")
Back to Journals » Cancer Management and Research » Volume 10
Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer
Authors Pan JH, Zhou H, Zhu SB, Huang JL, Zhao XX, Ding H, Pan YL
Received 4 April 2018
Accepted for publication 16 May 2018
Published 1 August 2018 Volume 2018:10 Pages 2289—2301
DOI https://doi.org/10.2147/CMAR.S170105
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Harikrishna Nakshatri
Jing-hua Pan,1 Hong Zhou,2 Sheng-bin Zhu,1 Jin-lian Huang,1 Xiao-xu Zhao,1 Hui Ding,1 Yun-long Pan1
1Department of General Surgery, The First Affiliated Hospital of Jinan University, Guangzhou 510632, China; 2Department of Gynecology, The First Affiliated Hospital of Jinan University, Guangzhou 510632, China
Abstract: RAF kinase is crucially involved in cell proliferation and survival in colorectal cancer (CRC). Patients with metastatic CRC (mCRC) harboring BRAF mutations (BRAFms) not only experience a poor prognosis but also benefit less from therapeutics targeting ERK signaling. With advances in RAF inhibitors and second-generation inhibitors including encorafenib and vemurafenib, which have been approved for treating BRAF-V600E malignancies, the combinatorial therapeutic strategies of RAF inhibitors elicit remarkable responses in patients with BRAF-V600E mCRC. However, the therapeutic efficacy is restricted by resistance, which might be due to RAF dimerization and reactivation of the MAPK pathway. In addition, the next-generation RAF inhibitors, which are characterized by varying structural and biochemical properties, have achieved preclinical and clinical advances. Herein, we summarize the existing mechanism of RAF kinases in CRC, including MAPK feedback reactivation of resistance to RAF inhibitors. We additionally summarize the development of three generations of RAF inhibitors and different therapeutic strategies including the combination of EGFR, BRAF, and PI3K inhibitors for BRAFm CRC treatment.
Keywords: BRAF, inhibitor, colorectal cancer, V600E, structure, resistance
Introduction
Colorectal cancer (CRC) is one of the most common malignancies both in men and women globally.1 In recent decades, great achievements have been made in decreasing both the incidence and mortality rates of CRC in developed countries through decreased exposure to risk factors (eg, reduced smoking and red meat consumption and enhanced uptake of aspirin) and the introduction and prevalence of screening examinations, as well as therapeutic progress.2 In addition, high-quality health care, as well as incentivization of healthier lifestyles, play a role in decreasing CRC incidence.3 However, especially among patients older than 50 years, where CRC incidence rate rose by 22% from 2000 to 2013,4 the different clinical and biological features of CRC lead to different therapeutic responses and subsequent prognoses, suggesting distinct etiologic mechanisms in CRC.5
Oncogenic mutations in B-type RAF kinase (BRAF) occur in 8–12% of metastatic CRC (mCRC), and the most universal BRAF mutation (BRAFm) is the missense mutation V600E, which is seen in ~90% of subjects.6 Additionally, these mutations are likely to be strongly associated with tumor aggressiveness and poor prognosis.7 The RAF kinase family (ARAF and BRAF, as well as CRAF [also known as RAF1]) consists of key components of the RAS–RAF–MEK–ERK signaling cascade (ERK signaling), which mediates signals from cell surface receptors to the nuclei during the modulation of cell growth and differentiation, as well as survival.8,9 As soon as the RAF kinases are activated, MEK1 and MEK2 are subsequently phosphorylated and activated, followed by the phosphorylation and activation of ERK1 and ERK2. Activated ERK enhances cell proliferation, as well as survival, via phosphorylation of numerous substrates both in the cytosol and the nucleus.10 The RAF family has been an important therapeutic target for CRC, and there have been great advances in the development of RAF inhibitors for clinical use.11
The RAS–RAF–MEK–ERK pathway is one of the most attractive pathways as the designed target of small-molecule drugs.12 Regarding RAF kinases, the first-generation inhibitors have been used in the clinic but the first-generation inhibitor was limited for further use due to BRAFm.13 Additionally, clinical results show a short resistance time when using second-generation RAF inhibitors because of dimer formation and feedback reactivation of RAS.14,15 Moreover, the next-generation inhibitors are currently undergoing preclinical and clinical trials, which focus on the inhibition of monomeric forms and dimeric forms of RAF but focus less on target toxicities.16 Therefore, to further understand the development of small-molecule therapeutics and strategies for targeting RAF kinase in CRC, we summarized the existing advancements in our understanding of RAF activation and the mechanisms of resistance to RAF inhibitors and aimed to propose a comprehensive understanding of the development of small-molecule drugs and future perspectives regarding BRAFm CRC.
The structure and mechanism of RAF kinase in BRAFm CRC
Structural insight into RAF activation and different types of RAF inhibitors
A deeper understanding of the three-dimensional structure of the RAF protein has played a very important role in the development of small molecules for RAF inhibition. The inactive RAF protein exhibits a closed and monomeric conformation in the cytoplasm because of the intramolecular interaction between the carboxyl and amino terminal domains.17–19 The structure of RAF harbors the typical C-terminal lobe (C-lobe) and N-terminal lobe (N-lobe) connected by a flexible hinge. There are three major active sites of RAF, including the nucleotide (ADP or ATP)-binding site, the magnesium-binding site (DFG [Asp–Phe–Gly] motif), and the phospho-acceptor site (activation segment [AS]) of the two lobes (Figure 1A).20 A closed conformation between the two lobes is necessary for the catalytic activity of the kinase domain.21 In inactive RAF, the AS exhibits a helical conformation, leading to the positioning of the αC helix in the “OUT” position (Figure 1A). However, in active RAF, the AS is transformed to an extended conformation, enabling the αC helix of the N-lobe to flex to the “IN” position (Figure 1B). Additionally, the dimerization of RAF potentiates its independent activation and limits the response to conventional RAF inhibitors because the αC helix is in the OUT position. Excluding the RAF monomer, RAF dimerization will confer resistance against the RAF inhibitors. Based on the structural insight into RAF dimerization, next-generation RAF inhibitors, such as TAK632, can stabilize the αC helix in the IN (active) position, which allows binding of a second TAK632 molecule to the second protomer (Figure 1C).22
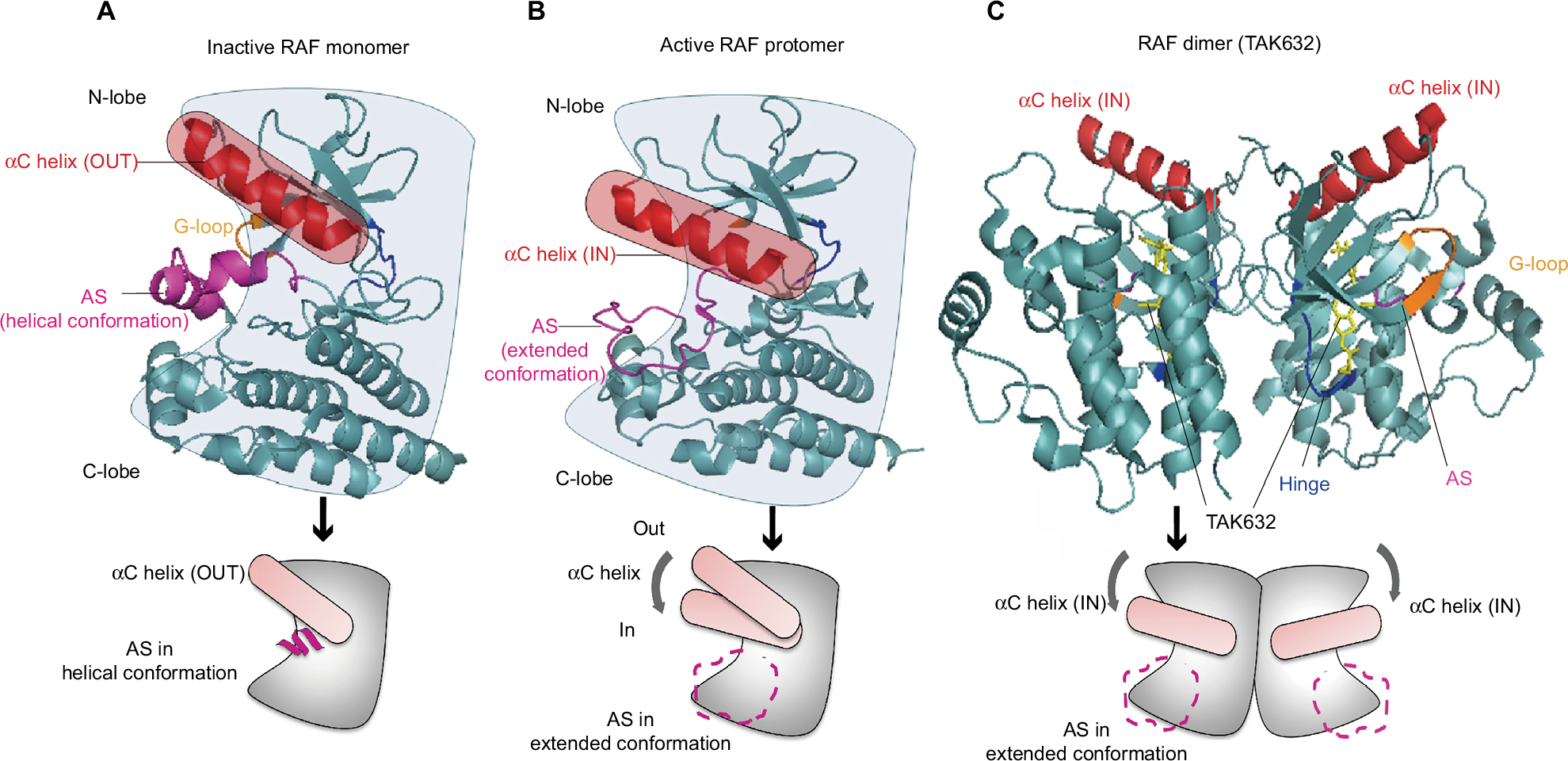 | Figure 1 Insight on the structural characteristics of RAF activation. Notes: (A) The inactive RAF is a monomer, and the kinase domain has the characteristic N-lobe and C-lobe linked by a hinge. The positioning of the αC helix of N-lobe maintains its “OUT” conformation, and the AS forms as a helical conformation (PDB ID: 4RZV). (B) After the RAF activation, the conformation of AS is extended, which induces the αC helix to move inward to the “IN” position. The active RAF displays a comprehensive static “closed” conformation (PDB ID: 4MNE). (C) The structural characteristics of typical RAF inhibitors (TKA632) bound to a BRAF dimer show that both αC helixes are in the IN position (PDB ID: 4KSP). Abbreviations: AS, activation segment; PDB, Protein Data Bank. |
The DFG motif is another important factor in BRAF activity. When the DFG motif maintains the IN position (Figure 2A), the Asp residue is adapted to provide a catalytically competent active conformation.23 Type II inhibitors can stabilize the DFG motif in the OUT position and enable the Asp residue to point away from the active site (Figure 2C). Based on the structural features of the αC helix and DFG in BRAF, there are four representative structural types of RAF inhibitors in current development for targeting BRAF: type I inhibitor (αC-IN/DFG-IN) (Figure 2A), type I1/2 inhibitor (αC-OUT/DFG-IN) (Figure 2B), type II inhibitor (αC-IN/DFG-OUT) (Figure 2C), and type I1/2 inhibitor with R506-OUT (Figure 2D).21
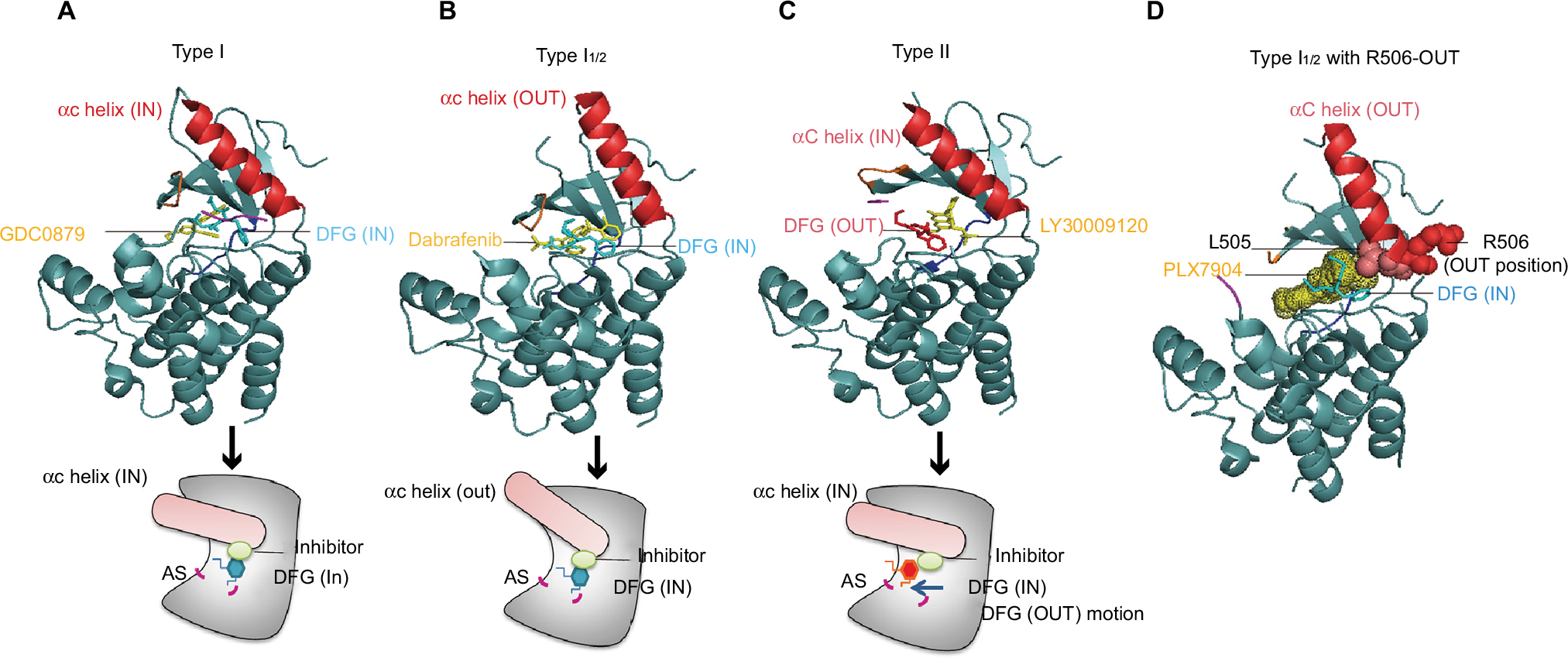 | Figure 2 The crystal structure of different types of RAF inhibitors binding to BRAF kinase. Notes: (A) The DFG motif (magnesium-binding site) is located at the N-lobe base of the AS, and the type I inhibitor (GDC0879) binding to BRAF displays the DFG motif of BRAF maintaining the “IN” position, as well as the αC helix (PDB ID: 4MMF). (B) The type I1/2 inhibitor (dabrafenib) binds to BRAF and stabilizes the αC helix in the “OUT” position, and the position of the DFG motif remains in the IN position (PDB ID: 4XV2). (C) The structure of a type II inhibitor (LY3009120)-binding BRAF shows the different DFG motif positions, and the DFG motif shows the OUT motion compared with the BRAF crystal structure binding with type I or I1/2 RAF inhibitors (PDB ID: 5C9C). (D) The IN position of the residue R506 of the αC helix associates with the interaction of RAF with RAS-GTP. PLX7940 is a type I1/2 RAF inhibitor, and except the αC helix OUT and DFG motif IN positions of BRAF, the residue R506 in the αC helix displays an outward movement upon binding of the type I1/2 RAF inhibitor, which reduces the paradoxical activation of ERK signaling. Abbreviations: AS, activation segment; PDB, Protein Data Bank. |
Despite numerous accessible crystal structures of wild-type (WT) BRAF or BRAF-V600E, the mechanism by which BRAF-V600 mutants activate BRAF remains poorly understood. Cell-based studies had shown that certain oncogenic BRAFm enhances spontaneous BRAF dimerization and activation by forming homodimers without RAS-GTP.24,25 Additionally, several mutations had been detected in residues in the AS or in the glycine-rich loop, which interplays with the former.13 The diversity of oncogenic mutations in BRAF limits the development of specific inhibitors based on structure. Therefore, the specific structural mechanism of different BRAFms still needs further study.
Feedback reactivation of MAPK signaling following BRAF inhibition
The feedback reactivation of MAPK is another critical factor in BRAF inhibitor resistance. Several mechanisms of feedback reactivation of MAPK had been described by some researchers.26–28 First, the constitutive activation of BRAFm drives downstream MAPK signaling and enhances ERK activation (Figure 3A). The activation of ERK leads to ERK-dependent negative feedback on receptor tyrosine kinase (RTK) activation and reduces RAS activity. However, when BRAF is suppressed by inhibitors, the ERK-dependent negative feedback is reduced, allowing enhanced activation of RTKs, as well as downstream RAS. Under these conditions, RAS activates CRAF, inducing reactivation of the MAPK signaling. Moreover, BRAF-V600 mutants are not affected by upstream feedback and they drive high levels of ERK signaling output, which profoundly inhibit intracellular RAS activity (Figure 3A). Therefore, BRAF-V600E predominantly exists as a drug-sensitive monomer and RAF inhibitors rapidly inhibit ERK signaling in BRAF-V600E cancer.26 It attenuates the feedback suppression of RAS, subsequently resulting in the induction of both WT BRAF and BRAF-V600E dimers. Additionally, RAF dimerization is able to effectively prevent RAF inhibition by current clinical αC-OUT RAF inhibitors, which is attributed to negative allostery. Because current RAF inhibitors selectively inhibit BRAF monomers and are much less potent inhibitors of RAF dimers, a rebound in ERK signaling ensues and attenuates the antitumor effects of these inhibitors.27 BRAF-V600E CRC is not sensitive to monotherapy with RAF inhibitors, and the ERK rebound after inhibition by RAF inhibitors is much greater than that observed in melanoma.28 Based on the mechanisms described earlier, several studies have reported that a combination of MEK and BRAF suppression reduces the rebound and is more effective than BRAF inhibition alone.29,30 Therefore, combinatorial strategies are being evaluated in clinical trials and might improve the response in BRAFm CRC (Figure 3B).
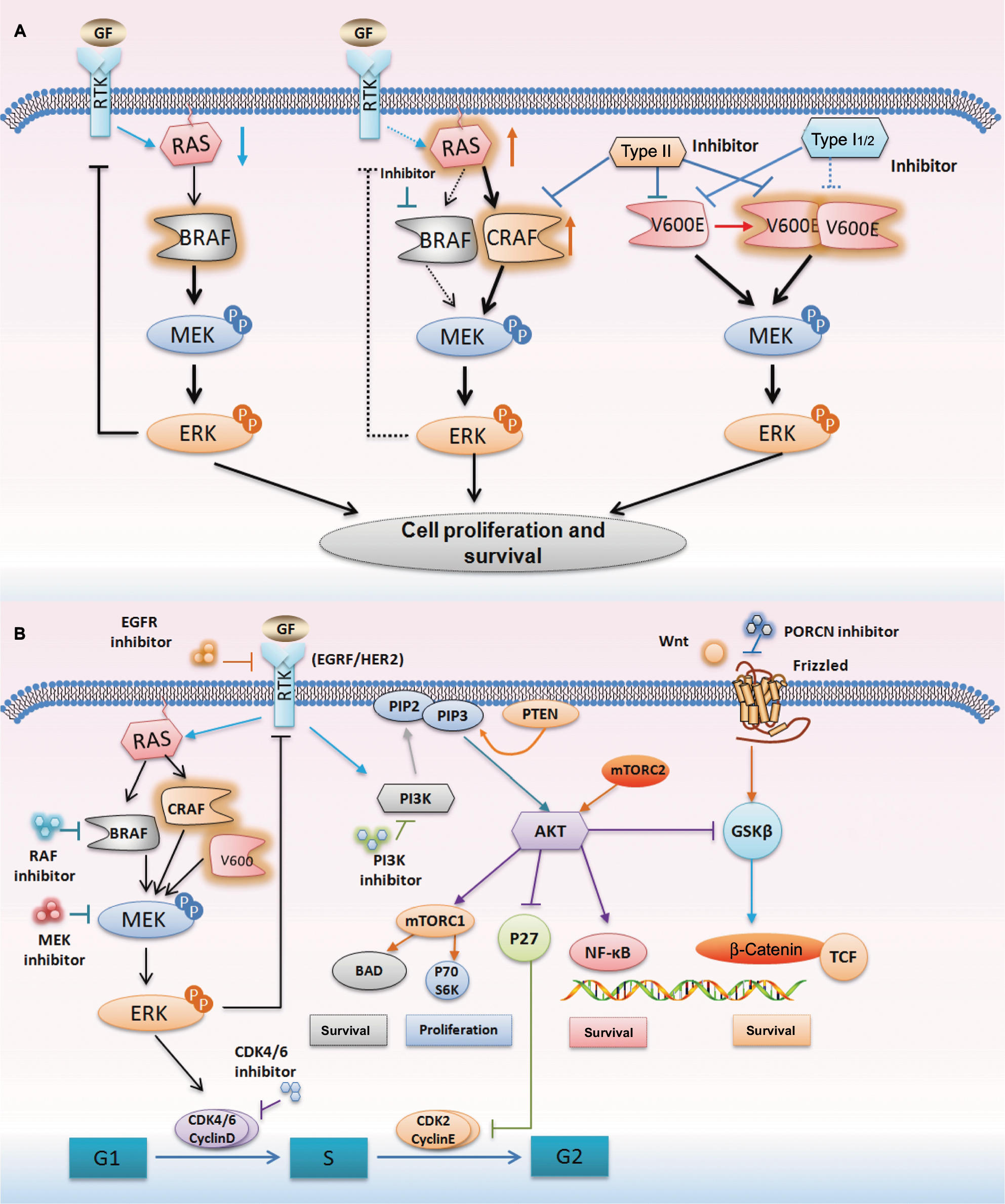 | Figure 3 The mechanism of feedback reactivation following BRAF inhibition and current combinatorial therapeutic targets. Notes: (A) The activation of BRAF drives downstream MAPK signaling, and the activation of ERK leads to negative feedback on RTK activation and reduces RAS activity. When BRAF is suppressed by inhibitors, the ERK feedback is reduced and the activation of RAS is enhanced. RAS activates CRAF, inducing reactivation of the MAPK signaling. However, BRAF with the V600E mutant can drive high levels of ERK signaling output independently. Type II inhibitors can inhibit not only BRAF and CRAF but also BRAF with V600E mutant. (B) Outline of current combinatorial therapeutic targets based on the MAPK, PI3K/AKT, and Wnt/β-catenin pathways. These pathways have been identified as a mechanism of resistance to BRAF inhibitors. Abbreviation: RTK, receptor tyrosine kinase. |
BRAFm as a prognostic factor in clinical studies of treatment of mCRC
Because BRAFm had been detected in mCRC samples, studies have suggested that BRAFm is a strong negative prognostic factor in mCRC. BRAFm is more commonly seen in subjects of an older age at diagnosis and in females, in terms of epidemiological characteristics.31 Regarding pathological characteristics, BRAFm is associated with larger tumor size, microsatellite instability (MSI), and mucinous histology, as well as poor differentiation, and BRAFm is more often detected in proximal colon cancers.32,33 BRAFm mCRCs are sensitive to none of the existing chemotherapies, and patients with stage II and III colon cancer accompanied by BRAF-V600E exhibit worse prognoses regardless of stage or therapy.34 In addition, there is a correlation between BRAF-V600E mutation in microsatellite-stable (MSS) stages II–IV colon cancer, while the prognosis of MSI cancers is not affected by the aforementioned mutation.35 BRAFm is commonly correlated with MSI cancers (regarded as a good prognostic factor at the early stage). The studies reporting BRAFm as a negative prognostic indicator in MSI included up to 15% of mCRC subjects, whereas research demonstrating no effect in the MSI subset included exclusively early-stage CRC.36,37 The pooled analysis of three large randomized trials (COIN, PICCOLO, and FOCUS)38 enrolled 231 mCRC subjects with BRAFm. The results demonstrated lower overall survival (OS) but strikingly similar progression-free survival (PFS) and disease control rate in the BRAFm population compared to that in the BRAF WT population. Therefore, it is gradually accepted that BRAF is a potential target to improve prognosis in the treatment of BRAFm mCRC.38
Development of RAF small-molecule inhibitors and application in CRC
The first-generation RAF inhibitors
When RAF was first introduced as a target for cancer treatment, before the discovery of BRAFm, the first-generation inhibitors of RAF were small-molecule ATP-competitive inhibitors, which were explored to target CRAF in cancer.39 ZM336372 was screened as the first compound-suppressing BRAF, as well as CRAF, in vitro, and the feedback loop for RAF appeared to suppress its own activation.39 Sorafenib (Nexavar®, BAY43-9006) is the only first-generation RAF inhibitor that has gained approval from the US FDA; however, sorafenib exhibits weak antitumor activity in cells with BRAF-V600E and its clinical efficacy in the abovementioned cancers with WT BRAF might be attributed to its multikinase profile. No PFS benefit was detected from the addition of sorafenib to first-line mFOLFOX6 for mCRC in the RESPECT trial.40 Additionally, the combination of sorafenib and irinotecan41 or cetuximab42 had been evaluated in a trial for KRAS-mutated CRC; however, low or no objective responses were observed. Other first-generation RAF inhibitors, such as SB-590885,43 GDC-0879,44 GW5074,45 and L779450,46 were not applied in clinical practice (Figure 4).
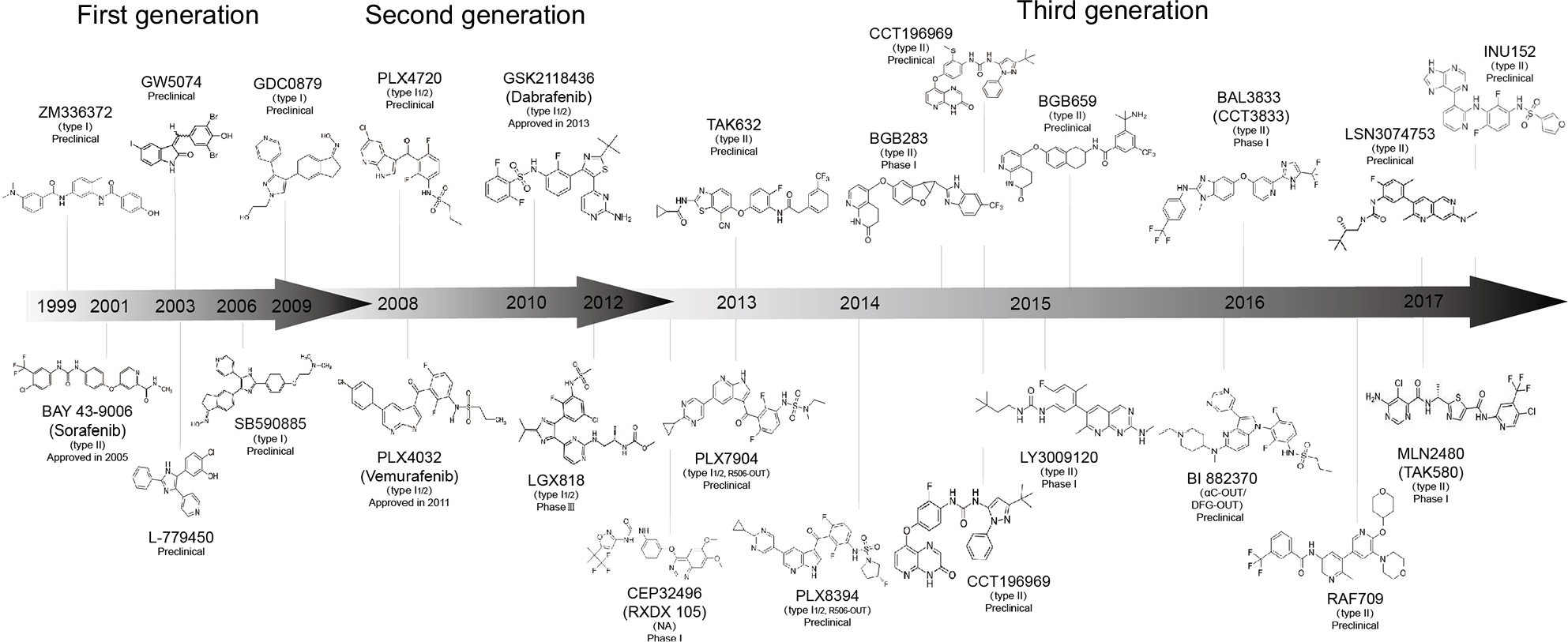 | Figure 4 Summary of the chronological development and development phase of the three generations of RAF inhibitors. |
The second-generation RAF inhibitors
After BRAFm was detected in 2002, and in consideration of BRAF (V600E) as the most prevalent oncogenic protein kinase mutation at present, multiple studies focus on evaluating second-generation compounds to selectively inhibit BRAF-V600E. PLX4720 was the first selective BRAF (V600E) inhibitor identified in 2008,47 and PLX4032 (vemurafenib) was developed by Plexxikon via a structure-guided discovery approach.48 As a αC-OUT/DFG-IN inhibitor, vemurafenib exhibited significantly prolonged OS and PFS in melanoma subjects with BRAF-V600E and was subsequently approved by US Food and Drug Administration (FDA) in 2011.49 Dabrafenib, another selective inhibitor for BRAFm,50 gained approval from the FDA in 2013 for treating BRAF (V600)-mutant metastatic melanoma.51 In 2015, the selective inhibitor for BRAFm was applied for treating BRAFm CRC. Vemurafenib monotherapy was insufficiently active in subjects with BRAF-V600-mutant CRC,52 while an antitumor effect was observed upon treatment with an anti-EGFR antibody (cetuximab) and vemurafenib.53 A combination of dabrafenib and trametinib, a selective MEK inhibitor, yielded a partial response (PR) or an improved response in 12% patients.54 Preliminary results suggested that the concurrent administration of selective BRAF (V600E) inhibitors as part of combinatorial therapeutic strategies may be required for meaningful clinical benefit.
Encorafenib, a second-generation RAF inhibitor, exhibits a pharmacologic profile that is distinct from that of other clinically active BRAF inhibitors.55 Preclinical evidence suggests that concomitant BRAF and EGFR inhibition triggers a continuous suppression of the MAPK pathway, as well as suppresses tumor growth in BRAF-V600E CRC models. Dual (encorafenib plus cetuximab) and triple (encorafenib plus cetuximab and alpelisib) combination treatments are tolerable and exhibit promising clinical efficacy in BRAFm mCRC patients.56 In addition, encorafenib shows longer residence time compared to vemurafenib or dabrafenib, which may prolong target inhibition and lead to positive effect.56 Phases I–IV clinical trials are ongoing to evaluate combinatorial therapeutic strategies for BRAF-V600E mCRC, which may promote further treatment with RAF inhibitors in CRC. However, RAF dimerization limits the response of second-generation RAF inhibitors in the treatment of BRAFm CRC.
The third-generation RAF inhibitors
Based on the discovery of RAF dimerization and the distinct allosteric mechanisms of RAF, third-generation RAF inhibitors are under development in current studies.26 One type of third-generation RAF inhibitor focuses on compounds to find effective and equipotent inhibitors of dimeric forms, as well as monomeric forms, of RAF and is anticipated to battle resistance resulting from RAF dimerization. The other type of third-generation RAF inhibitor uses “paradox breakers” by not inducing RAF paradoxical activation (referred to as pan-RAF inhibitors) and is derived from αC-OUT RAF inhibitors with diverse terminal sulfonamide, as well as sulfamide, substitutions.57 αC-OUT RAF inhibitors are anticipated to maintain broad therapeutic profiles and overcome on-target toxicities attributed to paradoxical ERK activation in nontumor cells.
Several third-generation RAF inhibitors have exhibited breakthrough effects in preclinical studies. The 7-cyano derivative 8B (TAK632) and MLN2480 (TAK580) are pan-RAF inhibitors whose binding patterns with BRAF were verified through co-crystal structures. Crystallographic data of TAK632 bound to BRAF show similar affinity for both protomers in RAF dimers based on the type II conformation.58 TAK632 persistently binds to both protomers of RAF dimers, consequently suppressing the kinase activity of the RAF dimer, and presents equipotent suppression of dimeric RAF and monomeric RAF in cells. TAK632 is anticipated to be effective in vemurafenib-resistant BRAF-V600E CRC cells.59 TAK580, a type II RAF inhibitor that serves as an equipotent antagonist of BRAF-V600E, demonstrates therapeutic effects in BRAF-mutant pediatric astrocytomas.60 LY3009120 (Eli Lilly and Company, Indianapolis, IN, USA), another pan-RAF inhibitor with minimal paradoxical activation, is currently being assessed in Phase I clinical trials. LY3009120 shows preclinical activity as a pan-RAF inhibitor for the BRAFm and KRAS-mutant CRC population.61 Moreover, LY3009120 represents a potential therapeutic choice for subjects burdened with malignancies with BRAF deletions or other BRAFms where BRAF acts as a dimer.62 LSN3074753, an analog of LY3009120 and a pan-RAF inhibitor, was combined with cetuximab in BRAF-CRC patient-derived xenograft (PDX) models, which suggested a synergistic antitumor activity.63 BAL3833, a pan RAF/SRC kinase inhibitor, blocks BRAF and CRAF and inhibits the SRC kinase family and is currently being explored as a daily oral administration in a Phase I dose-escalation clinical trial in adult subjects harboring advanced solid tumors.64 BGB659, a type II RAF inhibitor, functions as an ATP competitive inhibitor, binds to RAF dimers, and sufficiently suppresses tumor growth triggered by all RAF mutants in mice.25 In addition, BGB283, a relevant compound that functioned as a dual RAF and EGFR inhibitor in a Phase I clinical trial, has been shown to inhibit EGFR reactivation in BRAF-V600E CRC.25 Because drug resistance is generally modulated by pathway reactivation via RTK/SRC-family kinase (SFK) signaling or mutant NRAS, CCT196969 and CCT241161 were discovered as dual pan-RAF and SRC kinase inhibitors. Additionally, inhibitory effects on RAS-mutant melanomas, the CRC and PDX models of melanomas, were shown by treatment with CCT196969 and CCT241161.65 LXH254, an orally administered inhibitor of all members of the serine/threonine protein kinase RAF family, is undergoing a Phase I trial by Novartis in subjects harboring advanced solid tumors. Finally, RAF709, a kinase-selective and cellular potent inhibitor, was soluble, kinase selective, and efficacious in a KRAS-mutant xenograft model.66 To completely inhibit and suppress paradoxical activation in cancer cells with mutant RAS, it is critical for RAF inhibitors to exhibit a valid inhibitory efficacy against RAF isoforms. INU152 suppresses all RAF isoforms, as well as MAPK pathways, in BRAFm cells and exhibits minimal paradoxical pathway activation in melanoma cells with mutant RAS (Figure 4).67
The other type of third-generation RAF inhibitors showed “paradox breakers” without inducing RAF paradoxical activation to reduce on-target toxicities of second-site cancers attributed to paradoxical ERK activation in normal cells. PLX7904 and PLX8394 were initially reported as “paradox breakers” that had no effect on MAPK pathway activation when suppressing mutant BRAF cells57 and PLX8394 also exhibited precise suppression of BRAF-V600 mutation without paradoxically enhancing the MAPK pathway in CRC.68 Although Phases I and II trials of PLX8394 were ongoing for BRAFm tumors and unresectable solid tumors, acquired PLX8394-resistant cells eventually occurred in melanoma, as with other targeted therapies; the potential mechanism may include not only the enhancement of AKT activity and upregulation of platelet-derived growth factor receptors (PDGFRs) but also other drivers of resistance in these cells.69 Finally, BI 882370 is a highly selective and effective RAF inhibitor that acts via binding to the DFG-OUT (inactive) conformation of BRAF kinase; it has been proved to be effective via oral administration in numerous mouse models of BRAFm CRC, as well as in melanomas. Despite tumor regression induced by BI 882370 in tumor-bearing mice, drug resistance gradually occurred within 3 weeks. A combination of BI 882370 and trametinib led to a higher degree of tumor regression, and no resistance was detected over a 5-week second-line therapy.70
Other inhibitors, such as HM95573 (Hanmi Pharmaceutical, South Korea; Genentech, USA) and CEP32496 (RXDX-105; Ambit Biosciences/Ignyta, USA), are also in clinical trials for solid cancers bearing mutant NRAS, KRAS, or BRAF genes; however, these inhibitors cannot be classified due to the lack of structural data.71 These two inhibitors are under assessment in Phase I clinical studies for solid tumors and advanced or metastatic tumors. The development of RAF inhibitors expands the therapeutic window and reduces the on-target toxicities and resistance due to RAF dimerization. However, it is virtually impossible to demonstrate single-agent clinical activity in BRAF-mutant CRC patients and combinatorial approaches are being developed to maximize antitumor efficacy and minimize the development of drug resistance.72
The strategies of RAF inhibitors in BRAFm CRC
According to the current understanding of BRAFm CRC, the mechanism underlying the feedback reactivation of MAPK signaling and other relevant mechanisms were discovered to be involved in the resistance to BRAF inhibition in BRAFm CRC. We summarize the results of current combinatorial therapeutic strategies utilizing RAF inhibitors (Table 1) and future combinatorial therapeutic strategies in Figure 5.
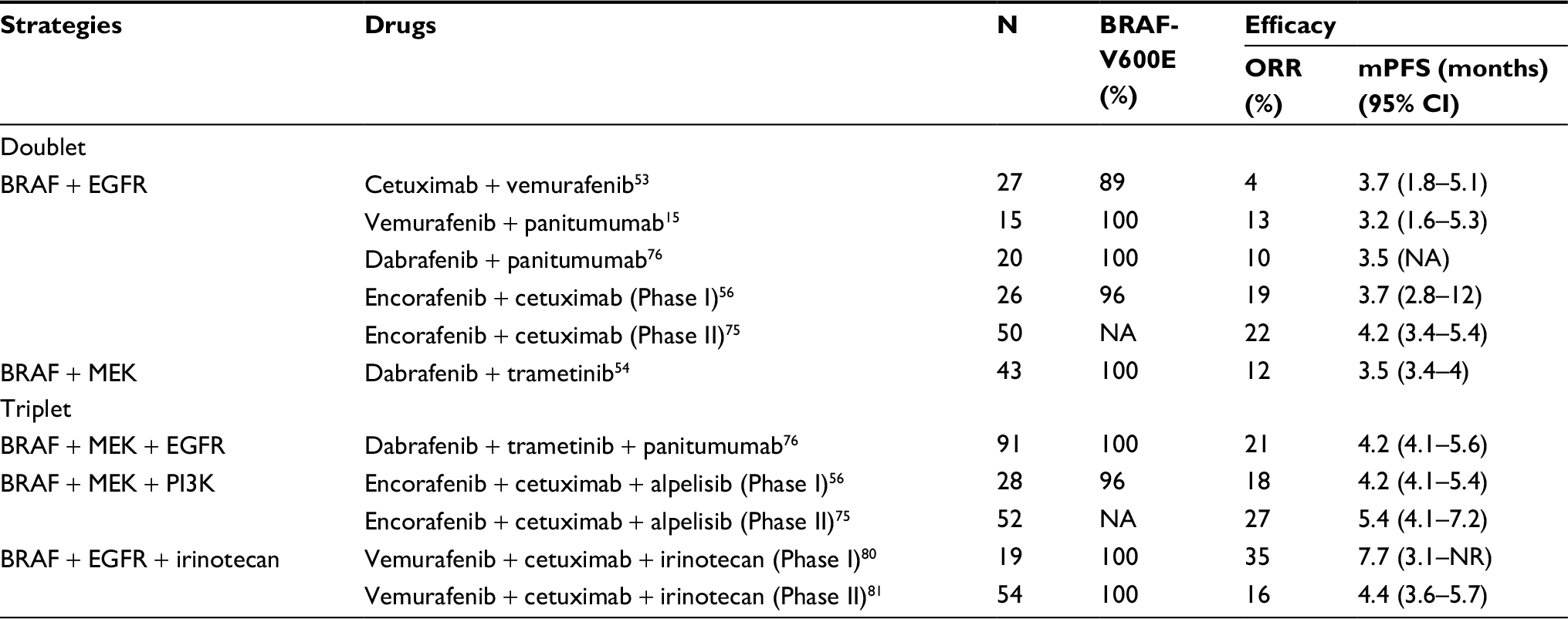 | Table 1 The efficacy and toxicities of combinatorial therapeutic strategies of RAF inhibitors in BRAFm CRC patients Note: aGrade 3/4 adverse events. Abbreviations: BRAFm, BRAF mutation; CI, confidence interval; CRC, colorectal cancer; mPFS, median progression-free survival; mOS, median overall survival; NA, not available; NR, not reached; ORR, objective response rate. |
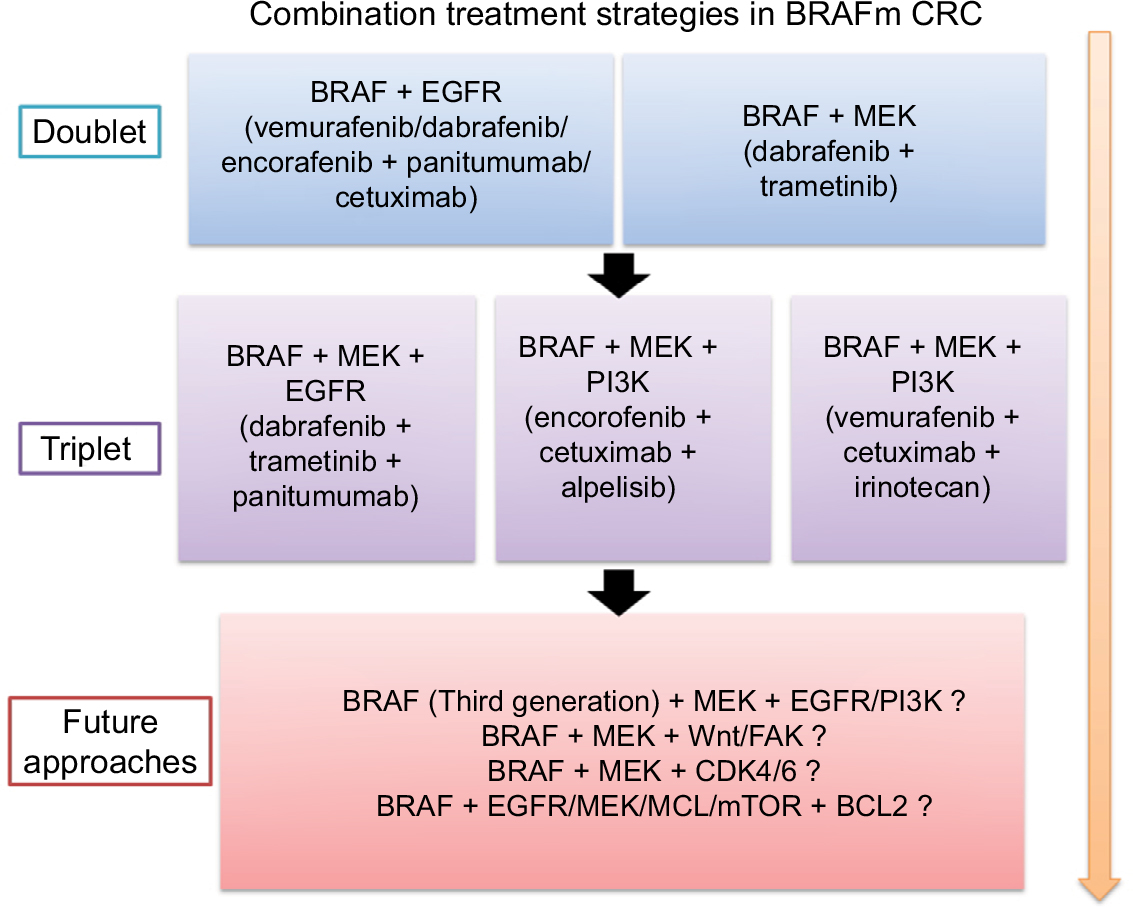 | Figure 5 A flowchart of current and future combination treatment strategies for BRAF-mutant CRC. Abbreviation: CRC, colorectal cancer. |
Doublet combinatorial therapeutic strategies
Despite successful outcomes of BRAF inhibitors in BRAFm melanoma, there have been disappointing results showing that monotherapy with an RAF inhibitor did not demonstrate any valuable clinical efficacy in CRC patients harboring BRAF-V600E, with the response rate (RR) ranging from 0 to 5%.52,53,73 However, a low response was observed when combined with an EGFR inhibitor (cetuximab) in BRAF-V600-mutated CRC.53 Therefore, combinatorial strategies are currently being evaluated for BRAF-V600-mutated CRC. BRAF with EGFR inhibition is one of the combinatorial therapeutic strategies. Additionally, vemurafenib and panitumumab were evaluated for BRAF-V600E-mutant CRC, aiming to reduce rebound in ERK signaling; the study revealed less cutaneous toxicity and modest clinical efficacy in highly aggressive and chemoresistant subgroups of CRC.15 A full dose of dabrafenib and panitumumab was associated with significant dermatologic toxicity, and the RR was only 10% in lower dose cohorts.74 Encorafenib, a selective RAF kinase inhibitor, combined with cetuximab demonstrated promising clinical efficacy and tolerability in mCRC patients with BRAFm; the overall RR was 19%, the median duration of response was 46 weeks in these subjects, and the overall safety profile was acceptable.56 A Phase II trial of encorafenib and cetuximab in mCRC revealed a 22% overall RR and 58% grade 3/4 adverse events.75
BRAF and MEK inhibitions represent another combinatorial therapeutic strategy. In 43 patients harboring BRAF-V600-mutant mCRC who received dabrafenib (150 mg twice daily) and trametinib (2 mg daily), the RR was 12% (consisting of one complete response [CR] and four PR), 24 (56%) patients achieved stable disease (SD), and 10 (23%) patients maintained the therapeutic regimens for >6 months. The MAPK pathway was suppressed in all assessed patients but was far from the impressive results in melanoma.54 The most common toxicities included pyrexia and diarrhea, as well as in patients undergoing combination therapy. Therefore, the MAPK signaling is confirmed to be a therapeutic target in BRAFm mCRC and it is potentially effective to target this pathway in clinical practice. Because of the suboptimal effects of doublet combinatorial therapies, more effective therapeutic strategies that inhibit the MAPK signaling pathway are worthy of further research in BRAFm CRC patients.
Triplet combinatorial therapeutic strategies
Due to EGFR overactivation induced by BRAF inhibition, targeting BRAF, MEK, and EGFR were the first triplet combinatorial therapeutic strategies against BRAFm mCRC. Dabrafenib (D), trametinib (T), and panitumumab (P) were combined to be used in 35 BRAFm mCRC patients. Of them, 24 patients were administered with the full dose (P 6 mg/kg every 2 weeks, D 150 mg bid, and T 2 mg od). The confirmed RR was 26%, including one CR, suggesting encouraging clinical efficacy with acceptable tolerability for the triplet BRAF + MEK + EGFR in BRAFm mCRC. A later trial enrolling 83 BRAFm mCRC patients who received DTP treatment showed that the verified (CR)/PR and SD were 18 and 67%, as well as exhibiting acceptable tolerability and activity in BRAFm mCRC.76
Moreover, activation of the PI3K/AKT pathway has been identified as a mechanism of resistance to BRAF inhibitors in BRAFm CRC cell lines;77 therefore, targeting BRAF + MEK + PI3K has been suggested to improve outcomes in patients with BRAFm mCRC (Figure 3B). To this end, encorafenib and cetuximab with a PI3K inhibitor (alpelisib) were compared with doublet combinatorial cetuximab and encorafenib. The confirmed overall RR of BRAF + MEK + PI3K was 17.9%, while that of BRAF + MEK was 19.2%, demonstrating that dual and triple combination treatments were tolerable and provided promising clinical efficacy in the difficult-to-treat patients with BRAFm mCRC.56 In a Phase II trial, 52 patients were enrolled in the triple therapy (encorafenib with cetuximab and alpelisib) and the planned interim analysis showed a median PFS of 5.4 months (primary endpoint). There was no statistical significance between the two therapeutic strategies. The overall RR of the doublet therapy was 27% (16–41%). The toxicity appeared to rise in the triple combination (79 vs 58% of adverse events).75 Therefore, the addition of alpelisib did not appear to improve RRs as predicted from preclinical studies, indicating that PI3K may be limited in driving associated resistant mechanisms.
A combination of EGFR suppression and vemurafenib, a BRAF inhibitor, leads to synergistic cytotoxicity. A combined therapy of irinotecan and cetuximab has gained approval for mCRC subjects with WT RAS.78 PDX models from BRAF-V600E CRC indicated improved RRs, as well as longer survival, in animals receiving vemurafenib, irinotecan, and cetuximab than in those receiving irinotecan and cetuximab or vemurafenib and cetuximab.79 In addition, six of the 17 (35%) assessed patients who were treated with vemurafenib in combination with irinotecan and cetuximab acquired a radiographic response, according to the Response Evaluation Criteria.80 In a Phase II randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-V600E mCRC patients, the RR was 16% compared with 4% and the disease control rate was 67% compared with 22%. The results demonstrate the clinical benefits of the triplet BRAF + EGFR + irinotecan in BRAF-V600E mCRC and support this strategy as a potential new treatment option in this molecular subset.81 The efficacy and major toxicities of published clinical trials, including doublet and triplet combinatorial therapeutic strategies, are summarized in Table 1.
Future perspectives
In the last 5 years, BRAFm CRC treatment has exhibited breakthrough progress, with increasing novel options moving directly from the bench to the bedside. Novel adaptive signaling mechanisms underlying resistance to BRAF inhibition in BRAFm CRC were discovered, such as the observation that the Wnt/β-catenin pathway82 and CDK4/683 were co-regulated when using a BRAF inhibitor (Figure 3B); additionally, other strategies combining a BRAF inhibitor (LGX818) with a Wnt-pathway inhibitor (WNT974) or a CDK4/6 inhibitor (LEE011) were in Phase I or II trials. Additionally, various MEK mutations were identified that diminish sensitivity to both single-agent RAF inhibitors and combined BRAF and MEK inhibition.84 Moreover, BCL-2 antiapoptotic complexes were overexpressed in some cases of BRAFm CRC.85 BCL-2 inhibitors such as ABT-737 and ABT-263 had shown synergistic effects when combined with MEK, MCL1, or mTOR inhibitors in BRAFm CRC.86–88 However, the effect of combined use of BRAF and BCL-2 inhibitors needs further evaluation in clinical trials. An ongoing study has combined BRAF, EGFR, and BCL-2 inhibitors for solid tumors including BRAFm CRC (NCT01989585). Importantly, identifying the novel underlying mechanism and regulation of the adaptive response to RAF inhibitors would be useful to optimize combinatorial therapeutic strategies, as well as to enhance the RR of BRAFm CRC treatment. Based on the new underlying mechanism, new and more effective inhibitors can also be designed for co-targeting RAF and other adaptive response pathway targets.
The BRAF dimerization is still an obstacle for resistance to RAF inhibitors in CRC, and there is a pressing need to identify structural and biochemical regulations of BRAF dimerization. Although third-generation RAF inhibitors are more effective for RAF dimers, their selectivity for WT or mutant BRAF dimer is not superlative. To prevent negative allostery effect, further research will be conducted on allosteric inhibitors that effectively suppress the WT or mutant BRAF dimer and/or BRAF monomer, while not enhancing the RAF priming.20 In addition, the structure of the BRAF–CRAF heterodimer in RAF oncogenic signaling is crucial for the development of strategies that block the formation of homodimers and heterodimers of BRAF–CRAF.89 Moreover, the interplay of the kinase domain and the N-terminal regulatory domain in either the inactive conformation or the active conformation is complicated. Other than the V600 mutation, non-V600 BRAFms have been reported in ~2.2% of mCRC subjects and these mutations exhibit a distinct clinical subtype of CRC, as well as excellent prognosis.90 Despite a previous description of >1000 unique BRAFms in patients with diverse malignant cancers,13 while non-V600 BRAFms, as naturally occurring models of BRAF signaling regulation, are likely to evade the paradoxical ERK reactivation that inevitably occurs with exogenous BRAF inhibitors, non-V600 BRAFms with damaged kinase activity are still more capable of phosphorylating ERK than WT BRAF.91 Therefore, a better understanding of the structures of non-V600 BRAFms would provide a novel insight to develop tailored BRAF inhibitors.
For the application of BRAF inhibitors in CRC therapy, although the doublet and triplet combinatorial therapeutic strategies have made remarkable progress, only second-generation RAF inhibitors have been reported on the market, including dabrafenib, encorafenib, and vemurafenib. These inhibitors exhibit no or very low effects as monotherapy for BRAF-V600E mt-CRC; third-generation RAF inhibitors should pass the clinical trials and gain market approval in the future. More novel mechanisms for CRC response will be discovered, and more therapeutic strategic trials can be designed based on third-generation RAF inhibitors. In addition, BRAFm CRC is associated with MSI-H and programmed death-1 (PD-1) inhibition has been shown to have a significant benefit and has been approved for treating MSI tumors, including mCRC.92 Immune checkpoint inhibition should be a combination regimen, which may be a logical next step in the development of BRAFm/MSI-H CRC combinatorial therapeutic strategies. Finally, current clinical trial data have showed that anticancer drugs exhibit variable efficacy within patient populations based on the heterogeneity.93 Therefore, optimizing drug independence represents a novel orientation for BRAFm CRC treatment and combination strategies should be based on patient heterogeneity and optimizing drug independence. Accurate biomarkers that aim to predict the optimization of drug response and disease progression will make it possible to use fewer drugs for precision combination therapy.
Conclusion
The oncogene BRAF is a recognized therapeutic target in CRC. The development of small-molecule inhibitors of RAF and the novel strategy for drug combinations have prolonged the survival of CRC subjects harboring BRAFm. However, the effectiveness is limited by resistance attributed to BRAF-V600 mutations and there are multiple unclarified underlying mechanisms of resistance. We have good understanding of the mechanisms of RAF kinases in BRAFm CRC, including the MAPK feedback reactivation of resistance to RAF inhibitors and the development of three generations of RAF inhibitors and different therapeutic strategies of RAF inhibitors in BRAF-V600E mCRC. In the future, more effective RAF inhibitor-based therapeutic strategies will progress and improve survival in BRAFm CRC patients.
Acknowledgments
The study was funded by an Overseas Study Fellowship from the China Scholarship Council to Jing-hua Pan. This study was supported by the National Natural Science Foundation of China (81472849), the Guangdong Natural Science Research (2014A030313383), and the Guangdong High-level University Construction Fund for Jinan University (88016013034).
Disclosure
The authors report no conflicts of interest in this work.
References
Yin D, Morris CR, Bates JH, German RR. Effect of misclassified underlying cause of death on survival estimates of colon and rectal cancer. J Natl Cancer Inst. 2011;103(14):1130–1133. | ||
Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544–573. | ||
Vogelaar I, van Ballegooijen M, Schrag D, et al. How much can current interventions reduce colorectal cancer mortality in the U.S.? Mortality projections for scenarios of risk-factor modification, screening, and treatment. Cancer. 2006;107(7):1624–1633. | ||
Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics. CA Cancer J Clin. 2017;67(3):177–193. | ||
Lee MS, Menter DG, Kopetz S. Right versus left colon cancer biology: integrating the consensus molecular subtypes. J Natl Compr Canc Netw. 2017;15(3):411–419. | ||
Fakih MG. Metastatic colorectal cancer: current state and future directions. J Clin Oncol. 2015;33(16):1809–1824. | ||
Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386–1422. | ||
Ritt DA, Abreu-Blanco MT, Bindu L, et al. Inhibition of Ras/Raf/MEK/ERK Pathway Signaling by a Stress-Induced Phospho-Regulatory Circuit. Molecular Cell. 2016;64:875–887. | ||
Chowdhury I, Thompson WE, Thomas K. Prohibitins role in cellular survival through Ras-Raf-MEK-ERK pathway. Journal of Cellular Physiology. 2014;229:998–1004. | ||
An S, Yang Y, Ward R, Liu Y, Guo XX, Xu TR. A-Raf: a new star of the family of raf kinases. Crit Rev Biochem Mol Biol. 2015;50(6):520–531. | ||
Neuzillet C, Tijeras-Raballand A, de Mestier L, et al. MEK in cancer and cancer therapy. Pharmacol Ther. 2013;141(2):160–171. | ||
Roskoski R Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res. 2015;100:1–23. | ||
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | ||
Martinelli E, Morgillo F, Troiani T, Ciardiello F. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat Rev. 2017;53:61–69. | ||
Yaeger R, Cercek A, O’Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21(6):1313–1320. | ||
Cavalieri S, Di Guardo L, Cossa M, Cimminiello C, Del Vecchio M. Unusual skin carcinomas induced by BRAF inhibitor for metastatic melanoma: a case report. J Clin Diagn Res. 2017;11(7):Xd06–Xd08. | ||
Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364(6435):352–355. | ||
Cutler RE Jr, Stephens RM, Saracino MR, Morrison DK. Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci U S A. 1998;95(16):9214–9219. | ||
Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74(1):205–214. | ||
Kornev AP, Taylor SS. Dynamics-driven allostery in protein kinases. Trends Biochem Sci. 2015;40(11):628–647. | ||
Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17(11):676–691. | ||
Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16(5):281–298. | ||
Palmieri L, Rastelli G. alphaC helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discov Today. 2013;18(7–8):407–414. | ||
Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20(6):963–969. | ||
Yao Z, Torres NM, Tao A, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28(3):370–383. | ||
Lito P, Pratilas CA, Joseph EW, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22(5):668–682. | ||
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–430. | ||
Hong A, Moriceau G, Sun L, et al. Exploiting drug addiction mechanisms to select against MAPKi-resistant melanoma. Cancer Discov. 2018;8(1):74–93. | ||
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | ||
Liu M, Yang X, Liu J, et al. Efficacy and safety of BRAF inhibition alone versus combined BRAF and MEK inhibition in melanoma: a meta-analysis of randomized controlled trials. Oncotarget. 2017;8(19):32258–32269. | ||
Gonsalves WI, Mahoney MR, Sargent DJ, et al. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J Natl Cancer Inst. 2014;106(7):dju106. | ||
Ogino S, Brahmandam M, Cantor M, et al. Distinct molecular features of colorectal carcinoma with signet ring cell component and colorectal carcinoma with mucinous component. Mod Pathol. 2006;19(1):59–68. | ||
Clancy C, Burke JP, Kalady MF, Coffey JC. BRAF mutation is associated with distinct clinicopathological characteristics in colorectal cancer: a systematic review and meta-analysis. Colorectal Dis. 2013;15(12):e711–e718. | ||
Popovici V, Budinska E, Tejpar S, et al. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol. 2012;30(12):1288–1295. | ||
Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65(14):6063–6069. | ||
Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst. 2013;105(15):1151–1156. | ||
Sinicrope FA, Mahoney MR, Smyrk TC, et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Clin Oncol. 2013;31(29):3664–3672. | ||
Sanz-Garcia E, Argiles G, Elez E, Tabernero J. BRAF mutant colorectal cancer: prognosis, treatment and new perspectives. Ann Oncol. 2017;28(11):2648–2657. | ||
Hall-Jackson CA, Eyers PA, Cohen P, et al. Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol. 1999;6(8):559–568. | ||
Tabernero J, Garcia-Carbonero R, Cassidy J, et al. Sorafenib in combination with oxaliplatin, leucovorin, and fluorouracil (modified FOLFOX6) as first-line treatment of metastatic colorectal cancer: the RESPECT trial. Clin Cancer Res. 2013;19(9):2541–2550. | ||
Samalin E, Bouche O, Thezenas S, et al. Sorafenib and irinotecan (NEXIRI) as second- or later-line treatment for patients with metastatic colorectal cancer and KRAS-mutated tumours: a multicentre phase I/II trial. Br J Cancer. 2014;110(5):1148–1154. | ||
Do K, Cao L, Kang Z, et al. A phase II study of sorafenib combined with cetuximab in EGFR-expressing, KRAS-mutated metastatic colorectal cancer. Clin Colorectal Cancer. 2015;14(3):154–161. | ||
Takle AK, Brown MJ, Davies S, et al. The identification of potent and selective imidazole-based inhibitors of B-Raf kinase. Bioorg Med Chem Lett. 2006;16(2):378–381. | ||
Hoeflich KP, Herter S, Tien J, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69(7):3042–3051. | ||
Johnson K, Liu L, Majdzadeh N, et al. Inhibition of neuronal apoptosis by the cyclin-dependent kinase inhibitor GW8510: identification of 3’ substituted indolones as a scaffold for the development of neuroprotective drugs. J Neurochem. 2005;93(3):538–548. | ||
Shelton JG, Moye PW, Steelman LS, et al. Differential effects of kinase cascade inhibitors on neoplastic and cytokine-mediated cell proliferation. Leukemia. 2003;17(9):1765–1782. | ||
Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105(8):3041–3046. | ||
Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. | ||
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. | ||
Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–1901. | ||
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | ||
Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–4038. | ||
Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–736. | ||
Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol. 2015;33(34):4023–4031. | ||
Huang T, Karsy M, Zhuge J, Zhong M, Liu D. B-Raf and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:30. | ||
van Geel R, Tabernero J, Elez E, et al. A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 2017;7(6):610–619. | ||
Zhang C, Spevak W, Zhang Y, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526(7574):583–586. | ||
Okaniwa M, Hirose M, Arita T, et al. Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: design, synthesis, and biological evaluation of C-7-substituted 1,3-benzothiazole derivatives. J Med Chem. 2013;56(16):6478–6494. | ||
Nakamura A, Arita T, Tsuchiya S, et al. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013;73(23):7043–7055. | ||
Sun Y, Alberta JA, Pilarz C, et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 2017;19(6):774–785. | ||
Vakana E, Pratt S, Blosser W, et al. LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget. 2017;8(6):9251–9266. | ||
Chen SH, Zhang Y, Van Horn RD, et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov. 2016;6(3):300–315. | ||
Yao YM, Donoho GP, Iversen PW, et al. Mouse PDX trial suggests synergy of concurrent inhibition of RAF and EGFR in colorectal cancer with BRAF or KRAS mutations. Clin Cancer Res. 2017;23(18):5547–5560. | ||
Saturno G, Lopes F, Girotti MR, et al. Therapeutic efficacy of the paradox-breaking panRAF and SRC drug CCT3833/BAL3833 in KRAS-driven cancer models. Eur J Cancer. 2016;61:S199. | ||
Girotti MR, Lopes F, Preece N, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. 2015;27(1):85–96. | ||
Nishiguchi GA, Rico A, Tanner H, et al. Design and discovery of N-(2-methyl-5’-morpholino-6’-((tetrahydro-2H-pyran-4-yl)oxy)-[3,3’-bipyridin]-5-y l)-3-(trifluoromethyl)benzamide (RAF709): a potent, selective, and efficacious RAF inhibitor targeting RAS mutant cancers. J Med Chem. 2017;60(12):4869–4881. | ||
Hong SP, Ahn SK. Discovery of a novel pan-RAF inhibitor with potent anti-tumor activity in preclinical models of BRAF(V600E) mutant cancer. Life Sci. 2017;183:37–44. | ||
Tutuka CSA, Andrews MC, Mariadason JM, et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer. 2017;16(1):112. | ||
Hartsough EJ, Kugel CH, Vido MJ, et al. Response and resistance to paradox breaking BRAF inhibitor in melanomas in vivo and ex vivo. Mol Cancer Ther. 2018;17(1):84–95. | ||
Waizenegger IC, Baum A, Steurer S, et al. A novel RAF kinase inhibitor with DFG-out-binding mode: high efficacy in BRAF-mutant tumor xenograft models in the absence of normal tissue hyperproliferation. Mol Cancer Ther. 2016;15(3):354–365. | ||
James J, Ruggeri B, Armstrong RC, et al. CEP-32496: a novel orally active BRAF(V600E) inhibitor with selective cellular and in vivo antitumor activity. Mol Cancer Ther. 2012;11(4):930–941. | ||
Corcoran RB, Andre T, Atreya CE, et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discovery. 2018;8:428–443. | ||
Sundar R, Hong DS, Kopetz S, Yap TA. Targeting BRAF-mutant colorectal cancer: progress in combination strategies. Cancer Discov. 2017;7(6):558–560. | ||
Atreya CE, Van Cutsem E, Bendell JC, et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC). J Clin Oncol. 2015;33(15_suppl):103. | ||
Tabernero J, Geel RV, Guren TK, et al. Phase 2 results: encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J Clin Oncol. 2016;34:3544. | ||
Corcoran RB, André T, Yoshino T, et al. Efficacy and circulating tumor DNA (ctDNA) analysis of the BRAF inhibitor dabrafenib (D), MEK inhibitor trametinib (T), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E–mutated (BRAFm) metastatic colorectal cancer (mCRC). Ann Oncol. 2016;27:455O. | ||
Mao M, Tian F, Mariadason JM, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2012;19(3):657–667. | ||
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345. | ||
Yang H, Higgins B, Kolinsky K, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012;72(3):779–789. | ||
Hong DS, Morris VK, El Osta B, et al. Phase IB study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov. 2016;6(12):1352–1365. | ||
Kopetz S, McDonough SL, Lenz H-J, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J Clin Oncol. 2017;35(15_suppl):3505. | ||
Chen G, Gao C, Gao X, et al. Wnt/beta-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol Cancer Ther. 2018;17(4):806–813. | ||
Pek M, Yatim S, Chen Y, et al. Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene. 2017;36(35):4975–4986. | ||
Emery CM, Monaco KA, Wang P, et al. BRAF-inhibitor associated MEK mutations increase RAF-dependent and -independent enzymatic activity. Mol Cancer Res. 2017;15(10):1431–1444. | ||
Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell. 2013;49(4):751–758. | ||
Jones JC, Renfro LA, Al-Shamsi HO, et al. (Non-V600) BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J Clin Oncol. 2017;35(23):2624–2630. | ||
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. | ||
Sehdev A, Cramer HM, Ibrahim AA, Younger AE, O’Neil BH. Pathological complete response with anti-PD-1 therapy in a patient with microsatellite instable high, BRAF mutant metastatic colon cancer: a case report and review of literature. Discov Med. 2016;21(117):341–347. | ||
Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell. 2017;171:1678–1691.e13. | ||
Jones JC, Renfro LA, Al-Shamsi HO, et al. (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. Journal of Clinical Oncology. 2017;35:2624–2630. | ||
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. | ||
Sehdev A, Cramer HM, Ibrahim AA, et al. Pathological complete response with anti-PD-1 therapy in a patient with microsatellite instable high, BRAF mutant metastatic colon cancer: a case report and review of literature. Discovery Medicine. 2016;21:341–347. | ||
Palmer AC, Sorger PK. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell. 2017;171:1678–1691.e1613. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.