chemotherapy regimens or histology-tailored therapies vs doxorubicin alone in the first-line setting of advanced/metastatic STSs failed to show any survival advantage.9,10 However, OS is a composite end point, influenced by all subsequent treatment administered in the metastatic setting, and the exact effect of various post-study treatments is not easy to define. This aspect will certainly require further investigation, also because PFS for patients enrolled in the study was only marginally influenced, indicating that the greatest benefit for patients enrolled in the combination treatment arm consisted mostly of survival benefits in post-progression survival (PPS).
Furthermore, PFS, particularly when assessed by RECIST criteria in patients treated with a drug that targets mainly angiogenic pathways, is usually not the best parameter to evaluate the survival gain of the population, as it can be usually observed also in other settings where antiangiogenic drugs are more extensively used, such as in colorectal cancer.63
Moreover, some issues, such as the reduced power of the study and the small number of patients included, remain and will be hopefully solved by the Phase III ANNOUNCE study, which has recently completed accrual.
Another unanswered question is the reason behind the disparity between PFS and OS study end points. Some interesting hypotheses could explain this topic, such as olaratumab may display a long-term modulatory effect on the tumor and stromal microenvironment or possess a synergistic antitumor effect with doxorubicin, even if supporting evidence is currently lacking. To date, studies investigating olaratumab mechanism of action, both as a single-agent therapy and in combination, are required to improve the efficacy of this drug. Several ongoing clinical trials are evaluating olaratumab in combination with other drugs, such as gemcitabine and docetaxel, which may help to clarify if the postulated synergic effect of olaratumab plus chemotherapy is restricted to anthracyclines or could be extended also to other chemotherapeutics.
Other concerns are raised by possible differences in disease biology between treatment groups, which may impact on the findings of relatively small studies. Some differences between cohorts in the study arms were noticed; for example, the proportion of patients with undifferentiated pleomorphic sarcoma, an aggressive STS subtype, was higher in the doxorubicin monotherapy arm vs the combination arm (21% vs 15%, respectively). Another imbalance also occurred in the “other” disease category (26% in the combination arm vs 9% in the monotherapy arm), and finally, more women were included in the combination arm (61% vs 51%, respectively). These differences between groups might account for the differences observed in terms of OS.
The effect of olaratumab on the PDGFR pathway also seems to be interesting, even if in the Phase Ib/II study, PDGFRα expression in tumor samples did not show any predictive value on efficacy and, indeed, the authors reported that negative expression of this marker was associated with a favorable outcome, which does not support PDGFRα inhibition in the tumor as the main mechanism of action. However, as confirmed by the authors, the immunohistochemistry assay used to assess PDGFRα expression was not specific; therefore, this issue would require further investigation.
Olaratumab represents a promising and well-tolerated new anticancer therapy that has demonstrated an unexpected survival benefit in combination with doxorubicin in patients with advanced or metastatic STSs who have poor prognosis. Although in the future this combination seems to have the potential to become the new standard of care for first-line treatment of STS patients, the results of the ANNOUNCE confirmatory Phase III trial are warranted in order to confirm these optimistic perspectives and solve the unanswered questions.
Disclosure
The authors report no conflicts of interest in this work.
References
Linch M, Miah AB, Thway K, Judson IR, Benson C. Systemic treatment of soft-tissue sarcoma-gold standard and novel therapies. Nat Rev Clin Oncol. 2014;11(4):187–202. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Clark MA, Fisher C, Judson I, Thomas JM. Soft-tissue sarcomas in adults. N Engl J Med. 2005;353(7):701–711. | ||
Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol. 1987;5(6):840–850. | ||
Knepper TC, Saller J, Walko CM. Novel and expanded oncology drug approvals of 2016-PART 1: new options in solid tumor management. Oncology (Williston Park). 2017;31(2):110–121. | ||
Shirley M. Olaratumab: first global approval. Drugs. 2017;77(1):107–112. | ||
von Mehren M, Randall RL, Benjamin RS, et al. Soft tissue sarcoma, version 2. 2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14(6):758–786. | ||
Bramwell VHC, Anderson D, Charette ML. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft-tissue sarcoma: a meta-analysis and clinical practice guideline. Sarcoma. 2000;4(3):103–112. | ||
Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415–423. | ||
Seddon BM, Whelan J, Strauss SJ, et al. GeDDiS: a prospective randomised controlled phase III trial of gemcitabine and docetaxel compared with doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft tissue sarcomas (EudraCT 2009-014907-29). J Clin Oncol. 2015;33(suppl):abstr10500. | ||
Ryan CW, Merimsky O, Agulnik M, et al. PICASSO III: a phase III, placebo-controlled study of doxorubicin with or without Palifosfamide in patients with metastatic soft tissue sarcoma. J Clin Oncol. 2016;34(32):3898–3905. | ||
Tap WD, Papai Z, Van Tine BA, et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2017;18(8):1089–1103. | ||
Montironi R, Cheng L, Mazzucchelli R, et al. Immunohistochemical detection and localization of somatostatin receptor subtypes in prostate tissue from patients with bladder outlet obstruction. Cell Oncol. 2008;30(6):473–482. | ||
Florio T, Montella L, Corsaro A, et al. In vitro and in vivo expression of somatostatin receptors in intermediate and malignant soft tissue tumors. Anticancer Res. 2003;23:2465–2471. | ||
Crespo-Jara A, González Manzano R, Lopera Sierra M, Redal Peña MC, Brugarolas Masllorens A. A patient with metastatic sarcoma was successfully treated with radiolabeled somatostatin analogs. Clin Nucl Med. 2016;41(9):705–707. | ||
George D. Platelet-derived growth factor receptors: a therapeutic target in solid tumors. Semin Oncol. 2001;28(5 suppl 17):27–33. | ||
Board R, Jayson GC. Platelet-derived growth factor receptor (PDGFR): a target for anticancer therapeutics. Drug Resist Updat. 2005;8(1–2):75–83. | ||
Ostman A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004;15(4):275–286. | ||
Ehnman M, Missiaglia E, Folestad E, et al. Distinct effects of ligand-induced PDGFRalpha and PDGFRbeta signaling in the human rhabdomyosarcoma tumor cell and stroma cell compartments. Cancer Res. 2013;73(7):2139–2149. | ||
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710. | ||
Mir O, Brodowicz T, Italiano A, et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17(12):1732–1742. | ||
George S, Merriam P, Maki RG, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J Clin Oncol. 2009;27(19):3154–3160. | ||
Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27(19):3133–3140. | ||
van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886. | ||
Kilvaer TK, Valkov A, Sorbye SW, et al. Platelet-derived growth factors in non-GIST soft-tissue sarcomas identify a subgroup of patients with wide resection margins and poor disease-specific survival. Sarcoma. 2010;2010:751304. | ||
Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft tissue sarcoma: an open-label phase Ib and randomised phase II trial. Lancet. 2016;388(10043):488–497. | ||
Heldin CH, Eriksson U, Ostman A. New members of the platelet-derived growth factor family of mitogens. Arch Biochem Biophys. 2002;398(2):284–290. | ||
Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15(4):197–204. | ||
Seifert RA, Hart CE, Phillips PE, et al. Two different subunits associate to create isoform-specific platelet-derived growth factor receptors. J Biol Chem. 1989;264(15):8771–8778. | ||
Svegliati S, Cancello R, Sambo P, et al. Platelet-derived growth factor and reactive oxygen species (ROS) regulate Ras protein levels in primary human fibroblasts via ERK1/2. Amplification of ROS and Ras in systemic sclerosis fibroblasts. J Biol Chem. 2005;280(43):36474–36482. | ||
Smaldone S, Olivieri J, Gusella GL, Moroncini G, Gabrielli A, Ramirez F. Ha-Ras stabilization mediates pro-fibrotic signals in dermal fibroblasts. Fibrogenesis Tissue Repair. 2011;4(1):8. | ||
Spadoni T, Svegliati Baroni S, Amico D, et al. A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol. 2015;67(6):1611–1622. | ||
Alvarez RH, Kantarjian HM, Cortes JE. Biology of platelet-derived growth factor and its involvement in disease. Mayo Clin Proc. 2006;81(9):1241–1257. | ||
Dong J, Grunstein J, Tejada M, et al. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 2004;23(14):2800–2810. | ||
Crawford Y, Kasman I, Yu L, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. 2009;15(1):21–34. | ||
Shah GD, Loizos N, Youssoufian H, Schwartz JD, Rowinsky EK. Rationale for the development of IMC-3G3, a fully human immunoglobulin G subclass 1 monoclonal antibody targeting the platelet-derived growth factor receptor α. Cancer. 2010;116(4 suppl):1018–1026. | ||
Lokker NA, O’Hare JP, Barsoumian A, et al. Functional importance of platelet-derived growth factor (PDGF) receptor extracellular immunoglobulin-like domains. Identification of PDGF binding site and neutralizing monoclonal antibodies. J Biol Chem. 1997;272(52):33037–33044. | ||
LaRochelle WJ, Jensen RA, Heidaran MA, et al. Inhibition of platelet derived growth factor autocrine growth stimulation by a monoclonal antibody to the human a platelet-derived growth factor receptor. Cell Growth Differ. 1993;4(7):547–553. | ||
Loizos N, Xu Y, Huber J, et al. Targeting the platelet-derived growth factor receptor alpha with a neutralizing human monoclonal antibody inhibits the growth of tumor xenografts: implications as a potential therapeutic target. Mol Cancer Ther. 2005;4(3):369–379. | ||
Dolloff NG, Russell MR, Loizos N, Fatatis A. Human bone marrow activates the Akt pathway in metastatic prostate cells through transactivation of the alpha-platelet-derived growth factor receptor. Cancer Res. 2007;67(2):555–562. | ||
Moroncini G, Grieco A, Nacci G, et al. Epitope specificity determines pathogenicity and detectability of anti-platelet-derived growth factor receptor alpha autoantibodies in systemic sclerosis. Arthritis Rheumatol. 2015;67(7):1891–1903. | ||
Moroncini G, Cuccioloni M, Mozzicafreddo M, et al. Characterization of binding and quantification of human autoantibodies to PDGFRalpha using a biosensor-based approach. Anal Biochem. 2017;528:26–33. | ||
Svegliati S, Amico D, Spadoni T, et al. Agonistic anti-PDGF receptor autoantibodies from patients with systemic sclerosis impact human pulmonary artery smooth muscle cells function in vitro. Front Immunol. 2017;8:75. | ||
Luchetti MM, Moroncini G, Jose Escamez M, et al. Induction of scleroderma fibrosis in skin-humanized mice by administration of anti-platelet-derived growth factor receptor agonistic autoantibodies. Arthritis Rheumatol. 2016;68:2263–2273. | ||
Tonra J, Deevi D, Carrick F, et al. Enhanced antitumour activity of anti-platelet derived growth factor receptor alpha antibody, IMC-3G3, in combination with doxorubicin against a human soft-tissue sarcoma xenograft model. AACR-NCI-EORTC MolecularTargets and Cancer Therapeutics International Conference; 2005 Nov 14–18. Philadelphia, PA: 2005:A67. | ||
Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. | ||
Chiorean EG, Sweeney C, Youssoufian H, et al. A phase I study of olaratumab, an anti-platelet-derived growth factor receptor alpha (PDGFRα) monoclonal antibody, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014;73(3):595–604. | ||
Doi T, Ma Y, Dontabhaktuni A, Nippgen C, Nippgen J, Ohtsu A. Phase I study of olaratumab in Japanese patients with advanced solid tumors. Cancer Sci. 2014;105(7):862–869. | ||
NIH [homepage on the Internet]. A Study of Doxorubicin Plus Olaratumab (LY3012207) in Participants with Advanced or Metastatic Soft Tissue Sarcoma. 2018. Available from: http://www.mayo.edu/research/clinical-trials/cls-20193937. Accessed January 17, 2018. | ||
Oakley GJ 3rd, Wijayawardana S, Marchal C. Expression of pdgfrα, ligands, and related genes versus clinical outcomes in a phase 1b/2 study of olaratumab plus doxorubicin in soft tissue sarcoma. CTOS 2017 Annual Meeting, Maui, Hawaii: Poster 217 #2768598. | ||
Barker S, Peterson P, Ilaria RL, et al. Olaratumab after treatment with olaratumab + doxorubicin: monotherapy (mono) outcomes from the jgdg phase 2 clinical trial. CTOS 2017 Annual Meeting, Maui, Hawaii: Poster 202 #2778170. | ||
NIH [homepage on the Internet]. A Study of Olaratumab (LY3012207) in Participants with Advanced Soft Tissue Sarcoma. 2018. Available from: http://www.utswmedicine.org/cancer/clinical-trials/fact-detail.html?primarypurpose=10&studyId=STU%20012016-032. Accessed January 17, 2018. | ||
Wagner AJ, Kindler H, Gelderblom H, et al. A phase II study of a human anti-PDGFRα monoclonal antibody (olaratumab, IMC-3G3) in previously treated patients with metastatic gastrointestinal stromal tumors. Ann Oncol. 2017;28(3):541–546. | ||
Gerber DE, Swanson P, Lopez-Chavez A, et al. Phase II study of olaratumab with paclitaxel/carboplatin (P/C) or P/C alone in previously untreated advanced NSCLC. Lung Cancer. 2017;111:108–115. | ||
FDA [webpage on the Internet]. Orphan Drug Designations and Approvals. 2018. Available from: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/. Accessed January 17, 2018. | ||
EPAR Summary for the Public. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004216/human_med_002036.jsp&mid=WC0b01ac058001d124. Accessed January 17, 2018. | ||
Lartruvo® (olaratumab) injection [FDA prescribing information] [homepage on the Internet]; 2016. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761038lbl.pdf. Accessed January 17, 2018. | ||
Lartruvo® (olaratumab) injection [EMA prescribing information] [homepage on the Internet]; 2016. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004216/WC500216869.pdf. Accessed January 17, 2018. | ||
Mo G, Baldwin JR, Luffer-Atlas D, et al. Population pharmacokinetic modeling of olaratumab, an anti-PDGFRa human monoclonal antibody, in patients with advanced and/or metastatic cancer. Clin Pharmacokinet. Epub 2017 Jun 15. | ||
Lowery CD, Blosser W, Dowless M, et al. Olaratumab exerts anti-tumor activity in preclinical models of pediatric bone and soft tissue tumors through inhibition of platelet-derived growth factor receptor & alpha. Clin Cancer Res. Epub 2017 Nov 30. | ||
Koos B, Jeibmann A, Lünenbürger H, et al. The tyrosine kinase c-Abl promotes proliferation and is expressed in atypical teratoid and malignant rhabdoid tumors. Cancer. 2010;116(21):5075–5081. | ||
Vincenzi B, Badalamenti G, Napolitano A, et al. Olaratumab: PDGFR-α inhibition as a novel tool in the treatment of advanced soft tissue sarcomas. Crit Rev Oncol Hematol. 2017;118:1–6. | ||
Giampieri R, Scartozzi M, Del Prete M, et al. The “angiogenetic ladder”, step-wise angiogenesis inhibition in metastatic colorectal cancer. Cancer Treat Rev. 2014;40(8):934–941. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.