")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Development of small bisquaternary cholinesterase inhibitors as drugs for pre-treatment of nerve agent poisonings
Authors Kuca K , Karasova JZ, Soukup O , Kassa J, Novotna E, Sepsova V, Horova A, Pejchal J, Hrabinova M, Vodakova E, Jun D , Nepovimova E, Valis M, Musilek K
Received 23 January 2017
Accepted for publication 21 November 2017
Published 9 March 2018 Volume 2018:12 Pages 505—512
DOI https://doi.org/10.2147/DDDT.S133038
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Kamil Kuca,1,2 Jana Zdarova Karasova,2,3 Ondrej Soukup,2 Jiri Kassa,3 Eva Novotna,2 Vendula Sepsova,2,3 Anna Horova,2 Jaroslav Pejchal,3 Martina Hrabinova,2,3 Eva Vodakova,2 Daniel Jun,2,3 Eugenie Nepovimova,1,2 Martin Valis,4 Kamil Musilek1,2
1Department of Chemistry, Faculty of Science, University of Hradec Kralove, 2Biomedical Research Center, University Hospital Hradec Kralove, 3Department of Toxicology and Military Pharmacy, Faculty of Military Health Sciences, University of Defence, 4Department of Neurology, University Hospital Hradec Kralove, Hradec Kralove, Czech Republic
Background: Intoxication by nerve agents could be prevented by using small acetylcholinesterase inhibitors (eg, pyridostigmine) for potentially exposed personnel. However, the serious side effects of currently used drugs led to research of novel potent molecules for prophylaxis of organophosphorus intoxication.
Methods: The molecular design, molecular docking, chemical synthesis, in vitro methods (enzyme inhibition, cytotoxicity, and nicotinic receptors modulation), and in vivo methods (acute toxicity and prophylactic effect) were used to study bispyridinium, bisquinolinium, bisisoquinolinium, and pyridinium-quinolinium/isoquinolinium molecules presented in this study.
Results: The studied molecules showed non-competitive inhibitory ability towards human acetylcholinesterase in vitro that was further confirmed by molecular modelling studies. Several compounds were selected for further studies. First, their cytotoxicity, nicotinic receptors modulation, and acute toxicity (lethal dose for 50% of laboratory animals [LD50]; mice and rats) were tested to evaluate their safety with promising results. Furthermore, their blood levels were measured to select the appropriate time for prophylactic administration. Finally, the protective ratio of selected compounds against soman-induced toxicity was determined when selected compounds were found similarly potent or only slightly better to standard pyridostigmine.
Conclusion: The presented small bisquaternary molecules did not show overall benefit in prophylaxis of soman-induced in vivo toxicity.
Keywords: AChE inhibitors, prophylaxis, pre-treatment, nerve agents, toxicity, soman
Introduction
Organophosphorus compounds (OP) are widely used (eg, in agriculture as pesticides, in industry as softening agents). Some of them were, unfortunately, developed for military purposes as chemical warfare agents. Sarin, soman or VX belong to the most known nerve agents (NA), especially among OP.1 NA mechanism of action is based on its ability to inhibit the enzyme acetylcholinesterase (AChE; EC 3.1.1.7) at peripheral and central cholinergic synapses. Afterward, generalized cholinergic crisis occurs and in severe cases may lead to death.2
To prevent the deleterious effect of NA, several prophylactic approaches were formerly developed.3 AChE protection against the OP using reversible cholinesterase (ChE) inhibitors is the oldest approach for ChE pre-treatment. In this case, the non-competitive or competitive reversible inhibitors of AChE are used to protect the irreversible binding of NA, and thus protect AChE function until the most of OP is biodegraded. This effect cannot be expected for administration of AChE reactivators (oximes) that are known to be very poor AChE inhibitors.4 Recently, enzymes scavenging or hydrolyzing OP are highly investigated.5,6
During last decade, some attention was turned back to the reversible ChE inhibitors including SAD-128, a bisquaternary compound, which was considered as pre-treatment drug candidate in eighties (Figure 1).7–10 In fact, it was the first bisquaternary compound used for this purpose and lacking oxime moiety that is common for bisquaternary reactivators (eg, trimedoxime, obidoxime). The major reason for use of charged reversible AChE inhibitor was challenge of rapidly aging organophosphates (eg, soman), which are very difficult to treat after intoxication, but might be prevented prior to intoxication by combining peripherally and centrally acting reversible AChE inhibitor. In this case, the charged inhibitor would serve as a peripherally acting one. With this hypothesis, several novel series of bisquaternary compounds were prepared and evaluated (Figures 2 and 3). In this article, we would like to summarize results, which were obtained by our group within the last seven years for bisquaternary reversible AChE inhibitors with possible impact on protection against OP compounds. The whole summary is divided into several sections that are presented consecutively step by step such as in the development process: synthesis, in vitro evaluation, cytotoxicity, nicotinic receptor evaluation, toxicity, pharmacokinetics and in vivo evaluation of the prophylactic effect.
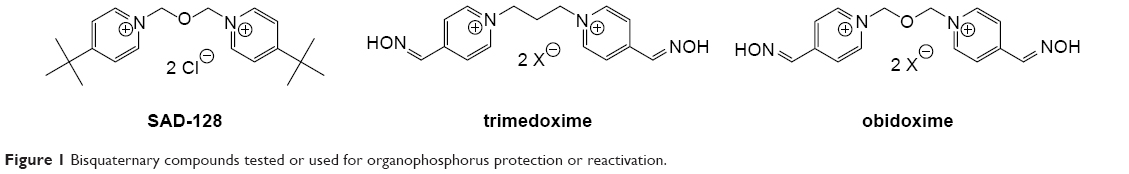 | Figure 1 Bisquaternary compounds tested or used for organophosphorus protection or reactivation. |
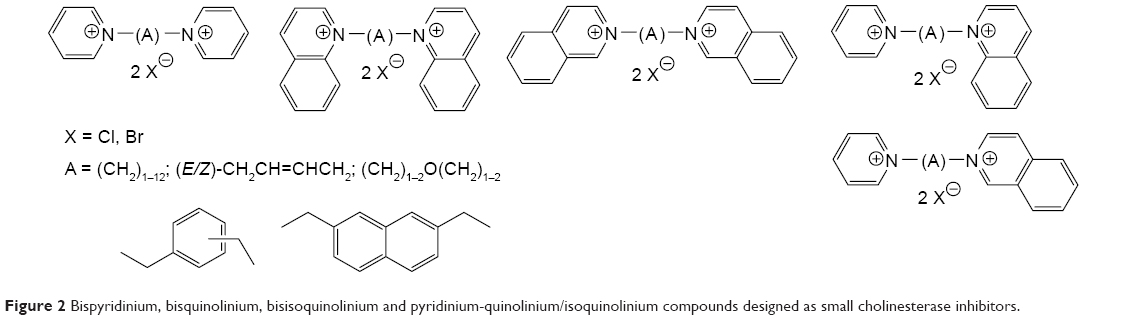 | Figure 2 Bispyridinium, bisquinolinium, bisisoquinolinium and pyridinium-quinolinium/isoquinolinium compounds designed as small cholinesterase inhibitors. |
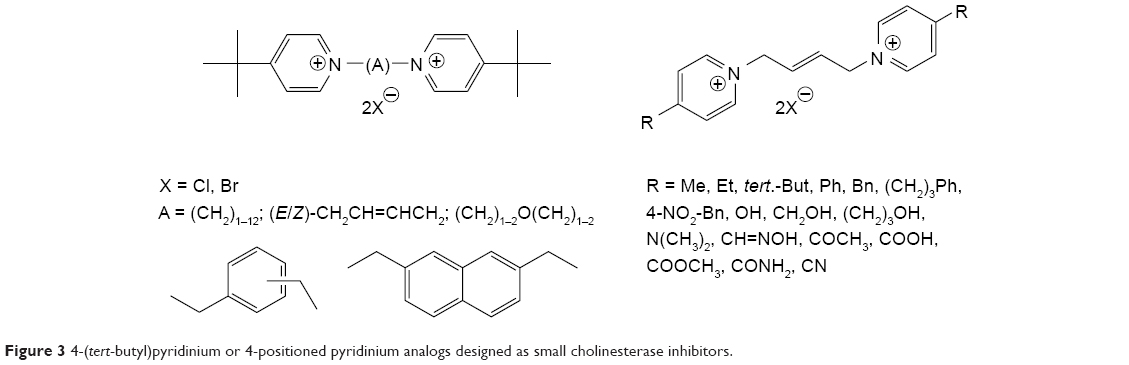 | Figure 3 4-(tert-butyl)pyridinium or 4-positioned pyridinium analogs designed as small cholinesterase inhibitors. |
Design and synthesis
The bisquaternary compounds were designed as simple molecules with two pyridinium, quinolinium, isoquinolinium or mixed pyridinium-quinolinium/isoquinolinium moieties connected by variety of linkers (Figure 2).11–14 The latter compounds were derived from 4-(tert-butyl)pyridinium or 4-positioned pyridinium analogs bridged also by varying linkage (Figure 3).15,16
The design of presented molecules originated from known compounds (eg, ambenonium dichloride, BW284c51 or SAD-128) that were known for excellent or selective cholinesterase inhibition in vitro (ambenonium dichloride, BW284c51) or in vivo (SAD-128). The heteroaromatic moieties were used to cover π-cationic interactions with the aromatic amino acid residues in the cholinesterase binding pocket (especially AChE). The varying linkage was adapted to possibly connect the AChE cation–π binding site with the peripheral aromatic binding site. In this case, the better reversible inhibition of AChE was hypothesized for some of the chosen linkers (one to 12 methylene units). Some heteroatoms, double bonds or aromatic rings were also introduced to the linkage to improve the other inhibitor-enzyme interactions within the AChE binding pocket including π-π interactions (double bonds, aromatic rings) of hydrogen bonding (ether fragment).
The synthesis of presented molecules was fairly simple (Figure 4). The heteroaromatic compound was dissolved in dimethylformamide (DMF) or acetonitrile (MeCN) upon addition of dihalogenated linking molecule and stirred at about 70°C for several hours/days. The products were obtained in good yield and purity, which was checked by standard techniques (NMR, ESI-MS).17
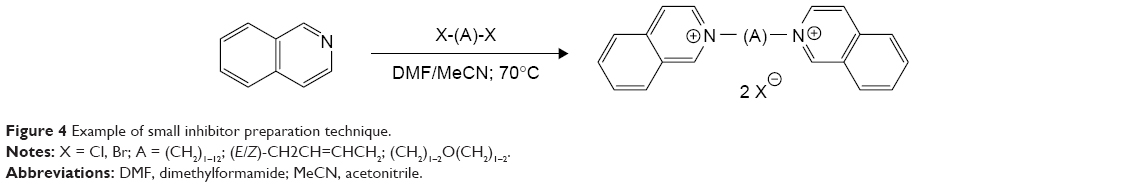 | Figure 4 Example of small inhibitor preparation technique. |
In vitro screening, molecular docking and structure–activity relationship
The in vitro screening of prepared molecules was done on human recombinant AChE and human plasmatic BChE (hBChE) to exclude the possible interspecies differences. The half maximal inhibitory concentration (IC50) was determined for all molecules and the selected compounds were further tested in kinetic experiments in vivo. The IC50 data were compared to pattern bisquaternary molecules with similar mechanism of action (ambenonium dichloride, BW284c51 or SAD-128).15,16 The selected promising compounds from all prepared series are presented in Figure 5 and Table 1.
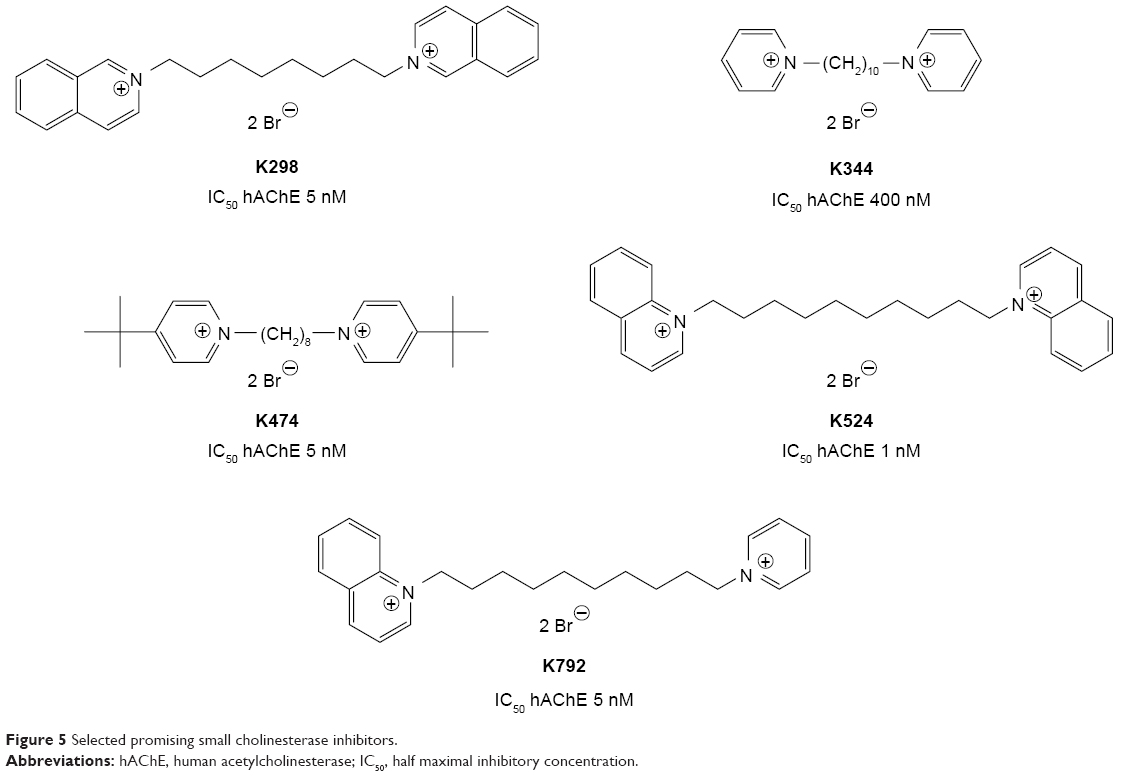 | Figure 5 Selected promising small cholinesterase inhibitors. |
 | Table 1 In vitro screening data for selected promising small cholinesterase inhibitors |
The in vitro results confirmed the hypothesis that the prepared compounds might be potent cholinesterase inhibitors. The best of them presented inhibition of human AChE (hAChE) on sub-μM (K344) or nM scale (K298, K474, K524, K792). They were able to exceed the inhibitory ability of SAD-128 or BW284c51 in vitro, but they were worse to ambenonium. Concerning the selectivity issues, the selectivity of newly prepared compounds was found to be worse to ambenonium or BW284c51 (selectivity standard), but better to SAD-128. However, the selectivity of inhibitor molecule K524 and most importantly non-symmetrical molecule K792 seemed to be improved with favorable hAChE inhibition. The kinetic experiments were performed to confirm the possible mechanism of actions of newly prepared cholinesterase inhibitors. From this point of view, they were found to be non-competitive hAChE inhibitors mostly comparable with used standards.
Moreover, the interactions with hAChE and hBChE active sites were further studied by molecular modeling experiments (Figure 6) to propose possible mechanism of action. The molecular modeling results confirmed several important interactions for each type of molecule and their non-competitive binding. The symmetrical molecules (K298, K344, K474, K524) presented closure of hAChE gorge entrance via π-π or π-cationic interactions, where one heteroarenium moiety was positioned into the gorge and the second one was located outside the gorge close to its entrance.11–13 Differently, the non-symmetrical compound K792 was found to be deep inside the gorge with pyridinium moiety located close to the active site (Ser203) and quinolinium moiety interacting with peripheral active site (Tyr124, Trp 286).14 Thus, the molecular modeling results confirmed the in vitro findings of high hAChE inhibition by symmetrical or non-symmetrical compounds plus disclosed the matter of higher selectivity of K792 for hAChE.
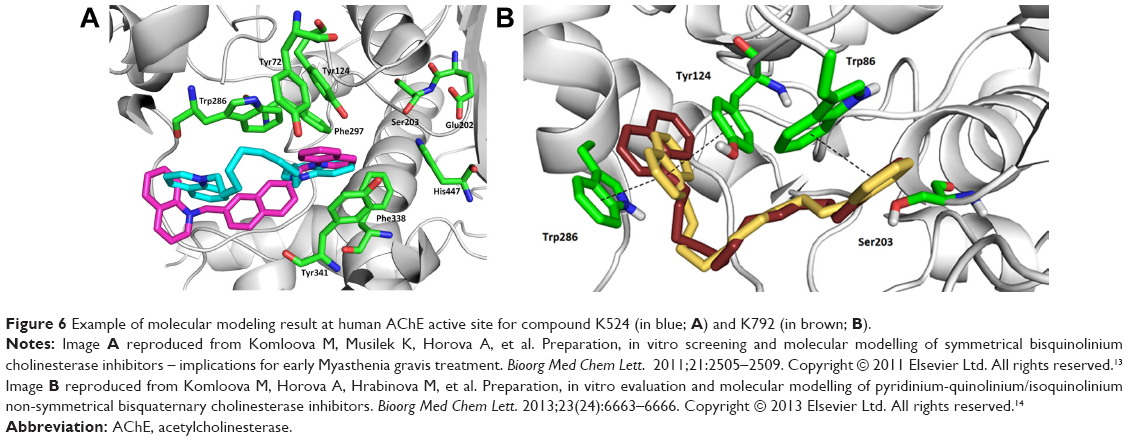 | Figure 6 Example of molecular modeling result at human AChE active site for compound K524 (in blue; A) and K792 (in brown; B). |
From the structure–activity relationship point of view, the better inhibitory ability toward hAChE was obtained for structurally similar compounds. Among these, two quaternary heteroaromatic rings connected by the linker in the length of 8–10 methylene units were found optimal within in vitro evaluation with AChE inhibition from μM to nM range. In this case, the highlighted compounds (K298, K344, K474, K524, K792) optimally connected the peripheral binding site and cation–π binding site, when cation–π interactions were confirmed by molecular docking results. Less favorably, naphthyl moiety in the connecting linker was found to inhibit AChE in μM range having additional π–π interaction with aromatic residues within the active site, but this linker was found too short to efficiently bind between the peripheral binding site and cation–π binding site.11–14 The phenyl or double bond containing linkers were found inefficient. The varying substitution on heteroaromatic moiety was another tested aspect. Firstly, the tert-butyl moiety from SAD-128 was used. Its combination with varying linker length (one to 12 methylene units) proved that compound K474 is hAChE inhibitor in nM range and is much more effective than SAD-128.16 The inhibitory ability of K474 was comparable with other heteroaromatic compounds without ring substitution (eg, K524, K792). Additionally, varying substitutions were used for two pyridinium rings connected by double bond.15 In this case, the phenyl propyl moiety extending the length of the molecule similarly to compounds K298 or K524 was found most favorable with nM inhibition of hAChE. Additional phenyl propyl scaffolds improved π–π binding and together with cation–π binding properly positioned the molecule within the active site. Among other substitutions, methyl and N,N-dimethyl amino moieties were found with inhibitory ability in low μM range.15 In both cases and in accordance with tert-butyl moiety results, the electron donating substituents were hypothesized to increase electron density in the heteroaromatic ring and thus strengthen the cation–π binding, whereas compounds possessing the electron withdrawing functional groups were found with medium to low inhibitory ability.
Cytotoxicity
As a pre-step for assessing acute toxicity in vivo, cytotoxicity assay was performed in vitro. Standard MTT test was used (all the cell lines were obtained from the collection of ATCC). This method was based on the spectrophotometric assessment of formazan that is produced by viable cells and has been widely used for research purposes for a long time.18 For the purposes of this cytotoxicity study, a comparison of compounds K298 and K524 with commercially available standards (pyridostigmine and BW284c51) on three different cell lines (HeLa – human cervical cancer cell line, BJ – fibroblasts and ACHN – renal carcinoma cell line) was performed.
Obtained results showed clear pattern of the cytotoxic effect on all the cell lines (Table 2) and can be considered as very low. Standard compound BW284c51 was found to be least toxic to all the cell lines. This fact is probably due to the presence of quaternary nitrogens in this structure, ensuring the low penetration across the cell membrane to the cytosol. The same can be postulated for newly prepared bisquaternary inhibitors; however, the cytotoxic effects are a bit higher probably due to higher lipophilicity gained by the long carbon linker. Thus, it may be concluded that newly synthesized compounds do not differ much from the investigated standards on used cell lines. They can also be considered as low toxic compounds and might proceed to in vivo examination.19
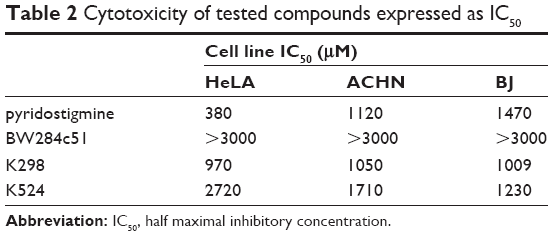 | Table 2 Cytotoxicity of tested compounds expressed as IC50 |
Nicotinic receptors modulation
Both newly prepared AChE inhibitors (K298, K524) were also compared with two standards (BW284c51, edrophonium) for their modulation of nicotinic receptors. All tested compounds showed a comparatively high reversible inhibition effect (IC50). When tested compounds were pre-applied (in different concentration) for 5 s and then co-applied with the agonist, acetylcholine (100 μM) responses were inhibited in a concentration-dependent manner. Newly synthesized inhibitor K524 showed the lowest IC50 value 570±5 nM that was approximately one or two orders of magnitude lower compared to used standards. K298 showed approximately 4-fold lower affinity (IC50=1900±200 nM). Potentiation of the muscle-type nicotinic receptors was not recorded.19 Such potent inhibition may provide protection of nicotinic receptors by the pre-treatment especially when combined by protection of the AChE. Furthermore, it might also be used as the post-exposure treatment via the antagonism of the overstimulated nicotinic receptors.20
Acute toxicity
In front of the pharmacokinetic and prophylactic evaluation of newly prepared ChE inhibitors, acute toxicity of selected compounds was tested in mice (K298, K344, K474) and rats (K298, K524) by the assessment of their lethal dose for 50% of laboratory animals (LD50) values and their 95% confidence limits using probit-logarithmical analysis of death occurring within 24 hours after intramuscular (im) administration of each inhibitor. According to the obtained results, tested quaternary ChE inhibitors were about one order of magnitude more toxic than commonly used AChE reactivators (LD50 for asoxime in mice 671 mg·kg−1, in rats 781 mg·kg−1) (Tables 3 and 4).21,22 Ethics Committee of the Faculty of Military Health Sciences in Hradec Kralove (Czech Republic) approved the animal experiments. The Guide for the Care and Use of Laboratory Animals was followed.
 | Table 3 Acute toxicity of reversible inhibitors of AChE in mice after intramuscular administration |
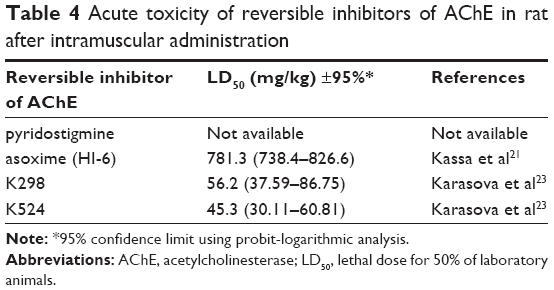 | Table 4 Acute toxicity of reversible inhibitors of AChE in rat after intramuscular administration |
Pharmacokinetics
Time-dependent concentration changes of selected AChE inhibitors (K298, K524) were measured in rat plasma to select appropriate time for administration as prophylactics. For this purpose, equitoxic doses (5% LD50) were applied intramuscularly. HPLC-MS instrumentation was selected as the most appropriate for determination of low inhibitor concentrations.
The maximal plasma concentrations were achieved in 30 min (K298) and 39 min (K524) and corresponded to 100 nM concentration (Table 5).23 The pyridostigmine maximal levels after i.m. administration in rats were formerly found in about 3 min with elimination half-life in about 25 min.24 The fast absorption and also fast elimination from blood stream were characteristic for tested compounds, when their elimination was partially slower with half-life of about 50–55 min.23 Obtained plasma curves were similar to typical plasma absorption curves obtained for AChE reactivators (HI-6, obidoxime, K027 and K203).25–27
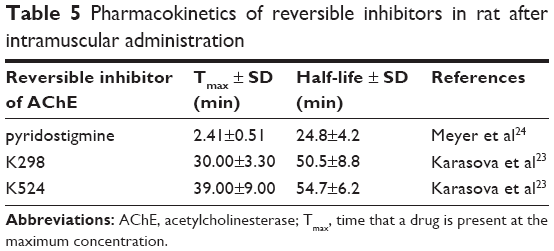 | Table 5 Pharmacokinetics of reversible inhibitors in rat after intramuscular administration |
In vivo prophylactic effect
The prophylactic effect of selected AChE reversible inhibitors pyridostigmine, K298, K344 and K474 was evaluated in vivo. They were administered by i.m. injection 30 minutes prior to i.m. soman challenge in dose corresponding to 2% of their LD50 values. Soman-induced toxicity was found via evaluation of its LD50 value and its 95% confidence limit using probit-logarithmical analysis of death that was observed within 24 h after its administration.28 The effect of selected prophylactic compounds was calculated as protective ratio (LD50 value of soman in pretreated mice/LD50 value of soman in non-pretreated mice).
Further, the inhibitors pyridostigmine, K298, K344 and K474 were evaluated for the therapeutic effect when antidotal treatment was tested. All selected inhibitors were administered 30 min prior to soman injection, while the antidotal treatment (5% LD50 asoxime dichloride in combination with atropine 21 mg/kg) was carried out by i.m. injection 1 min after the soman challenge. Again, soman-induced toxicity was found via evaluation of its LD50 value and its 95% confidence limit using probit-logarithmical analysis of death that was observed within 24 h after its administration.28 The influence of tested prophylactic compounds on the antidotal treatment within soman poisoning was calculated as protective ratio A (LD50 value of soman in pretreated mice with antidotal treatment/LD50 value of soman in non-pretreated mice without antidotal treatment) and protective ratio B (LD50 value of soman in pretreated mice with antidotal treatment/LD50 value of soman in non-pretreated mice with antidotal treatment). The differences between LD50 values were considered to be significant when p<0.05.29
The results of the study showed that none of the tested ChE inhibitors was able to decrease acute soman-induced toxicity in mice (Table 6).28 The standard in the field (pyridostigmine) was found to be poorly effective. This finding was most probably caused by its i.m. administration 30 min prior to intoxication, when its half-life was about 25 min. Thus, its plasma level in time of intoxication was apparently low and it cannot increase the protection of rats against the soman challenge. The prophylactic administration of reversible AChE inhibitors K298 and K344 was found not to influence the therapeutic effect of used antidotal treatment in soman-poisoned mice, when their pharmacokinetics was properly adjusted in this case. Only inhibitor K474 slightly increased the effect of the antidotal treatment (asoxime in combination with atropine) in soman-poisoned mice and was slightly better than pyridostigmine, although the differences between treated soman-poisoned mice with or without pre-treatment for K474 were found not significant.28
 | Table 6 Prophylactic effect of reversible AChE inhibitors with one LD50 of soman in mice |
Discussion
In this summary, whole pipeline for development of novel prophylactic means was shown. It consists of design and synthesis of novel AChE inhibitors, their in vitro evaluation, cytotoxicity tests, animal toxicity evaluation and finally in vivo pharmacodynamic (prophylactic activity) measurement. So, all steps that were performed during the development of novel small quaternary inhibitors of ChE were shown. Although more than 100 novel compounds were prepared, only a few of them were selected according to their in vitro ability and safety (cytotoxicity, acute toxicity) in more sophisticated experiments such as pharmacokinetics or in vivo prophylactic evaluation. As shown according to the in vivo results, promising compound was unfortunately not found in the final step (all tested ChE inhibitors could be considered as poor pre-treatment), which could replace standard prophylactic means (pyridostigmine). On the contrary, this study provided lots of novel information, which could be used in the development of new generation of small inhibitors (eg, possible toxicity, expected pharmacokinetic profile).
There are several ways to progress in future investigations. First, novel non-symmetric analogs should be prepared to show the differences between already known compounds and the novel ones. There could be similar differences found as in the case of oxime reactivators such as obidoxime (symmetric compound) and HI-6 (non-symmetric compound).22 Moreover, introduction of several functional groups such as carbamoyl group could decrease ChE inhibitors toxicity as in the case of AChE reactivators.17 Finally, preparation of non-quaternary inhibitors derived from Alzheimer disease drugs could be considered as appropriate pre-treatment with peripheral and central antiChE activity.29–32
Results obtained in this work could be utilized also in a different way. Many structurally different compounds were prepared and tested (differing in length and shape of the linker, heteroarenium moiety). These compounds form a relatively large dataset, which could be used in future for a structure-activity study enabling prediction of more promising compounds using different computational methods.
Acknowledgments
This work was supported by the Ministry of Defence of the Czech Republic (Long Term Development Plan-1011), the University Hospital in Hradec Kralove (number 00179906) and the University of Hradec Kralove (number 2201-2017).
Disclosure
The authors report no conflicts of interest in this work.
References
Marrs TC. Organophosphate anticholinesterase poisoning. Toxic Subst Mech. 1996;15:357–388. | ||
Bajgar J, Fusek J, Kuca K, Bartosova L, Jun D. Treatment of organophosphate intoxication using cholinesterase reactivators: facts and fiction. Mini Rev Med Chem. 2007;7(5):461–466. | ||
Patocka J, Jun D, Bajgar J, Kuca K. Prophylaxis against nerve agent intoxications. Def Sci J. 2006;56:775–784. | ||
Sepsova V, Karasova JZ, Zemek F, Bennion B, Kuca K. Oximes as inhibitors of acetylcholinesterase – a structure-activity relationship (SAR) study. Mil Med Sci Lett. 2011;80:178–186. | ||
Masson P, Josse D, Lockridge O, Viguie N, Taupin C, Buhler C. Enzymes hydrolyzing organophosphates as potential catalytic scavengers against organophosphate poisoning. J Physiol Paris. 1998;92(5–6):357–362. | ||
Masson P, Nachon F. Cholinesterase reactivators and bioscavengers for pre- and post-exposure treatments of organophosphorus poisoning. J Neurochem. 2017;142 (Suppl 2):26–40. | ||
Harris LW, Heyl WC, Stitcher DL, Broomfield CA. Effects of 1,1′-oxydimethylene bis-(4-tert-butylpyridinium chloride) (SAD-128) and decamethonium on reactivation of soman-inhibited and sarin-inhibited cholinesterase by oximes. Biochem Pharmacol. 1978;27(5):757–761. | ||
Alkondon M, Albuquerque EX. The nonoxime bispyridinium compound SAD-128 alters the kinetic properties of the nicotinic acetylcholine receptor ion channel: a possible mechanism for antidotal effects. J Pharmacol Exp Ther. 1989;250(3):842–852. | ||
Kloog Y, Sokolovsky M. Bisquaternary pyridinium oximes as allosteric inhibitors of rat brain muscarinic receptors. Mol Pharmacol. 1985;27:418–428. | ||
Lundy PM, Tremblay KP. Ganglion blocking properties of some bispyridinium soman antagonists. Eur J Pharmacol. 1979;60(1):47–53. | ||
Musilek K, Komloova M, Zavadova V, et al. Preparation and in vitro screening of symmetrical bispyridinium cholinesterase inhibitors bearing different connecting linkage – initial study for Myasthenia gravis implications, Bioorg Med Chem Lett. 2010;20:1763–1766. | ||
Musilek K, Komloova M, Holas O, et al. Preparation and in vitro screening of symmetrical bis-isoquinolinium cholinesterase inhibitors bearing various connecting linkage – implications for early Myasthenia gravis treatment. Eur J Med Chem. 2011;46:811–818. | ||
Komloova M, Musilek K, Horova A, et al. Preparation, in vitro screening and molecular modelling of symmetrical bisquinolinium cholinesterase inhibitors – implications for early Myasthenia gravis treatment. Bioorg Med Chem Lett. 2011;21:2505–2509. | ||
Komloova M, Horova A, Hrabinova M, et al. Preparation, in vitro evaluation and molecular modelling of pyridinium-quinolinium/isoquinolinium non-symmetrical bisquaternary cholinesterase inhibitors. Bioorg Med Chem Lett. 2013;23(24):6663–6666. | ||
Musilek K, Pavlikova R, Komloova M, et al. Preparation, in vitro screening and molecular docking of symmetrical bisquaternary cholinesterase inhibitors bearing but-(2E)-en-1,4-diyl connecting linkage. J Enzym Inhib Med Chem. 2011;26:245–253. | ||
Musilek K, Roder J, Komloova M, et al. Preparation, in vitro screening and molecular modelling of symmetrical 4-tert-butylpyridinium cholinesterase inhibitors – analogues of SAD-128. Bioorg Med Chem Lett. 2011;21(1):150–154. | ||
Musilek K, Kucera J, Jun D, Dohnal V, Opletalova V, Kuca K. Monoquaternary pyridinium salts with modified side chain–synthesis and evaluation on model of tabun- and paraoxon-inhibited acetylcholinesterase. Bioorg Med Chem. 2008;16(17):8218–8223. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65(1–2):55–63. | ||
Sepsova V, Krusek J, Zdarova-Karasova J, et al. The interaction of quaternary reversible acetylcholinesterase inhibitors with the nicotinic receptor. Physiol Res. 2014;63(6):771–777. | ||
Kassa J, Korábečný J, Nepovimová E. The evaluation of benefit of newly prepared reversible inhibitors of acetylcholinesterase and commonly used pyridostigmine as pharmacological pretreatment of soman-poisoned mice. Acta Medica (Hradec Kralove). 2017;60(1):37–43. | ||
Kassa J, Kuca K, Bartosova L, Kunesova G. The development of new structural analogues of oximes for the antidotal treatment of poisoning by nerve agents and the comparison of their reactivating and therapeutic efficacy with currently available oximes. Curr Org Chem. 2007;11(3):267–283. | ||
Musilek K, Jun D, Cabal J, Kassa J, Gunn-Moore F, Kuca K. Design of a potent reactivator of tabun-inhibited acetylcholinesterase – synthesis and evaluation of (E)-1-(4-carbamoylpyridinium)-4-(4-hydroxyiminomethylpyridinium)-but-2-ene dibromide (K203). J Med Chem. 2007;50(22):5514–5518. | ||
Karasova JZ, Hroch M, Musilek K, Kuca K. Small quaternary inhibitors K298 and K524: cholinesterases inhibition, absorption, brain distribution, and toxicity. Neurotox Res. 2016;29(2):267–274. | ||
Meyer HG, Lukey BJ, Gepp RT, Corpuz RP, Lieske CN. A radioimmunoassay for pyridostigmine. J Pharmacol Exp Ther. 1988;247:432–438. | ||
Karasova JZ, Hnidkova D, Pohanka M, Musilek K, Chilcott RP, Kuca K. Pharmacokinetics of acetylcholinesterase reactivator K203 and consequent evaluation of low molecular weight antioxidants/markers of oxidative stress. J Appl Biomed. 2012;10:71–78. | ||
Karasova JZ, Chladek J, Hroch M, Fusek J, Hnidkova D, Kuca K. Pharmacokinetic study of two acetylcholinesterase reactivators, trimedoxime and newly synthesized oxime K027, in rat plasma. J Appl Toxicol. 2013;33(1):18–23. | ||
Karasova JZ, Novotny L, Antos K, Zivna H, Kuca K. Time-depend changes in concentration of two clinically used acetylcholinesterase reactivators (HI-6 and obidoxime) in rat plasma determined by HPLC techniques after in vivo administration. Anal Sci. 2010;26:63–67. | ||
Kassa J, Musilek K, Koomlova M, Bajgar J. A comparison of the efficacy of newly developed reversible inhibitors of acetylcholinesterase with commonly used pyridostigmine as pharmacological pre-treatment of soman-poisoned mice. Bas Clin Pharm Tox. 2012;110(4):322–326. | ||
Tallarida R, Murray R. Manual of Pharmacological Calculation with Computer Programs. New York: Springer-Verlag; 1987. | ||
Lorke DE, Nurulain SM, Hasan MY, Kuca K, Petroianu GA. Prophylactic administration of non-organophosphate cholinesterase inhibitors before acute exposure to organophosphates: assessment using terbufos sulfone. J Appl Toxicol. 2014;34(10):1096–1103. | ||
Lorke DE, Hasan MY, Nurulain SM, Shafiullah M, Kuca K, Petroianu GA. Acetylcholinesterase inhibitors as pretreatment before acute exposure to organophosphates: assessment using methyl-paraoxon. CNS Neurol Disord Drug Targets. 2012;11(8):1052–1060. | ||
Petroianu GA, Nurulain SM, Shafiullah M, Hasan MY, Kuca K, Lorke DE. Usefulness of administration of non-organophosphate cholinesterase inhibitors before acute exposure to organophosphates: assessment using paraoxon. J Appl Toxicol. 2013;33(9):894–900. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.