")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Development of Taiwan’s strategies for regulating nanotechnology-based pharmaceuticals harmonized with international considerations
Authors Guo J, Lee Y, Huang HW, Tzou M, Wang Y, Tsai J
Received 21 May 2014
Accepted for publication 19 July 2014
Published 15 October 2014 Volume 2014:9(1) Pages 4773—4783
DOI https://doi.org/10.2147/IJN.S68134
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jiun-Wen Guo,1 Yu-Hsuan Lee,2 Hsiau-Wen Huang,3 Mei-Chyun Tzou,3 Ying-Jan Wang,2 Jui-Chen Tsai1,4
1Institute of Clinical Pharmacy and Pharmaceutical Sciences, College of Medicine, National Chung Kung University, Tainan, Taiwan; 2Department of Environmental and Occupational Health, College of Medicine, National Cheng Kung University, Tainan, Taiwan; 3Food and Drug Administration, Ministry of Health and Welfare, Taiwan; 4Center for Pharmaceutical Regulatory Science, National Cheng Kung University, Tainan, Taiwan
Abstract: Nanotechnology offers potential in pharmaceuticals and biomedical developments for improving drug delivery systems, medical imaging, diagnosis, cancer therapy, and regenerative medicine. Although there is no international regulation or legislation specifically for nanomedicine, it is agreed worldwide that considerably more attention should be paid to the quality, safety, and efficacy of nanotechnology-based drugs. The US Food and Drug Administration and the European Medicines Agency have provided several draft regulatory guidance and reflection papers to assist the development of nanomedicines. To cope with the impact of nanotechnology and to foster its pharmaceutical applications and development in Taiwan, this article reviews the trends of regulating nanotechnology-based pharmaceuticals in the international community and proposes strategies for Taiwan’s regulation harmonized with international considerations. The draft regulatory measures include a chemistry, manufacturing, and controls (CMC) review checklist and guidance for CMC review of liposomal products. These have been submitted for discussion among an expert committee, with membership comprised of multidisciplinary academia, research institutions, the pharmaceutical industry, and regulators, and are currently approaching final consensus. Once a consensus is reached, these mechanisms will be recommended to the Taiwan Food and Drug Administration for jurisdiction and may be initiated as the starting point for regulating nanotechnology-based pharmaceuticals in Taiwan.
Keywords: CMC review, nanomedicine, nanotechnology, pharmaceuticals, regulatory guidance
Nanotechnology and its application in medicine
Since the introduction of nanotechnology and nanoscience, nanoscale materials and components have developed rapidly and been applied in many fields. According to the US National Nanotechnology Initiative, nanotechnology is defined as a technology that works at the atomic, molecular, and supramolecular levels over a length scale of approximately 1–100 nm range; creates and uses structures, devices, and systems that have novel properties and functions because of their small and/or intermediate size; and can control or manipulate on the atomic scale.1–3 Worldwide government funding of nanotechnology has increased since 1997 and was approximately $10 billion in 2005. It is estimated that the global value of nanotechnology products will exceed $2.5 trillion in 2014.1,2 The Taiwan Government has supported the National Nanotechnology Project with about NTD$3 billion (about US $100 million) each year since 2003.4
Benefiting from the rapid development of nanotechnology, many nanotech materials are widely used for commercial purposes and in everyday life, eg, in applications such as electronics, agriculture, and energy technology, as well as in the medical fields – the last referred to as nanomedicine. In this article, the terms “nanodrugs”, “nanopharmaceuticals”, and “nanomedicines” are used depending on their scope, and in some instances may be used interchangeably.5
Nanotechnology offers novel potential in pharmaceutical and biomedical developments for improving drug delivery systems, medical imaging, diagnosis, cancer therapy, regenerative medicine, implantable materials, and the development of nanodrugs.6,7 Compared with conventional, poorly stable, or poorly soluble drugs, nanodrugs may improve drug solubility, stability, and permeability, resulting in enhanced bioavailability and efficiency. For highly toxic drugs, they may offer tissue selectivity and thus reduce unwanted side effects. The pharmacokinetic properties of a nanodrug often differ from its conventional pharmaceutical composition. This is due mainly to nanotechnology changing the physical and chemical properties of the drug. These changes can involve particle size, charge, and surface characteristics, with profound effects on the efficacy and safety of nanodrugs.7–9 A well-known example is the first US-approved nanodrug, the anticancer Doxil®, in which the use of pegylated (PEG) nanoliposome avoids the clearance of doxorubicin by reticuloendoplasmic system and prolongs drug circulation time. Owing to the enhanced permeability and retention effect, Doxil is also passively targeted to tumors, and its doxorubicin is released and becomes available to tumor cells.5
Research in nanomedicine and pharmaceutical development is active. For instance, Europe’s recent Seventh Framework Programme for Research and Technological Development (FP7, 2007–2013), mainly supported by Nanotechnology and Nanosciences, Knowledge-based Multifunctional Materials and New Production Processes and Devices (NMP) (EU), has invested €4/15/2015300 million in nanomedicine projects.10 Currently, at least 30 nanomedicines have been approved in the global market.6,7,11,12 Eleven of the approved nanomedicines are available in Taiwan13 and one liposomal generic drug was developed by a local pharmaceutical company in 1998 (Table 1). In addition, approximately 70, 48, and ten nanomedicines are currently under clinical development in the US, Europe, and Taiwan, respectively.6,14,15 Reports indicated that the market value of nanomedicines in the US was around $53 billion in 2011, with the demand expected to increase by >17% per year to $110 billion in 2016.7,16 Because of the rapidly growing market demand and the current limited understanding of nanomedicines, it is important that we clarify the physical and chemical characteristics of nanomedicines as well as their effectiveness and functions.17 Accompanied nanotoxicology concerns require studies that investigate a variety of physicochemical aspects of nanomaterials, including 1) structure and morphology, 2) chemical composition, 3) surface area, 4) particle size, 5) size distribution, 6) surface charge, and 7) degree of hydrophilicity/hydrophobicity. These physicochemical properties have been associated with questions regarding the safety of nanomaterials.18 For instance, it was shown that 1.4 nm gold particles inhibited the growth of fibroblasts, epithelial cells, macrophages, and melanoma cells, but not the 15 nm particles.19 Compared in animals, carbon nanotubes apparently had greater pulmonary toxicity than fine-scale carbon graphite.20 Due to a high surface area to volume ratio, nanoparticles can efficiently interact with biological systems and deposit throughout the respiratory tract after administration, potentially causing vascular or lymphatic blockage.21
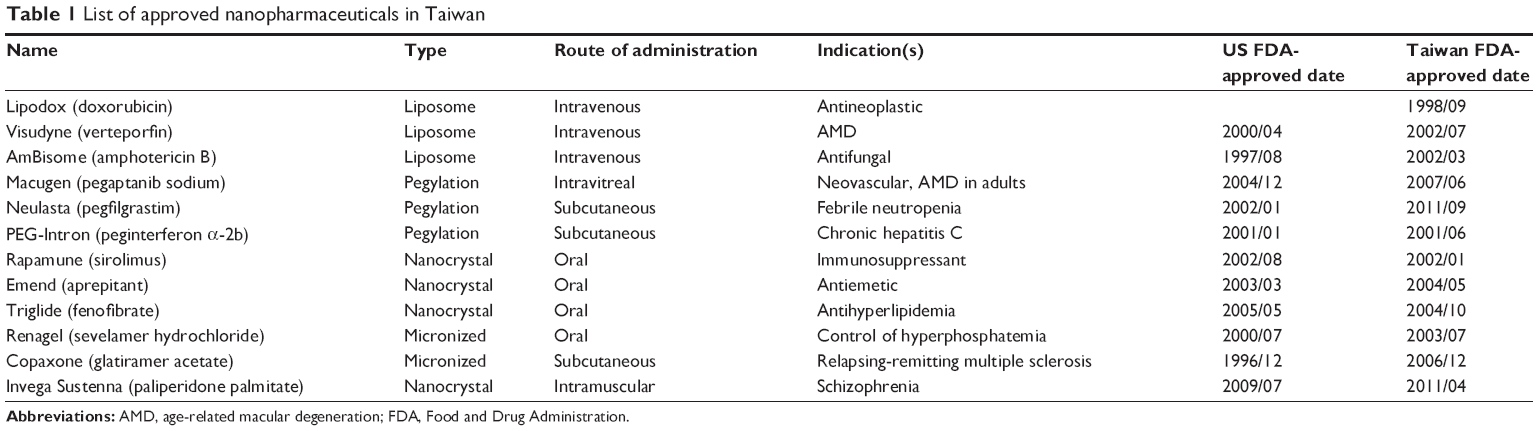 | Table 1 List of approved nanopharmaceuticals in Taiwan |
The impact of nanomaterials on biological systems may be the result of physical or chemical mechanisms.22 In the case of physical mechanisms, it is known that nanoparticles have sizes comparable with proteins and thus may find entry into biological compartments, including the cardiovascular system and the brain. Such mechanisms may induce genotoxic effects or neurotoxicity.23,24 Also, nanosize materials may induce disruption of the cell membrane, which may include the formation of holes, membrane thinning, and/or membrane erosion.25 They may also provoke alterations in membrane activity,26 regulate intracellular signaling pathways,27 increase the uptake of nanoparticles by interacting with the functional groups of membrane protein,28 disrupt the cytoskeleton,29 and form intranuclear protein aggregates.30 As a result, the small size of nanomaterials can induce cellular injury due to particle–cell interactions.31
The agglomeration and aggregation of nanoparticles also play an important role in biological systems. The degree of aggregation changes the size and the protein adsorption of nanoparticles in biological systems, thereby influencing the cellular internalization pathways and possibly leading to different cellular responses.18 The agglomeration state of nanoparticles influences the site of particle deposition, thus affecting cellular mechanisms and possibly inducing different toxicities.32–36 In the case of chemical mechanisms, nanoparticles have a larger surface area to undergo adsorption or reactions, thus manifesting increased chemical reactivity compared with the same mass of conventional materials.31 Some physicochemical properties have been found to be relevant to toxicity, such as reactive oxygen species generation and free radical formation. These processes can increase oxidative stress, induce mitochondrial perturbation, enhance protein denaturation, and promote the inflammation process. Finally, reactive oxygen species may alter cell cycle regulation, result in deoxyribonucleic acid damage, and lead to cell injury.18,31 Because the unique physical and chemical properties of nanoscale drugs could strongly affect their efficacy, safety, and pharmacokinetic properties, regulatory guidance to meet the needs of nanomedicine-specific properties during the developmental phase is very important.7,14
Development and management trends in nanopharmaceutical regulations
In the 1960s, pharmaceutical researchers started to utilize nanotechnology and created a novel lipid-based vesicle called a liposome, which was composed of lipid bilayers enclosing aqueous compartments, for drug delivery. Extensive liposome research has been conducted during the last five decades and several liposomal-based drugs have been approved in the US since 1995. This led to the recognition of the importance of regulatory issues in liposomal-based medicines and to the US Food and Drug Administration’s (FDA) publishing of the draft Guidance for Industry: Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation in 2002.37 This document provided recommendations to applicants with regard to the documentation required for liposome drug products submitted in new drug applications. However, considering the significant complexity and relatively novel nature of different liposomal or nanoscale drugs, a case-by-case or product class-specific approach for their evaluation and regulation would seem to be necessary.8,14 The US FDA thus published its Draft Guidance on Doxorubicin Hydrochloride in 2010,38 providing recommendations for the test and reference PEG liposome products when they: 1) have the same drug product composition, 2) are manufactured by an active liposome loading process with an ammonium sulfate gradient, and 3) have equivalent liposome characteristics, including liposome composition, state of encapsulated drug, internal environment of liposome, liposome size distribution, number of lamella, grafted PEG at the liposome surface, electrical surface potential or charge, and in vitro leakage rates.38
In the early 21st century, because the development of nanotechnology-based drugs was still in its infancy, there were no established standards for the study or regulatory evaluation of these products. In response to this, the US FDA formed a Nanotechnology Task Force to determine regulatory approaches that would enable continuance of the development of innovative, safe, and effective FDA-regulated products that use nanoscale materials. In 2007, the task force released its first nanotechnology report in which it highlighted scientific and regulatory issues involving products containing nanoscale materials. In 2008, following the task force’s recommendations, the Office of Pharmaceutical Science (OPS) within the Center for Drug Evaluation and Research (CDER) convened an advisory committee meeting where one of the topics was the use of nanotechnology in drug manufacturing, drug delivery, and drug products. To that end, OPS started to develop a comprehensive database of products containing nanomaterials that were submitted to CDER for drug applications. In 2010, CDER developed a format, the Manual of Policies and Procedures (MAPP) – Reporting Format for Nanotechnology-related Information in CMC Review, to help reviewers document in their reviews relevant information when an application is made for a product containing nanomaterials. The MAPP provided chemistry, manufacturing, and controls (CMC) reviewers within OPS with a framework by which relevant information about nanotechnology-related drugs or nanomaterial-containing drugs could be captured in CMC reviews of current and future CDER drug application submissions. These products may include nanoparticles, dendrimers, liposomes, micelles, nanoemulsions, nanocrystals, and metal colloids. This information would be entered into a nanotechnology database under construction and ultimately be used to develop policy regarding these products.39
Nanotechnology provides a new direction for development of nanodrug entities and drug delivery systems. Currently existing are the first-generation nanomedicines, including liposomal formulations, iron-based preparations, and drug nanocrystal technologies in oral dosage forms. Although the first-generation nanomedicines have been in successful routine clinical use for many years, the “follow-on” nanoscale drugs called “nanosimilar”, as well as the ongoing development of second- or next-generation nanomedicines, is underway. The latter employs the latest nanotechnology or manufacturing processes to create even smaller, more complex, and increasingly hybrid structures – drugs that have not been regulated for quality, safety, and efficacy. For example, PEG gold nanoparticles covalently linked to tumor necrosis factor-α as an anticancer agent have been in early clinical trials, and many multicomponent systems involving two or more components have been described.40 Therefore, it is important now for regulatory agencies to provide appropriate regulatory guidance and assist the development of nanomedicines.14 As early as 2011, the European Medicines Agency (EMA) released a reflection paper on nonclinical studies for generic nanoparticle iron-containing medicinal product applications. Later in 2013, EMA further provided another four draft reflection papers with a framework for discussion or clarification, particularly in areas where scientific knowledge is quickly evolving or experience is limited. These included block copolymer micelle medicinal products,41 intravenous liposomal products,42 surface-coated nanomedicine products,43 and intravenous iron-based nanocolloidal products.44 These draft reflection papers focused mainly on the establishment and confirmation of the physicochemical properties of nanomedicines, and detailed the supplementary nonclinical and clinical data requirement to ensure the ultimate quality, safety, and efficacy of the final nanoscale products.14 A pharmacovigilance/risk management plan may also be necessary. Last but not least, the European Commission recommended that the definition of nanomaterial be based on the size 1–100 nm. However, it was acknowledged that an upper limit had no reasonable scientific basis for all nanomaterials, especially with regard to pharmaceuticals. The definition of nano still needs extensive discussion in the international regulatory community.14
In April 2013, the Organisation for Economic Co-operation and Development (OECD) Working Party on Nanotechnology (WPN) began a project called Regulatory Frameworks for Nanotechnology in Food and Medical Products. This project was based on the active participation of 12 WPN delegations from 2011 to 2012 and provided an overview of some of the regulatory frameworks applicable to food and medical products that may contain nanomaterials or may otherwise involve the application of nanotechnology. The survey report of the medical products showed that ten delegations responded to the regulatory frameworks survey component, while seven delegations responded to the government-sponsored regulatory science research and other research activities survey component.45 Although the OECD survey report showed that most of the WPN delegations established the regulatory frameworks for nanomedicines, the regulation still needs to be seriously discussed. Previous reports,8,14 including the US FDA document Considering Whether an FDA-regulated Product Involves the Application of Nanotechnology in 2011,46 all suggested that the evaluation and regulation of nanoscale drugs should be considered in a product-specific approach, meaning a case-by-case process. However, the US FDA has not clearly identified whether products containing nanomaterials or involving the application of nanotechnology are harmful or not. Thus, the future might see further regulations by following or expanding Principles for Regulation and Oversight of Emerging Technologies47 and Policy Principles for the US Decision-making Concerning Regulation and Oversight of Applications of Nanotechnology and Nanomaterials.48 Currently, when the FDA considers whether a product contains nanomaterials or involves the application of nanotechnology, it asks the following questions: 1) whether the manufacture of artificial materials or final products has at least one dimension in the nanosize range (approximately 1–100 nm) and 2) whether the manufacture of artificial materials or final products exhibits the specific nanoscale nature (including physical, chemical, or biological effects), even though the size of the materials or products is in excess of the nanosize range up to 1 μm.
Development and suggestions for Taiwan’s nanopharmaceutical regulation
Although confronted with an increasingly rapid development of nanodrugs and similar products, Taiwan’s Food and Drug Administration (Taiwan FDA) has not yet established specific nanotechnology-based drug-related regulations. Currently, there is only general regulation for liposomal drug products without emphasis on the requirement for CMC review. There appeared to be a gap in regulating nanomedicines between Taiwan and US/EU/Japan (Table 2). In order to ensure the quality and safety of nanomedicines, the Taiwan FDA needs to establish the mechanisms to regulate nanomedicines. Accordingly, the Taiwan FDA started a 6-year project entitled Nanotechnology in Biomedical Applications and its Regulations under Taiwan’s National Nanoscience and Nanotechnology Program in 2009. One of the subprojects focused on the regulation of nanopharmaceuticals aims 1) to compare international legislation and practical implementation, 2) to clarify the current regulatory frameworks and status of the pharmaceutical industry in Taiwan, and 3) to publish draft guidance for regulating nanopharmaceuticals. By means of expert panel discussions and public hearings, the outcome of this project is expected to promote Taiwan’s nanotechnological research and to assist pharmaceutical applications and developments, harmonized with general international standards.
 | Table 2 Comparison of nanopharmaceutical regulation between Taiwan and US/EU/Japan |
For the execution of the project, we formed an expert committee and CMC review group. The expert committee included members from academia with multiple disciplines, from research institutions and from the pharmaceutical industry, with experience in nanodrug development and regulatory agencies. Through routinely based meetings, experts discussed the progress of international regulations relevant to nanomedicines and attempted to establish a cooperative direction of Taiwan’s nanomedicine regulations. The CMC review group invited senior reviewers from Taiwan’s Center for Drug Evaluation (Taiwan CDE) and technical representatives from Taiwan’s Center for Pharmaceutical Technology Development and the pharmaceutical industry. The mission of this group was to discuss the technical aspects of CMC review for nanomedicines.
Through implementation of the project by means of routine discussion of the expert committee and CMC review group, visits of pharmaceutical companies developing and/or manufacturing nanodrugs, organized symposiums related to nanotechnology and regulatory sciences, and, finally, briefings and hearings for the pharmaceutical industry, we progressively established a direction and reached a consensus for regulating nanomedicine as follows.
- The international consensus for nanomaterial scales can be summarized as about 1–100 nm. With respect to nanomedicine regulation, most countries have their own premarketing drug review processes. There has not been a clear-cut definition for the dimensions of pharmaceutical nanoproducts, and there is no specific legislation for such products. A case-by-case or product class-specific approach for evaluation and regulation should be considered.
- No matter whether a new drug product contains nanomaterials or involves the application of nanotechnology, the regulation should focus on the quality, safety, and efficacy of the final product, not on the specific applications of nanotechnology. However, a clearly defined review process will provide direction for the pharmaceutical industries and the regulatory agencies to follow.
- The safety and risks of nanotechnology-based drugs vary among the different types of products, eg, nanoparticles, dendrimers, liposomes, micelles, nanoemulsions, nanocrystal, and metal colloids. The regulatory requirements should be different for products applying novel nanotechnologies as opposed to products already on the market with substantial experience – products such as liposomal drugs and nanoemulsions.
- The rapid development of nanotechnology leads to new drug products with more complex structures and properties. It is increasingly difficult to classify a nanotechnology-based drug as being of a new chemical entity or of a new formulation. It was recommended that we utilize an approach similar to the US CDER reporting format for CMC reviews, thereby establishing our own database for nanotechnology-based drug products. Detailed information for CMC reviews will help pharmaceutical industries, CMC reviewers, and regulatory agencies to understand the nature and characteristics of nanotechnology-based drugs. The establishment of a domestic database of nanoproducts that is similar to the US system will improve international harmonization for further reviews as well as the subsequent monitoring and supervision of these products by the administration.
- Different reviewers may have different points of view on the technical specifications and reviews of new nanosized formulations with existing ingredients. It is recommended to follow the international approach, ie, to set up individual guidance and test requirements for different products. This will expedite the development of new drugs and the process of registration.
- Site visiting and exchanging opinions with four pharmaceutical companies involved in developing and/or manufacturing nanodrug products assisted us in understanding the ongoing concerns that occur during the processes of drug development, manufacturing, inspection, and registration. The current Good Manufacturing Practice regulation is still applicable for the manufacturing of nanodrug products. However, considering the rapid development of nanotechnology, it should be kept in mind that the current Good Manufacturing Practice guidance may still need to be amended.
- Continued attention should be paid to the international regulatory progress of safety issues, risk management, regulation, and development of analytical technology for nanotechnology-based drug products. An updated information database should be readily available as a reference for regulatory agencies, pharmaceutical companies, and research institutions.
In accordance with those recommendations and consensus, the following regulatory mechanisms are proposed and drafted.
- With all new drug applications involving nanotechnology, including dendrimers, liposomes, micelles, nanocrystals, polymer–drug conjugates, solid nanoparticles, metal colloids, and others, the applicants should provide a CMC checklist for technical review (see Supplementary material).
- Guidance for technical review of CMC of the liposomal drug products was drafted, with an attachment of the current regulations for liposomal drug products, including new chemical entities, new administration routes, new indications, new combinations, new dosage forms, new doses, new dose units, and generic formulations.
Liposomal drug guidance was considered in the first place since there are several products on the market and also several under development at different phases by local pharmaceutical companies. A couple of micellar products are also in development. However, experience remains lacking at present to set up regulatory standards for such products. Although there is no urgent need, careful attention should still be paid to the progress of international regulations, the technical levels of the latest and pending development of nanoproducts, and the needs of the local pharmaceutical industry. When the updated information has been assembled and adequately considered, appropriate responses can be made.
Taiwan’s CMC review checklist was adapted and modified from Reporting Format for Nanotechnology-related Information in CMC Review of the US CDER, 2010. The format was evaluated by both the CMC reviewers of the Taiwan CDE and the pharmaceutical applicants using several examples of products already approved or under development. After several discussions to accommodate the status of domestic pharmaceutical development, the checklist was accepted by all the parties at large. The CMC review checklist is expected not only to help the administrator set up the CMC review process for nanomedicine and a database for nanomedicine here in Taiwan but also to provide postmarket monitoring of these products in the future.
Despite the majority of the approved nanopharmaceuticals being liposomal, with several in development, the assessment criteria of CMC reviews for drug applications have not been explicitly stated. To meet the needs of drug applicants and regulatory reviewers, guidance for technical review of CMC of the liposomal drug products was proposed and adapted from both the US FDA’s draft Guidance for Industry: Liposome Drug Products (2002) and Draft Guidance on Doxorubicin Hydrochloride (2010), as well as EMA’s Reflection Paper on the Data Requirements for Intravenous Liposomal Products Developed With Reference to an Innovator Liposomal Product (2013). At the time of this paper’s writing, the draft guidance has been submitted to the expert committee for discussion and will be open to the public’s comments and suggestions soon afterwards. We expect a final consensus to be reached by the end of 2014.
Conclusion
There is no international regulation or legislation specifically for nanomedicine, but it is agreed worldwide that considerably more attention should be paid to the quality, safety, and efficacy of nanotechnology-based drugs. Meanwhile, we should also establish new assessment methods or revise existing assessment methods to cope with the impact of nanotechnology. Immediate consideration should be given to liposomal drug products, followed by metal colloids and block copolymer micelles. Because of the specific and sometimes unique physicochemical properties of each new nanomaterial and drug product, a case-by-case or product class-specific approach for evaluation and regulation seems to be necessary. We need to ensure that nanomedicines enter clinical development and consequently the market in a safe way that benefits people’s health. CMC review is a very important gate control for quality consistency and hence the utility of the final products. The proposed contemporary strategies of Taiwan include a CMC review checklist and draft guidance for CMC review of liposomal products. Once a consensus is reached among the expert committee, the industries, and the regulators, these mechanisms will be recommended to the Taiwan FDA for jurisdiction and may be initiated as the starting point for regulating nanotechnology-based pharmaceuticals in Taiwan.
Acknowledgment
This work was supported by grants DOH101-FDA-41301, DOH102-FDA-41702, and MOHW103-FDA-41407 from the Food and Drug Administration, Ministry of Health and Welfare, Taiwan.
Disclosure
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Food and Drug Administration, Ministry of Health and Welfare, Taiwan. The authors report no conflicts of interest in this work.
References
Roco MC. Nanotechnology: convergence with modern biology and medicine. Curr Opin Biotechnol. 2003;14(3):337–346. | ||
Shah RB, Khan MA. Nanopharmaceuticals: challenges and regulatory perspective. In: de Villiers MM, Aramwit P, Kwon GS, editors. Nanotechnology in Drug Delivery. New York: Springer New York; 2009:621–646. | ||
National Nanotechnology Initiative. Nanotechnology resources page. 2004; http://www.nano.gov/nanotech-101/what/definition. Accessed March 24, 2014. | ||
Board of Science and Technology, Executive Yuan. Available from: http://www.bost.ey.gov.tw/Default.aspx. Accessed August 21, 2014. | ||
Tinkle S, McNeil SE, Muhlebach S, et al. Nanomedicines: addressing the scientific and regulatory gap. Ann N Y Acad Sci. 2014;1313:35–56. | ||
Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine. 2013;9(1):1–14. | ||
Kumar A, Chen F, Mozhi A, et al. Innovative pharmaceutical development based on unique properties of nanoscale delivery formulation. Nanoscale. 2013;5(18):8307–8325. | ||
Chan VS. Nanomedicine: an unresolved regulatory issue. Regul Toxicol Pharmacol. 2006;46(3):218–224. | ||
Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. | ||
Gabellieri C, Frima H. Nanomedicine in the European Commission policy for nanotechnology. Nanomedicine. 2011;7(5):519–520. | ||
Zolnik BS, Sadrieh N. Regulatory perspective on the importance of ADME assessment of nanoscale material containing drugs. Adv Drug Deliv Rev. 2009;61(6):422–427. | ||
Bawa R. FDA and nanotech: baby steps lead to regulatory uncertainty. In: Bagchi D, Moriyama H, Shahidi F, editors. Bio-Nanotechnology: A Revolution in Food, Biomedical and Health Sciences. Oxford, UK: Blackwell Publishing Ltd; 2013. | ||
Taiwan Food and Drug Administration-Approved Drug Database. Available from: http://www.fda.gov.tw/MLMS/(S(xuro5q55k2sldgeu3smyi3qv))/H0001.aspx. Accessed August 21, 2014. | ||
Ehmann F, Sakai-Kato K, Duncan R, et al. Next-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Nanomedicine. 2013;8(5): 849–856. | ||
Clinical Trials Network in Taiwan. Available from: http://www1.cde.org.tw/ct_taiwan. Accessed August 21, 2014. | ||
Allhoff F. The coming era of nanomedicine. Am J Bioeth. 2009;9(10): 3–11. | ||
Linkov I, Satterstrom FK, Corey LM. Nanotoxicology and nanomedicine: making hard decisions. Nanomedicine. 2008;4(2):167–171. | ||
Fubini B, Ghiazza M, Fenoglio I. Physico-chemical features of engineered nanoparticles relevant to their toxicity. Nanotoxicology. 2010;4:347–363. | ||
Sharma A, Madhunapantula SV, Robertson GP. Toxicological considerations when creating nanoparticle-based drugs and drug delivery systems. Expert Opin Drug Met. 2012;8(1):47–69. | ||
Tsuji JS, Maynard AD, Howard PC, et al. Research strategies for safety evaluation of nanomaterials. Part IV: Risk assessment of nanoparticles. Toxicol Sci. 2006;89(1):42–50. | ||
Oberdorster G. Safety assessment for nanotechnology and nanomedicine: concepts of nanotoxicology. J Intern Med. 2010;267(1):89–105. | ||
Nel AE, Madler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–557. | ||
Sharma HS, Sharma A. Neurotoxicity of engineered nanoparticles from metals. CNS Neurol Disord Drug Targets. 2012;11(1):65–80. | ||
Guo YY, Zhang J, Zheng YF, Yang J, Zhu XQ. Cytotoxic and genotoxic effects of multi-wall carbon nanotubes on human umbilical vein endothelial cells in vitro. Mutat Res. 2011;721(2):184–191. | ||
Leroueil PR, Berry SA, Duthie K, et al. Wide varieties of cationic nanoparticles induce defects in supported lipid bilayers. Nano Lett. 2008;8(2):420–424. | ||
Navarro E, Baun A, Behra R, et al. Environmental behavior and ecotoxicity of engineered nanoparticles to algae, plants, and fungi. Ecotoxicology. 2008;17(5):372–386. | ||
Ovrevik J, Lag M, Schwarze P, Refsnes M. p38 and Src-ERK1/2 pathways regulate crystalline silica-induced chemokine release in pulmonary epithelial cells. Toxicol Sci. 2004;81(2):480–490. | ||
Hauck TS, Ghazani AA, Chan WC. Assessing the effect of surface chemistry on gold nanorod uptake, toxicity, and gene expression in mammalian cells. Small. 2008;4(1):153–159. | ||
Wu YL, Putcha N, Ng KW, et al. Biophysical responses upon the interaction of nanomaterials with cellular interfaces. Acc Chem Res. 2013;46(3):782–791. | ||
Chen M, von Mikecz A. Formation of nucleoplasmic protein aggregates impairs nuclear function in response to SiO2 nanoparticles. Exp Cell Res. 2005;305(1):51–62. | ||
Bakand S, Hayes A, Dechsakulthorn F. Nanoparticles: a review of particle toxicology following inhalation exposure. Inhal Toxicol. 2012; 24(2):125–135. | ||
Noel A, Charbonneau M, Cloutier Y, Tardif R, Truchon G. Rat pulmonary responses to inhaled nano-TiO2: effect of primary particle size and agglomeration state. Part Fibre Toxicol. 2013;10(1):48. | ||
Grassian VH, Adamcakova-Dodd A, Pettibone JM, O’Shaughnessy PI, Thorne PS. Inflammatory response of mice to manufactured titanium dioxide nanoparticles: Comparison of size effects through different exposure routes. Nanotoxicology. 2007;1(3):211–226. | ||
Balasubramanian SK, Poh KW, Ong CN, Kreyling WG, Ong WY, Yu LE. The effect of primary particle size on biodistribution of inhaled gold nano-agglomerates. Biomaterials. 2013;34(22):5439–5452. | ||
Muhlfeld C, Mayhew TM, Gehr P, Rothen-Rutishauser B. A novel quantitative method for analyzing the distributions of nanoparticles between different tissue and intracellular compartments. J Aerosol Med. 2007;20(4):395–407. | ||
Muhlfeld C, Rothen-Rutishauser B, Blank F, Vanhecke D, Ochs M, Gehr P. Interactions of nanoparticles with pulmonary structures and cellular responses. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L817–L829. | ||
Guidance for Industry: Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and: Bioavailability; and Labeling Documentation. Draft Guidance. US Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); 2002. | ||
Draft Guidance on Doxorubicin Hydrochloride. US United States Food and Drug Administration; 2010. | ||
Reporting Format for Nanotechnology-related Information in CMC Review. US Food and Drug Adminitration, Center for Drug Evaluation, Office of Pharmaceutical Science; 2010. | ||
Duncan R, Gaspar R. Nanomedicine(s) under the Microscope. Mol Pharm. 2011;8(6):2101–2141. | ||
Joint MHLW/EMA Reflection Paper on the Development of 4 Block Copolymer Micelle Medicinal Products. Draft. European Union, European Medicines Agency; 2013. | ||
Reflection Paper on the Data Requirements for Intravenous Liposomal Products Developed With Reference to an Innovator Liposomal Product. Draft. European Union, European Medicines Agency; 2011. | ||
Reflection Paper on Surface Coatings: General Issues for Consideration Regarding Parenteral Administration of Coated Nanomedicine Products. European Union, European Medicines Agency; 2012. | ||
Reflection Paper on the Data Requirements for Intravenous Iron-based Nano-colloidal Products Developed With Reference to an Innovator Medicinal Product. Draft. European Union, European Medicines Agency; 2013. | ||
Organisation for Economic Co-operation and Development. Regulatory Frameworks for Nanotechnology in Foods and Medical Products: Summary Results of a Survey Activity. OECD Science, Technology and Industry Policy Papers, No. 4; 2013. | ||
Considering Whether an FDA-regulated Product Involves the Application of Nanotechnology. Guidance for Industry. Draft. US Department of Health and Human Services, Food and Drug Administration, Office of the Commissioner; 2011. | ||
Principles for Regulation and Oversight of Emerging Technologies. US White House Emerging Technologies Interagency Policy Coordination Committee; 2011. | ||
Policy Principles for the US Decision-making Concerning Regulation and Oversight of Applications of Nanotechnology and Nanomaterials. US Office of Science and Technology Policy, White House; 2011. |
Supplementary materials
(This is English translation from its original Chinese version)
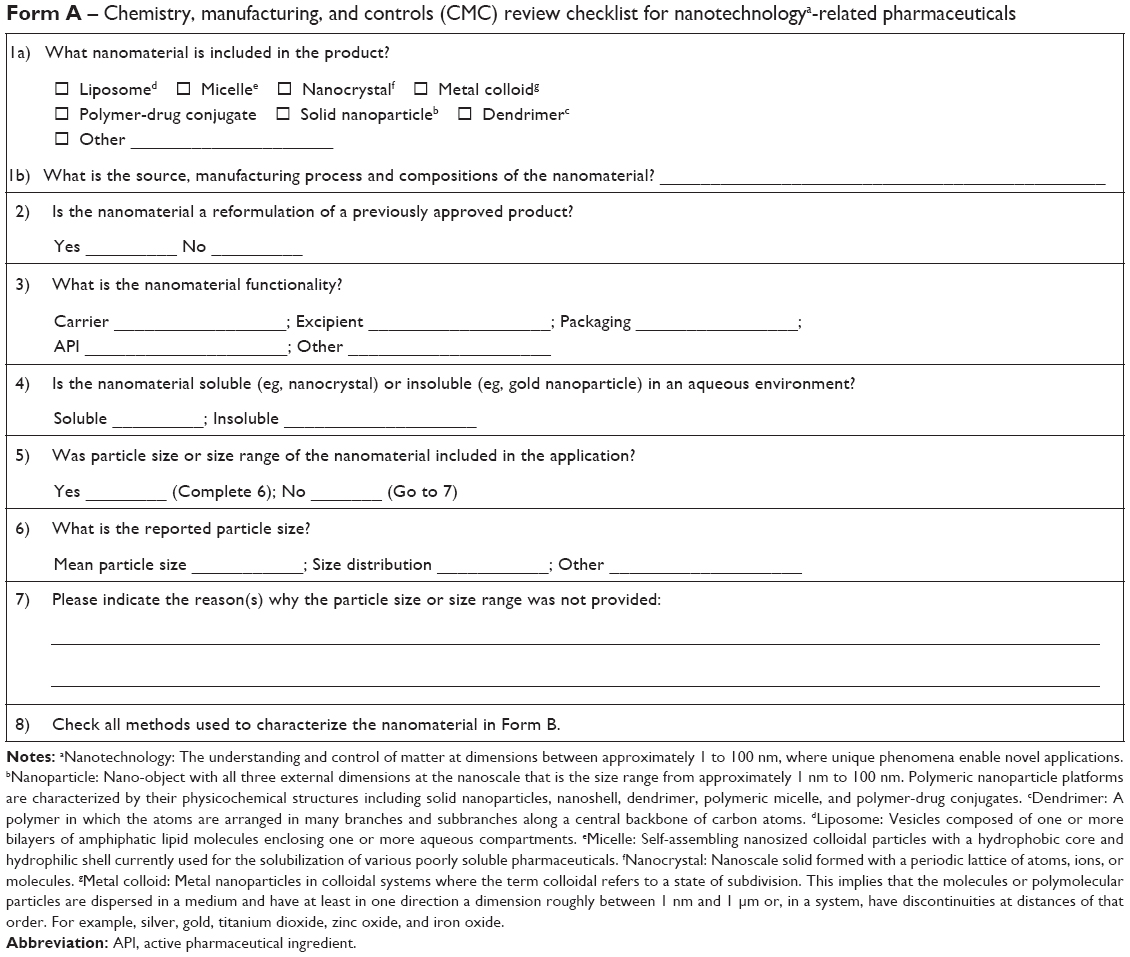 | Form A –Chemistry, manufacturing, and controls (CMC) review checklist for nanotechnologya-related pharmaceuticals |
 | Form B –Methods used to characterize the nanomaterial* |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.