")
Back to Journals » International Journal of Nanomedicine » Volume 15
Development of Nanosome-Encapsulated Honokiol for Intravenous Therapy Against Experimental Autoimmune Encephalomyelitis
Authors Hsiao YP , Chen HT, Liang YC, Wang TE, Huang KH, Hsu CC , Liang HJ, Huang CH, Jan TR
Received 3 May 2019
Accepted for publication 21 October 2019
Published 6 January 2020 Volume 2020:15 Pages 17—29
DOI https://doi.org/10.2147/IJN.S214349
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Yai-Ping Hsiao,1 Hui-Ting Chen,1 Yu-Chih Liang,2 Tse-En Wang,1 Kai-Hung Huang,3 Cheng-Chih Hsu,3 Hong-Jen Liang,4 Chung-Hsiung Huang,5 Tong-Rong Jan1
1Department of Veterinary Medicine, School of Veterinary Medicine, National Taiwan University, Taipei 10617, Taiwan; 2School of Medical Laboratory Science and Biotechnology, College of Medical Science and Technology, Taipei Medical University, Taipei 10617, Taiwan; 3Department of Chemistry, National Taiwan University, Taipei 10617, Taiwan; 4Department of Food Science, Yuanpei University, Hsinchu 30015, Taiwan; 5Department of Food Science, National Taiwan Ocean University, Keelung 20224, Taiwan
Correspondence: Tong-Rong Jan
Department and Graduate Institute of Veterinary Medicine, School of Veterinary Medicine, National Taiwan University, No.1, Sec. 4, Roosevelt Road, Taipei 10617, Taiwan, ROC
Tel +886-2-33661287
Fax +886-2-23661475
Email [email protected]
Chung-Hsiung Huang
Department of Food Science, National Taiwan Ocean University, No.2, Beining Road, Keelung 20224, Taiwan
Tel +886-2-24622192#5115
Email [email protected]
Background: Honokiol has been reported to possess anti-inflammatory and neuroprotective activities. However, the poor aqueous solubility of honokiol limits its clinical application for systemic administration.
Purpose: This study aims to develop a novel formulation of nanosome-encapsulated honokiol (NHNK) for intravenous therapy against mouse experimental autoimmune encephalomyelitis (EAE) that mimics human multiple sclerosis.
Methods: Nanosomes and NHNK were prepared by using an ultra-high pressure homogenization (UHPH) method. Mice were treated with NHNK or empty nanosomes during the peak phase of EAE symptoms. Symptoms of EAE were monitored and samples of the spinal cord were obtained for histopathological examinations.
Results: The stock of NHNK containing honokiol in the nanosome formulation, which showed the structure of single phospholipid bilayer membranes, was well formulated with the particle size of 48.0 ± 0.1 nm and the encapsulation efficiency 58.1 ± 4.2%. Intravenous administration of NHNK ameliorated the severity of EAE accompanied by a significant reduction of demyelination and inflammation in the spinal cord. Furthermore, NHNK decreased the number of IL-6+, Iba-1+TNF +, Iba-1+IL-12 p40+, and CD3+IFN-γ+ cells infiltrating the spinal cord.
Conclusion: The UHPH method simplified the preparation of NHNK with uniformly distributed nanosize and high encapsulation efficiency. Intravenous administration of NHNK ameliorated the severity of EAE by suppressing the infiltration of activated microglia and Th1 cells into the spinal cord. Collectively, these results suggest that the formulation of NHNK is a prospective therapeutic approach for inflammatory CNS diseases, such as multiple sclerosis.
Keywords: experimental autoimmune encephalomyelitis, honokiol, nanosome, neuroinflammation, ultra-high pressure homogenization
Introduction
Multiple sclerosis is a debilitating disease of the central nervous system (CNS) characterized by multifocal inflammation, demyelination, and neuronal damage. The mouse model of experimental autoimmune encephalomyelitis (EAE), which mimics clinical, immunological, and histopathological features of multiple sclerosis (MS), is commonly used for the investigation of neuroprotective agents against multiple sclerosis.1 Microglia and T lymphocytes are considered central to the pathophysiology of MS and its mouse model EAE.2–4 As the major resident immune cells in the CNS, microglia contribute to neuroinflammation by promoting the recruitment of inflammatory cells from the peripheral blood into the inflamed site and by producing pro-inflammatory cytokines, including IL-1β, IL-6, and TNF.5 In human CNS, the interaction of activated T cells and microglia in the presence of CD40 engagement resulting in IL-12 production is required for persistent and recurrent pro-inflammatory Th1 immune responses.6–8 Notably, the secretion of IL-12 by microglia is also regulated by both TNF and IFN-γ stimuli.7,9 So far, there are some disease-modifying therapies for multiple sclerosis, such as mitoxantrone, fingolimod, and siponimod. These treatments are in an attempt to facilitate recovery from attacks, retard the disease progression, and relieve symptoms.10 However, the therapies have the considerable risk of side effects.11 Therefore, emerging therapies are urgent to be developed for the management of multiple sclerosis.
Honokiol, a natural hydrophobic phytochemical extracted from the Chinese herb Houpu, has been traditionally used to treat many mental and neurological disorders.12 It has been well documented that honokiol exerts several pharmacological activities, including anti-anxiety,13 anti-hyperalgesia,14 anti-inflammation,15 anti-oxidation,16 anti-senescence,17 and neuroprotection.18 In mouse models, honokiol treatment could ameliorate the symptoms of allergic asthma, postoperative ileus and arthritis by reducing the production of inflammation-related factors, such as TNF, IL-6, and IFN-γ.19–21 Intraperitoneal administration of honokiol could attenuate motor dysfunction in hemi-parkinsonian mice and neuronal degeneration in mice with traumatic spinal cord injury.15,22 Additionally, mechanistic investigation revealed that honokiol influenced the activation of microglial cells by modulating the production of nitric oxide (NO), pro-inflammatory and anti-inflammatory cytokines.23 These results suggest that honokiol is a promising phytochemical to be developed as a potential anti-inflammatory and neuroprotective agent against inflammatory CNS diseases. However, the clinical application of honokiol is hampered by its low aqueous solubility and poor feasibility for intravenous administration.
Drug delivery system has received significant attention due to the potential to improve the therapeutic efficiency of drug candidates by increasing their aqueous solubility, bioavailability, and stability.24 One of the common drug delivery vehicles is liposomes, which is composed of several lipid bilayers enclosing aqueous compartments and are able to encapsulate both hydrophilic and hydrophobic agents.25 The size, surface charge and moieties, lipid constituents, and cholesterol contents of liposomes can be manipulated to produce liposomal drug formulations with various physicochemical properties and thus dramatically improve the bioavailability and toxicity of drug candidates.26 For example, liposomes can cross the blood–brain barrier or transport larger amounts of therapeutic agents to a specific site by adding specific targeting moieties on their surface.27,28 However, multiple-layer liposomes without any modifications are strait to pass through narrow blood vessels. Therefore, the materials entrapped in the inner layers of liposomes are hardly accessible and releasable for practical applications.29 Recently, lipid-based nanoparticles have been rapidly developed for the delivery of drugs, proteins, and genes.30 Nano-sized liposomes (nanosomes) could penetrate into small blood vessels via intravenous injection, and the encapsulated materials are easily transported and delivered to target cells compared to multiple-layer liposomes.31 Nonetheless, nanosomes with a single-bilayer structure are difficult to manufacture. Typically, subjecting large- and multiple-layer liposomes to ultrasonic energy is required for the manufacturing process of nanosomes. The process is time-consuming, tedious, extremely delicate, and mostly done in small batches.
Compared to other administration routes, one of the major advantages of intravenous injection is that the entire administered dose immediately reaches the systemic circulation. Other advantages of intravenous injection include minimal volume constraints, reducing injection site reaction, precise control of the dose and administration rate, dependable and reproducible effects, and more rapid recovery.32,33 Moreover, multiple doses can also be administered through an intravenous infusion, with a high degree of versatility and control.34,35 However, low aqueous solubility is one of the major restrictions for drugs to be administrated via intravenous route. Herein, we developed nanosomes using a UHPH method to simplify the manufacturing process and employed nanosomes as delivery vehicles for the intravenous administration of honokiol. The physicochemical properties of nanosomes and NHNK were measured, and the therapeutic effect of NHNK on EAE was investigated in mice. Furthermore, the neuroprotective and anti-inflammatory effects of NHNK were addressed by a mechanistic analysis of the activation of microglia and T cells.
Materials and Methods
Chemicals and Reagents
All chemicals and reagents were purchased from Sigma Chemical (St. Louis, Missouri, USA) unless otherwise stated. Honokiol was purchased from Selleck Chemical (Houston, Texas, USA). Antibodies used for immunohistochemical (IHC) were purchased from Abcam Inc. (Cambridge, Massachusetts, USA), or Aviva Systems Biology (San Diego, California, USA). Enzymes and reagents used for IHC staining were purchased from BioGenex (San Ramon, California, USA).
Preparation of Nanosome-Encapsulated Honokiol (NHNK)
Nanosome-encapsulated honokiol (NHNK) was prepared by an ultra-high pressure homogenization method. Briefly, the lipid components (soybean phosphatidylcholine and cholesterol) were dissolved at the molar ratio of 10:1 in ethanol. Honokiol was then added in the lipid/ethanol mixture at a concentration of 2 mg/mL. After removing ethanol, the mixture containing honokiol was subsequently homogenized with phosphate-buffered saline by using an Ultra-Turrax T25 (IKA, Staufen im Breisgau, Germany) at 5000 rpm for 1 min. The obtained pre-emulsion was then homogenized by an ultra-high pressure homogenizer (Impact Mixer GIM-03A; Taipei, Taiwan) applying four cycles at 20,000 psi. The preparation of empty nanosomes was the same as that of NHNK without the addition of honokiol. The structure of nanosomes was observed by field emission gun electron microscopy (FEI-TECNAI G2 F20 S-TWIN; Hillsboro, Oregon, USA) and the size of nanosomes and NHNK was measured by dynamic light scattering (Malvern Zetasizer Nano; Malvern, UK).
Determination of Encapsulation Efficiency of NHNK
Encapsulation efficiency of NHNK was determined by external standard methods using high-performance liquid chromatography (HPLC; Agilent 1260 Fluorescence Spectrophotometer; Santa Clara, California, USA). The HPLC mobile phase was water (with 0.1% formic acid) and acetonitrile (with 0.1% formic acid). The detected absorption wavelength was set at 254 nm. The triplicate injections (20 μL for each) were performed using the autosampler Waters SunFire C18 column (4.6 × 250 mm, 5 µm). A calibration plot of honokiol standard solutions was built with a series of purified honokiol concentrations of 0.005–0.1 ppm. Freshly prepared NHNK samples were centrifuged to separate different fractions of honokiol, following with the protocol described in the previous study.36 Each fraction was re-dissolved in methanol and analyzed using HPLC. The amount of honokiol in each fraction was estimated based on corresponding concentrations from the absorption response in the calibration curve, and the encapsulation efficiency was estimated to be 58.1 ± 4.2%.
MOG-Induced EAE and NHNK Treatment
Female C57BL/6 mice (11–13 weeks) were purchased from the National Laboratory Animal Center (Ilan, Taiwan) and housed in a temperature (25 ± 2°C), humidity (60% ± 20%), and light (12-hrs light/dark cycle)-controlled environment. Mice were supplied with standard food and water ad libitum. All animal experiments were approved by the Institutional Animal Care and Use Committee at the National Taiwan University (NTU107-EL-00005). All studied animals were cared and handled in compliance with the Guideline for the Care and Use of Laboratory Animals issued by the Council of Agriculture Executive Yuan, Taiwan.
A commercially available EAE induction kit (Hooke Laboratories Inc., Lawrence, Massachusetts, USA) was used following the supplier’s instructions. Briefly, mice were either left unimmunized (naïve, NA; n = 13) or injected subcutaneously with 0.2 mL myelin oligodendrocyte glycoprotein peptide (MOG)35–55 in complete Freund’s adjuvant containing 4 mg/mL heat-inactivated Mycobacterium tuberculosis (H37RA) on day 0 (the day of immunization). Then, the immunized mice were intraperitoneally injected with 200 ng of pertussis toxin at 3 hrs and 24 hrs post-immunization (Figure 1). The symptoms of EAE were observed and the clinical scores were recorded daily for 37 days as follows: 0, no disease signs; 0.5, reduced tail tonus; 1, limp tail; 1.5, ataxia; 2, hindlimb weakness; 2.5, at least one hind limb paralysis; 3, both hind limbs paralysis; 3.5, forelimb weakness; 4, paralysis until hip; 5, moribund or death. According to the effective therapeutic dose range of honokiol reported in previous studies, a dose of 20 mg/kg (equal to 0.18–0.25 mL of NHNK depending on the weight of individual mouse) was employed in the present study.15,37 As the encapsulation rate of NHNK was 58.1%, the realistic dose of honokiol carried by nanosomes was 11.6 mg/kg. The immunized mice were randomly distributed into different treatment groups on day 18 and intravenously injected with NHNK (n = 19) or empty nanosomes (vehicle control, NSE; n = 19) twice a week for 3 weeks. All mice were sacrificed on day 37, and the samples of spinal cords were harvested for further experiments.
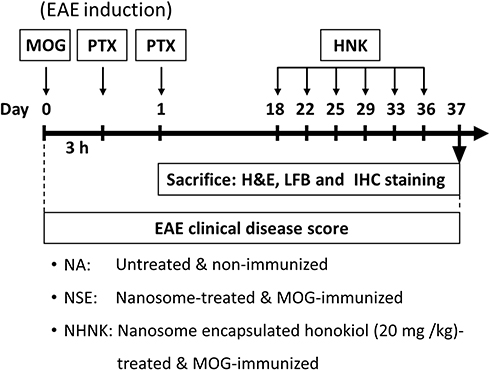 |
Figure 1 Protocol of EAE induction and NHNK administration. Female C57BL/6 mice were either left unimmunized (naïve, NA) or immunized with myelin oligodendrocyte glycoprotein (MOG)35–55 emulsion to induce EAE. The EAE mice were intravenously injected with 20 mg/kg NHNK or NSE (vehicle control) from day 18–36 for a total of six doses. Clinical symptoms of EAE were daily monitored. All mice were sacrificed on day 37, and the spinal cords were harvested for further experiments. |
Histological Examinations and Neuronal Demyelination
Tissue sections of the spinal cords were stained with H & E and luxol fast blue (LFB) to assess the infiltration of inflammatory cells and neuronal demyelination, respectively. The slides were visualized using an inverted microscope (Olympus IX83, Tokyo, Japan). Pathological scores were examined in H & E-stained sections and evaluated in a blind manner using the standard scoring from 0 to 3 as follows: 0, no inflammation; 1, small number of inflammatory cells; 2, numerous infiltrating cells; and 3, widespread infiltration. The density of LFB-positive signals was measured using ImageJ image processing and analysis program (Bethesda, Maryland, USA). The percentage of demyelination was calculated as follows:

Immunohistochemistry (IHC)
Tissue sections of the spinal cords were dewaxed, rehydrated, and then antigen-retrieved in Trilogy™ (Cell Marque, AR, USA) at 121°C for 15 mins. The sections were separately incubated with ice methanol containing 3% H2O2 and blocked with normal horse serum to reduce endogenous peroxidase activity and nonspecific reactions. The slides were incubated with anti-mouse CD3, Iba-1 (GeneTex; Irvine, California, USA), IFN-γ, IL-6, IL-12 p40, or TNF antibody at 4°C overnight, treated with Super enhancer reagent for 1 hr, and then treated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hr. For visualization, slides were incubated with the HRP substrate 3,3ʹ-diaminobenzidine for 3−7 mins followed by hematoxylin counterstaining for 5 mins in the dark. The dark-brown positive signals were counted manually.
To determine whether the cytokines were released by activated microglia or T cells, a dual-color immunochemical staining was performed. Tissue sections stained for Iba-1 or CD3 were prepared as described above without counterstaining. Subsequently, these slides were again incubated with the secondary antibody, e.g. anti-TNF, anti-IL-12 p40 or anti-IFN-γ antibody at 4°C overnight and then treated with alkaline phosphatase (AP)-conjugated secondary antibody for 1 hr. For visualization, the slides were incubated with the AP substrate 5-bromo-4-chloro-3-indolyphosphate/nitroblue tetrazolium (BCIP/NBT) for 30 mins. Cells showing both brown and dark red colors were enumerated manually as double-positive cells.
Statistical Analysis
Data are expressed as mean ± standard error (SE) for each experimental group. Statistical significance for the disease course and the percentage of demyelinated areas were analyzed using the Mann–Whitney test. Cumulative and mean clinical scores were analyzed using the Kruskal–Wallis test with Dunn’s multiple comparison as post hoc tests. All others were analyzed using one-way ANOVA and Student’s t-test. p < 0.05 was defined as statistical significance.
Results and Discussion
Characterization of NHNK
The prepared nanosomes observed by field emission gun high-resolution electron microscopy revealed a sphere bilayer structure and homogeneously distributed around 50 nm (Supplementary Figure 1A), which was consistent with the data of size distribution obtained from dynamic light scattering analysis (Supplementary Figure 1B). The mean diameter of nanosomes was 54.0 ± 0.7 nm, and the polydispersity index was 0.27. The diameter of NHNK was 48.0 ± 0.1 nm, and the polydispersity index was 0.28 (Table 1). The encapsulation efficiency of NHNK was 58.1 ± 4.2%, indicating that the realistic dose of honokiol carried by nanosome was about 11.6 mg/kg (Table 1). These results demonstrated that NHNK was well formulated with the properties of uniform size distribution and high encapsulation efficiency.
 |
Table 1 Characterization of NHNK Formulation |
Intravenous Administration of NHNK Ameliorated the Clinical Signs of EAE
We next addressed the therapeutic effect of NHNK on the disease severity of EAE. Clinical signs of EAE were daily monitored throughout the entire experimental period. Mice immunized with MOG35-55 exhibited a typical course of EAE and began to show clinical signs on day 12 post immunization, whereas no clinical signs could be observed in the NA group. The clinical scores of EAE mice progressively increased and reached the peak phase on day 18. On the same day, NHNK or empty nanosomes (NSE) was intravenously administered to EAE mice via the tail vein. NHNK treatment markedly decreased the scores of clinical signs from day 22 to day 37 compared to the NSE group (Figure 2A). Concordantly, the mean and cumulative clinical scores from the first day of intravenous therapy to the end of experimental period also revealed the same trend (Figure 2B and C), indicating a potential therapeutic effect of NHNK on EAE.
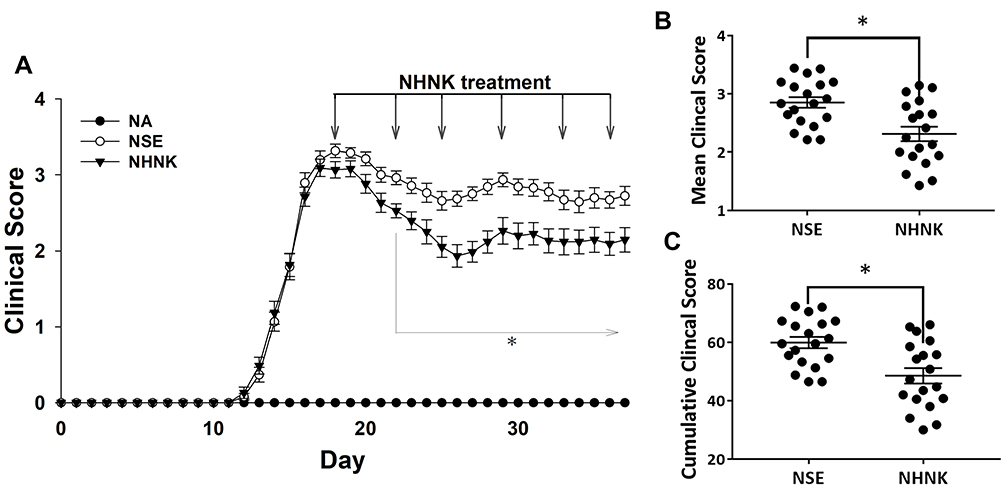 |
Figure 2 NHNK treatment ameliorated the disease severity of EAE. (A) Daily clinical scores of the NA (n = 13), NSE (n = 19), and NHNK (n = 19) groups are shown. Data are expressed as the mean ± SE of 13–19 samples pooled from three independent experiments. (B) The distribution of mean clinical score and (C) cumulative clinical score of individual mice in the NSE and NHNK groups from day 18–37 are shown. *p < 0.05 compared to the NSE group. |
Demyelination is an important hallmark of EAE, which is commonly detected in patients with long-standing multiple sclerosis.38 The severity of demyelination in the spinal cord of EAE mice was examined by LFB staining. Compared to that of the NA group, signals of LFB at the area of white matter were markedly lost in EAE mice (Figure 3A), indicating that the induction of EAE indeed resulted in marked demyelination. Notably, the percentage of demyelinated areas in the NHNK group was significantly decreased compared to that in the NSE group (Figure 3A and B), revealing the protective effect of NHNK against EAE-induced demyelination.
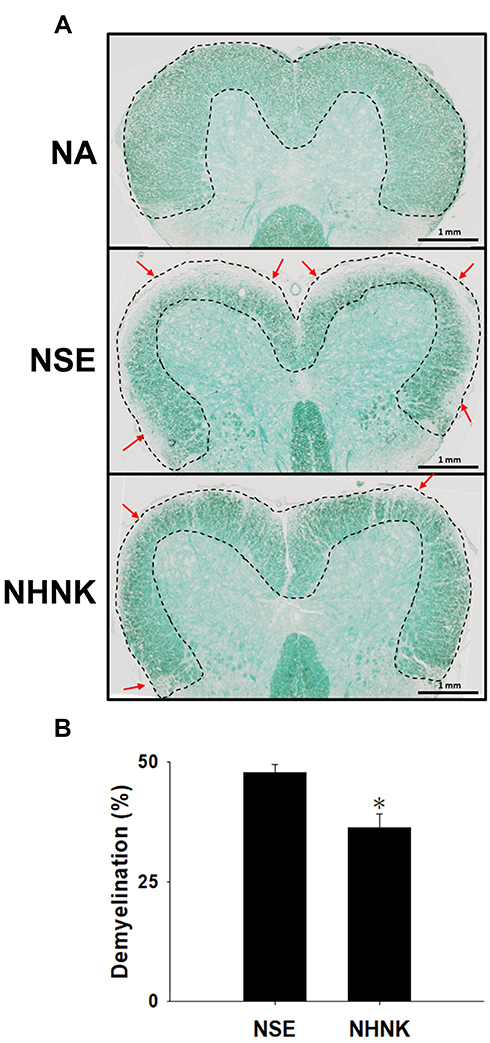 |
Figure 3 NHNK treatment attenuated demyelination in the spinal cords of EAE mice. (A) Representative tissue sections stained with LFB are shown. Areas marked by the dashed line show normal myelinated regions. Red arrows indicate demyelination. (B) The percentage of demyelination was quantified using ImageJ software as described in the Materials and methods. The data are expressed as the mean ± SE of 8−11 samples per group. *p < 0.05 as compared to the NSE group. The results are a representative of three independent experiments. |
NHNK Attenuated EAE-Induced Neuroinflammation
Neuroinflammation is the major pathogenesis causing neuron damage and demyelination in EAE mice.1 Therefore, we further examined the effects of NHNK on EAE-induced inflammation in the spinal cord. An obvious infiltration of inflammatory cells was observed in the myelin region of spinal cords harvested from EAE mice (Figure 4A). Notably, NHNK treatment reduced the number of infiltrated cells and the level of vacuolization (Figure 4A). Concordantly, the pathological score of the NHNK group was lower than that of the NSE group (Figure 4B), indicating the ameliorative effect of NHNK on neuroinflammation and neuronal damage.
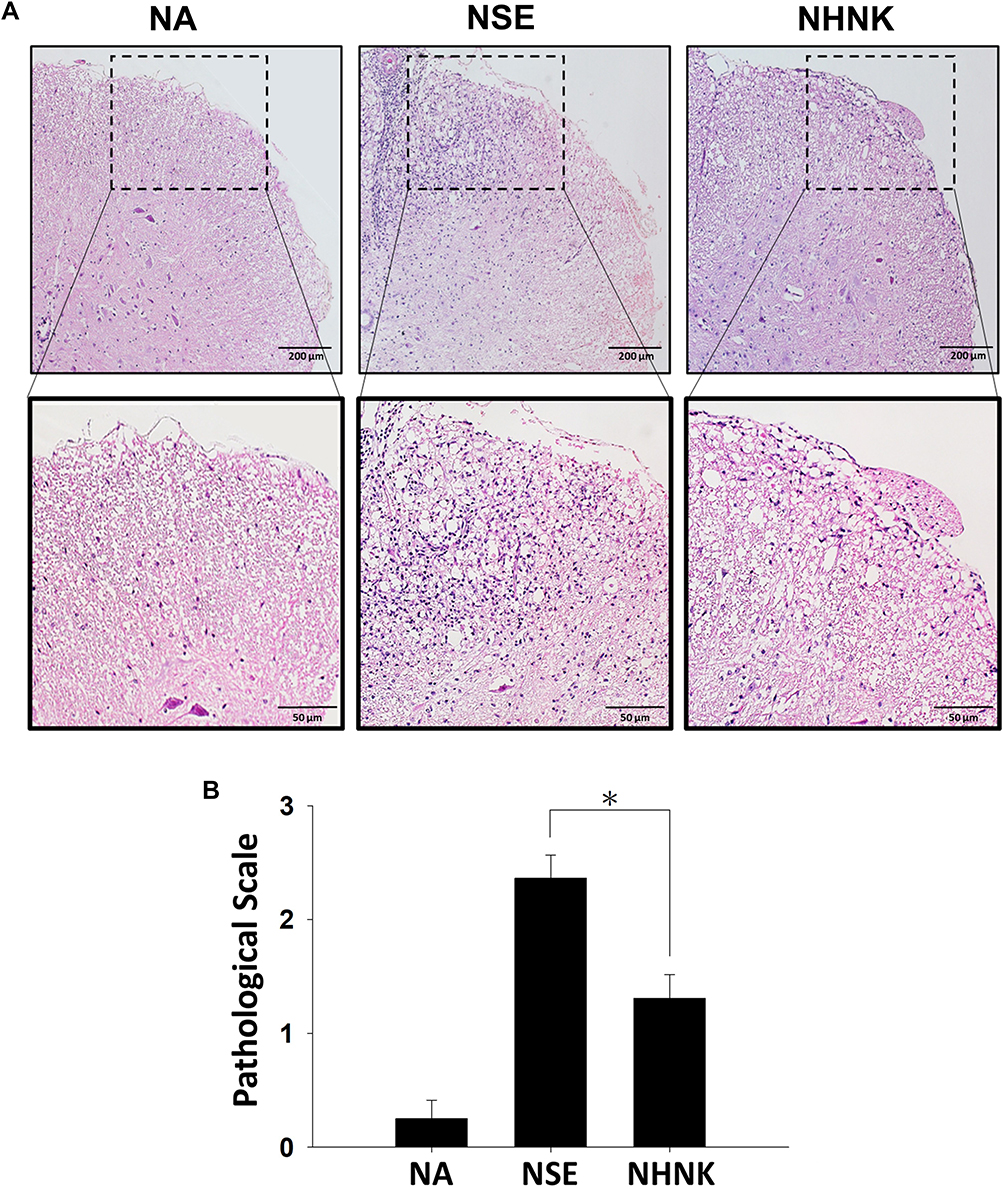 |
Figure 4 NHNK treatment reduced the infiltration of inflammatory cells into the spinal cords of EAE mice. (A) Representative tissue sections stained with H & E are shown. The lower panels are enlarged images of the area of dashed boxes. (B) Pathological scale of inflammation was assessed as described in the Materials and methods. The scores were expressed as the mean ± SE of 8−11 samples per group. *p < 0.05 as compared to the NSE group. The results are a representative of three independent experiments. |
NHNK Attenuated the Infiltration of Activated Microglial into the Spinal Cord of EAE Mice
Microglial activation is the major pathogenic contribution in the development of EAE through the release of pro-inflammatory cytokines.39 Hence, we examined the activation of microglia in the spinal cord using IHC staining. As shown in Table 2, a notable infiltration of IL-6+, TNF+, and Iba-1+ cells was observed in the spinal cords harvested from EAE mice, which was significantly reduced by the NHNK treatment. As Iba-1 is an activation marker for microglia, we next investigated whether the pro-inflammatory cytokines were expressed by activated microglia using IHC double staining. The infiltration of Iba-1+TNF+ cells was observed in the spinal cords harvested from EAE mice (Figure 5A and B), whereas NHNK treatment diminished the number of Iba-1+TNF+ cells (Figure 5A and B). These results indicate that NHNK treatment suppressed the infiltration of activated microglial cells, which are capable of expressing pro-inflammatory cytokines, into the CNS of EAE mice.
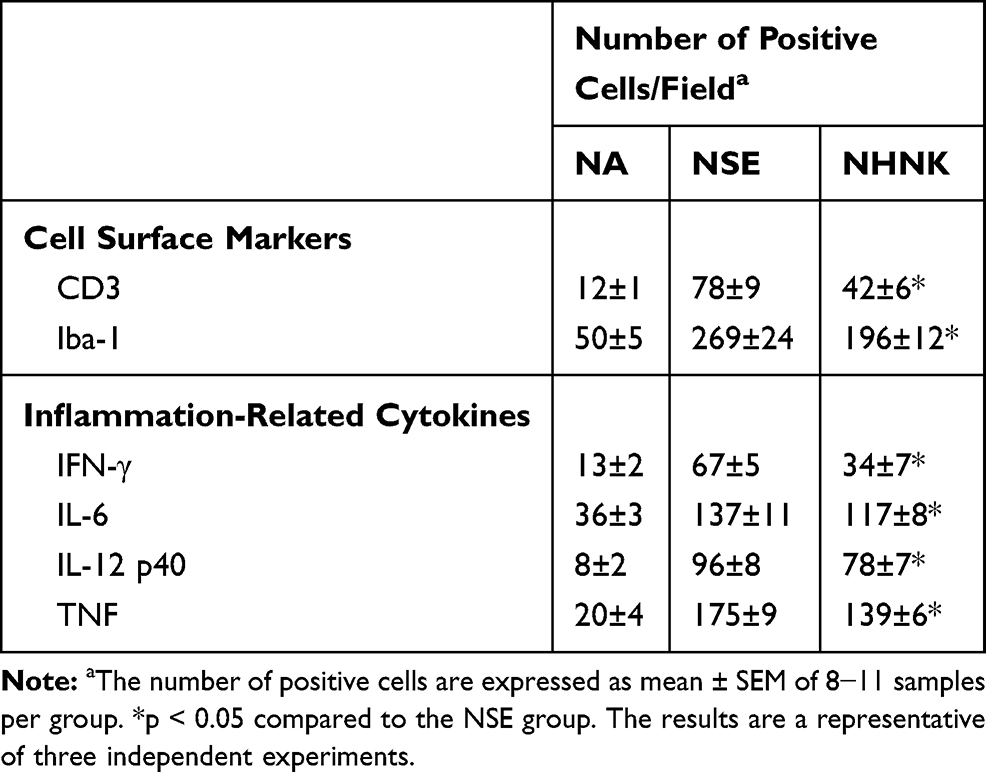 |
Table 2 Immunohistochemical Staining of CD3, Iba-1 and Inflammation-Related Cytokines in the Spinal Cord of EAE Mice |
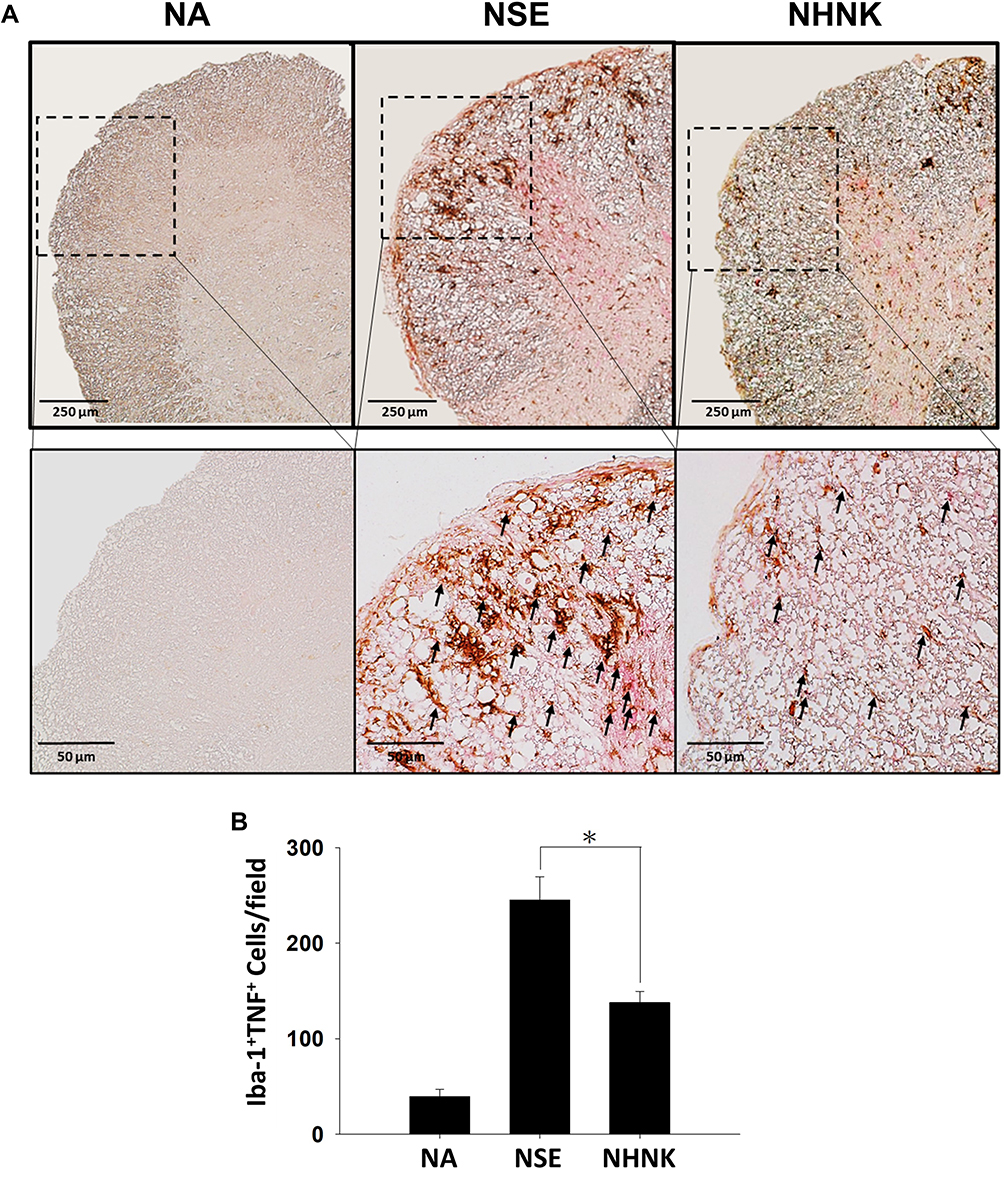 |
Figure 5 NHNK treatment decreased the infiltration of Iba-1+TNF+ cells into the spinal cords of EAE mice. (A) Representative tissue sections double stained for Iba-1 (brown) and TNF (red) are shown. The lower panels are enlarged images of the area of dashed boxes. Arrows indicate Iba-1+TNF+ cells. (B) The number of double-positive cells are expressed as the mean ± SE of 8−11 samples per group. *p < 0.05 as compared to the NSE group. The results are a representative of three independent experiments. |
NHNK Alleviated the Infiltration of Th1 Cells into the Spinal Cord of EAE Mice
As the proliferation of Th1 cells aggravates autoimmune reactions and neurological alterations associated with EAE, we next explored the influence of NHNK on T cell responses in EAE mice.40 First, the expression of IL-12 p40, which has been well known to promote the differentiation of Th1 cells,41 was measured by IHC staining. As shown in Figure 6 and Table 2, a remarkable infiltration of IL-12 p40+ and Iba-1+IL-12 p40+ cells was observed in the spinal cords harvested from EAE mice which was significantly reduced by the NHNK treatment (Figure 6 and Table 2), suggesting that NHNK may suppress the infiltration of microglia with the activity to intensify Th1 immune responses. To test our hypothesis, the infiltration of CD3+ T cells and CD3+IFN-γ+ functional Th1 cells into the spinal cord was investigated. As shown in Figure 7 and Table 2, the number of CD3+ and CD3+IFN-γ+ cells was obviously increased in the spinal cords harvested from EAE mice compared to that from the NA mice, whereas NHNK treatment markedly decreased the number of CD3+ and CD3+IFN-γ+ cells (Figure 7 and Table 2). These results showed that NHNK treatment could attenuate microglia-induced Th1 dominant neuroinflammation, providing a potential immunological mechanism for the therapeutic effect of NHNK on EAE.
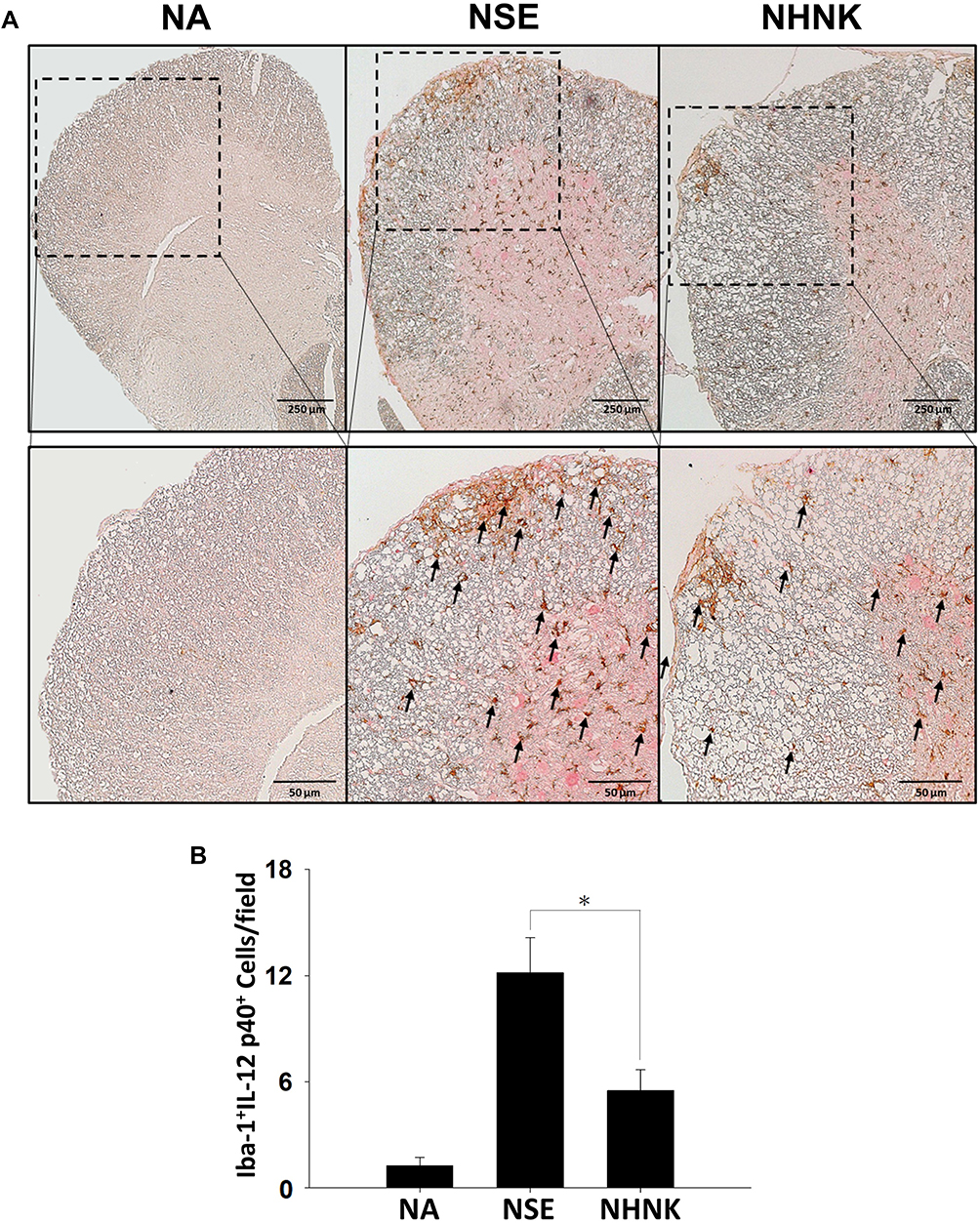 |
Figure 6 NHNK treatment limited the infiltration of Iba-1+IL-12 p40+ cells into the spinal cord of EAE mice. (A) Representative tissue sections double stained for Iba-1 (brown) and IL-12 p40 (red) are shown. The lower panels are enlarged images of the area of dashed boxes. Arrows indicate Iba-1+IL-12 p40+ cells. (B) The number of double-positive cells are expressed as the mean ± SE of 8−11 samples per group. *p < 0.05 as compared to the NSE group. The results are a representative of three independent experiments. |
 |
Figure 7 NHNK treatment alleviated the infiltration of CD3+IFN-γ+ cells in the spinal cord of EAE mice. (A) Representative tissue sections double stained for CD3 (brown) and IFN-γ (red) are shown. The lower panels are enlarged images of the area of dashed boxes. Arrows indicate CD3+IFN-γ+ cells. (B) The number of double-positive cells are expressed as the mean ± SE of 8−11 samples per group. *p < 0.05 as compared to the NSE group. The results are a representative of three independent experiments. |
Due to the low aqueous solubility, limited information pertaining to the anti-inflammatory effect of honokiol via intravenous administration is available. To date, intraperitoneal injection is the most common route to evaluate the effect of honokiol in animal models. For example, Liu et al reported that honokiol attenuated neuroinflammation in a rat model of spinal cord injury. After the operation of spinal cord injury, rats received intraperitoneal injection of honokiol (20 mg/kg) showed a reduction of pro-inflammatory cytokine production, microglial activation, and inflammatory cell infiltration in the spinal cord.15 Recently, Chen et al also reported the therapeutic effects of honokiol on motor impairment in hemiparkinsonian mice. Intraperitoneal injection with honokiol (0.1–5 mg/kg/day) for 7 days improved motor functions and reversed neurodegeneration.22 In the present study, EAE mice intravenously administered with NHNK (with a realistic dose of honokiol as 11.6 mg/kg) showed milder clinical signs and attenuation of neuroinflammation. These results demonstrated that our nanonized formulation could exert the anti-inflammatory effect of honokiol administered by a more clinical relevant dosing regimen, the intravenous route, suggesting NHNK is a promising formulation for intravenous therapy against EAE. In the same EAE model, fingolimod and mitoxantrone are commonly used as positive-control drugs to evaluate the efficacy of drug candidates. Daily treatment with fingolimod (0.1 mg/kg) by gavage alleviated mean clinical score and pathological score by about 20%.42 In the study of Alves et al, intraperitoneal injection with mitoxantrone (1 mg/kg/day) for 7 days decreased clinical score by 50–60% and reduced the production of IFN-γ and IL-12 approximately by 25% and 60%, respectively.43 In the present study, intravenous injection with NHNK (twice a week for 3 weeks) declined mean clinical score by 20% and reduced the production of IFN-γ and IL-12 by 50% and 20%, respectively. On the basis of these results, we speculate that the efficacy of intravenous NHNK therapy against EAE may be comparable to that of fingolimod and mitoxantrone. Further studies are warranted to more comprehensive compare the efficacy and dosimetry of these different agents.
Conclusion
To the best of our knowledge, nanosomes were employed at the first time for the encapsulation of honokiol, a hydrophobic phytochemical with neuroprotective and anti-inflammatory activities, to improve its feasibility for intravenous administration. The structure of nanosomes is composed of phosphatidylcholine and cholesterol, which is similar to that of the human cellular membrane, allowing it easily transported into cell and increasing its resistance to serum.44,45 However, organic solvents are often employed in traditional manufacturing processes of liposomes to solubilize either the lipids or the drugs. The manufacturing process of liposomes into nanosomes is time-consuming, tedious, extremely delicate, and mostly done in small batches. Accordingly, a gap between research and clinical application still exists due to the concerned adverse effects from residual solvents and the manufacturing practicality. As shown in Scheme 1, a UHPH method was employed in this study to simplify the manufacturing process of NHNK with the properties of uniformly distributed nanosize and high encapsulation efficiency. Intravenous administration of NHNK ameliorated the clinical and histopathological signs of EAE by suppressing neuroinflammation and the subsequent demyelination. Additionally, an inhibitory effect of NHNK on the infiltration of activated microglia and Th1 cells into the spinal cord was elucidated. Collectively, these results demonstrated that NHNK could target the immunopathology of EAE in the CNS via intravenous injection, offering insight into the design and development of nanosome-based therapeutic agents for inflammatory CNS diseases.
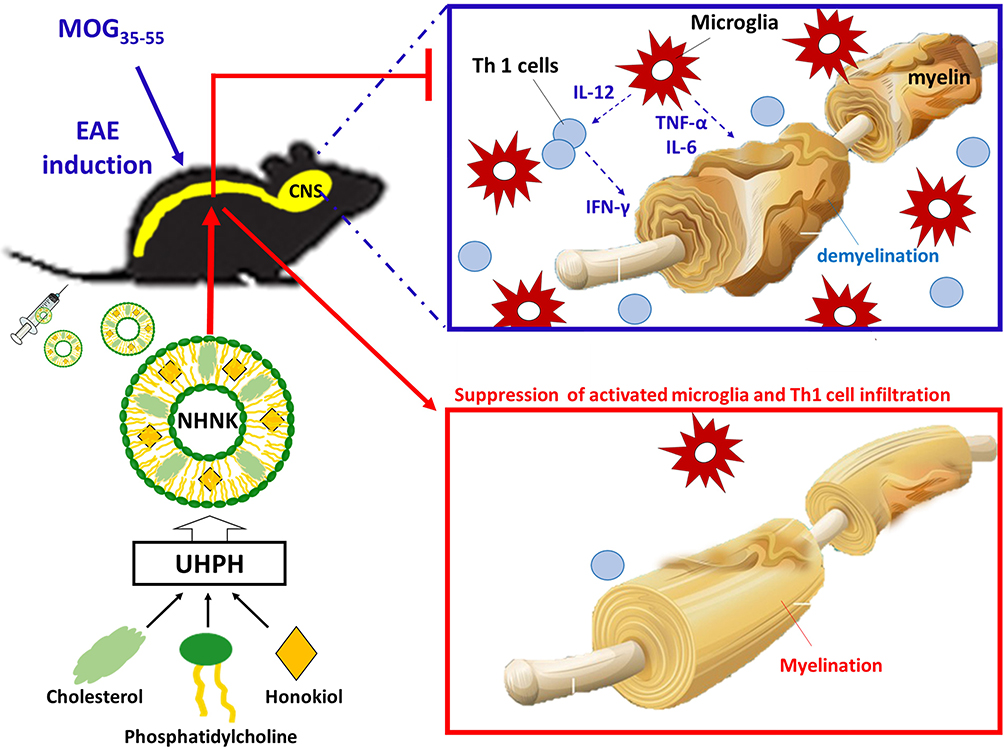 |
Scheme 1 Ultra-high pressure homogenization (UHPH) was employed to formulate nanosome-encapsulated honokiol (NHNK) to improve its feasibility for intravenous therapy against mouse experimental autoimmune encephalomyelitis. Intravenous administration of NHNK, with the properties of uniformly distributed nanosize and high encapsulation efficiency, ameliorated the severity of EAE accompanied by a significant reduction of demyelination and neuroinflammation. Mechanistic investigation revealed that the unique physicochemical properties of NHNK allow it to target activated microglia and T cells in the spinal cord, offering insight into the design and development of nanosome-based therapeutic agents for inflammatory CNS diseases. |
Acknowledgement
This work was supported by grant 107-2320-B-002-020, 107-2320-B-019-003, and 108-2320-B-019-006 from the Ministry of Science and Technology, Executive Yuan, Taiwan.
Disclosure
The authors report no conflicts of interests in this work.
References
1. Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079–1106.
2. Pyka-Fosciak G, Stasiolek M, Litwin JA. Immunohistochemical analysis of spinal cord components in mouse model of experimental autoimmune encephalomyelitis. Folia Histochem Cytobiol. 2018;56(3):151–158. doi:10.5603/FHC.a2018.0018
3. Gao Z, Tsirka SE. Animal models of MS reveal multiple roles of microglia in disease pathogenesis. Neurol Res Int. 2011;2011:383087. doi:10.1155/2011/383087
4. Herrero-Herranz E, Pardo LA, Gold R, Linker RA. Pattern of axonal injury in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neurobiol Dis. 2008;30(2):162–173. doi:10.1016/j.nbd.2008.01.001
5. Na KS, Jung HY, Kim YK. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2014;48:277–286. doi:10.1016/j.pnpbp.2012.10.022
6. Sedgwick JD, Ford AL, Foulcher E, Airriess R. Central nervous system microglial cell activation and proliferation follows direct interaction with tissue-infiltrating T cell blasts. J Immunol. 1998;160(11):5320–5330.
7. Becher B, Dodelet V, Fedorowicz V, Antel JP. Soluble tumor necrosis factor receptor inhibits interleukin 12 production by stimulated human adult microglial cells in vitro. J Clin Invest. 1996;98(7):1539–1543. doi:10.1172/JCI118946
8. Becher B, Antel JP. Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia. 1996;18(1):1–10. doi:10.1002/(ISSN)1098-1136
9. Becher B, Blain M, Antel JP. CD40 engagement stimulates IL-12 p70 production by human microglial cells: basis for Th1 polarization in the CNS. J Neuroimmunol. 2000;102(1):44–50. doi:10.1016/S0165-5728(99)00152-6
10. Feinstein A, Freeman J, Lo AC. Treatment of progressive multiple sclerosis: what works, what does not, and what is needed. Lancet Neurol. 2015;14(2):194–207. doi:10.1016/S1474-4422(14)70231-5
11. Jones DE. Early relapsing multiple sclerosis. Continuum (Minneap Minn). 2016;22(3):744–760. doi:10.1212/CON.0000000000000329
12. Talarek S, Listos J, Barreca D, et al. Neuroprotective effects of honokiol: from chemistry to medicine. Biofactors. 2017. doi:10.1002/biof.1385
13. Sulakhiya K, Kumar P, Gurjar SS, Barua CC, Hazarika NK. Beneficial effect of honokiol on lipopolysaccharide induced anxiety-like behavior and liver damage in mice. Pharmacol Biochem Behav. 2015;132:79–87. doi:10.1016/j.pbb.2015.02.015
14. Khalid S, Ullah MZ, Khan AU, et al. Antihyperalgesic properties of honokiol in inflammatory pain models by targeting of NF-kappaB and Nrf2 signaling. Front Pharmacol. 2018;9:140. doi:10.3389/fphar.2018.00140
15. Liu J, Zhang C, Liu Z, Zhang J, Xiang Z, Sun T. Honokiol downregulates Kruppel-like factor 4 expression, attenuates inflammation, and reduces histopathology after spinal cord injury in rats. Spine (Phila Pa 1976). 2015;40(6):363–368. doi:10.1097/BRS.0000000000000758
16. Yu Y, Li M, Su N, et al. Honokiol protects against renal ischemia/reperfusion injury via the suppression of oxidative stress, iNOS, inflammation and STAT3 in rats. Mol Med Rep. 2016;13(2):1353–1360. doi:10.3892/mmr.2015.4660
17. Costa A, Facchini G, Pinheiro A, et al. Honokiol protects skin cells against inflammation, collagenolysis, apoptosis, and senescence caused by cigarette smoke damage. Int J Dermatol. 2017;56(7):754–761. doi:10.1111/ijd.2017.56.issue-7
18. Chen HH, Chang PC, Chen C, Chan MH. Protective and therapeutic activity of honokiol in reversing motor deficits and neuronal degeneration in the mouse model of Parkinson’s disease. Pharmacol Rep. 2018;70(4):668–676. doi:10.1016/j.pharep.2018.01.003
19. Munroe ME, Businga TR, Kline JN, Bishop GA. Anti-inflammatory effects of the neurotransmitter agonist Honokiol in a mouse model of allergic asthma. J Immunol. 2010;185(9):5586–5597. doi:10.4049/jimmunol.1000630
20. Wang XD, Wang YL, Gao WF. Honokiol possesses potential anti-inflammatory effects on rheumatoid arthritis and GM-CSF can be a target for its treatment. Int J Clin Exp Pathol. 2015;8(7):7929–7936.
21. Mihara T, Mikawa S, Kaji N, et al. Therapeutic action of honokiol on postoperative ileus via downregulation of iNOS gene expression. Inflammation. 2017;40(4):1331–1341. doi:10.1007/s10753-017-0576-7
22. Chen HH, Chang PC, Wey SP, Chen PM, Chen C, Chan MH. Therapeutic effects of honokiol on motor impairment in hemiparkinsonian mice are associated with reversing neurodegeneration and targeting PPARgamma regulation. Biomed Pharmacother. 2018;108:254–262. doi:10.1016/j.biopha.2018.07.095
23. Rickert U, Cossais F, Heimke M, et al. Anti-inflammatory properties of Honokiol in activated primary microglia and astrocytes. J Neuroimmunol. 2018;323:78–86. doi:10.1016/j.jneuroim.2018.07.013
24. Teixeira MC, Carbone C, Souto EB. Beyond liposomes: recent advances on lipid based nanostructures for poorly soluble/poorly permeable drug delivery. Prog Lipid Res. 2017;68:1–11. doi:10.1016/j.plipres.2017.07.001
25. Abu Lila AS, Ishida T. Liposomal delivery systems: design optimization and current applications. Biol Pharm Bull. 2017;40(1):1–10. doi:10.1248/bpb.b16-00624
26. Bozzuto G, Molinari A. Liposomes as nanomedical devices. Int J Nanomedicine. 2015;10:975–999. doi:10.2147/IJN
27. Herringson TP, Altin JG. Effective tumor targeting and enhanced anti-tumor effect of liposomes engrafted with peptides specific for tumor lymphatics and vasculature. Int J Pharm. 2011;411(1–2):206–214. doi:10.1016/j.ijpharm.2011.03.044
28. Rip J. Liposome technologies and drug delivery to the CNS. Drug Discov Today Technol. 2016;20:53–58. doi:10.1016/j.ddtec.2016.07.005
29. Agrawal M, Ajazuddin, Tripathi DK, et al. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J Control Release. 2017;260:61–77. doi:10.1016/j.jconrel.2017.05.019
30. Alavi M, Karimi N, Safaei M. Application of various types of liposomes in drug delivery systems. Adv Pharm Bull. 2017;7(1):3–9. doi:10.15171/apb.2017.002
31. Kapoor M, Lee SL, Tyner KM. Liposomal drug product development and quality: current US experience and perspective. Aaps J. 2017;19(3):632–641. doi:10.1208/s12248-017-0049-9
32. Parker SE, Davey PG. Pharmacoeconomics of intravenous drug administration. Pharmacoeconomics. 1992;1(2):103–115. doi:10.2165/00019053-199201020-00007
33. Auerbach M, Macdougall I. The available intravenous iron formulations: history, efficacy, and toxicology. Hemodial Int. 2017;21(Suppl 1):S83–S92. doi:10.1111/hdi.12560
34. Wu XJ, Zhang J, Guo BN, et al. Pharmacokinetics and pharmacodynamics of multiple-dose intravenous nemonoxacin in healthy Chinese volunteers. Antimicrob Agents Chemother. 2015;59(3):1446–1454. doi:10.1128/AAC.04039-14
35. Liu D, Geng T, Wang Y, Ding L. Pharmacokinetic profile of cefbuperazone in healthy Chinese volunteers after single and multiple drip intravenous infusion by HPLC-MS/MS. J Pharm Biomed Anal. 2016;129:28–33. doi:10.1016/j.jpba.2016.06.029
36. Ong SG, Ming LC, Lee KS, Yuen KH. Influence of the encapsulation efficiency and size of liposome on the oral bioavailability of griseofulvin-loaded liposomes. Pharmaceutics. 2016;8(3). doi:10.3390/pharmaceutics8030025
37. Wang D, Dong X, Wang C. Honokiol ameliorates amyloidosis and neuroinflammation and improves cognitive impairment in Alzheimer’s disease transgenic mice. J Pharmacol Exp Ther. 2018;366(3):470–478. doi:10.1124/jpet.118.248674
38. Horstmann L, Schmid H, Heinen AP, Kurschus FC, Dick HB, Joachim SC. Inflammatory demyelination induces glia alterations and ganglion cell loss in the retina of an experimental autoimmune encephalomyelitis model. J Neuroinflammation. 2013;10:120. doi:10.1186/1742-2094-10-120
39. Mancini A, Tantucci M, Mazzocchetti P, et al. Microglial activation and the nitric oxide/cGMP/PKG pathway underlie enhanced neuronal vulnerability to mitochondrial dysfunction in experimental multiple sclerosis. Neurobiol Dis. 2018;113:97–108. doi:10.1016/j.nbd.2018.01.002
40. Domingues HS, Mues M, Lassmann H, Wekerle H, Krishnamoorthy G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS One. 2010;5(11):e15531. doi:10.1371/journal.pone.0015531
41. Lim HX, Hong HJ, Jung MY, Cho D, Kim TS. Principal role of IL-12p40 in the decreased Th1 and Th17 responses driven by dendritic cells of mice lacking IL-12 and IL-18. Cytokine. 2013;63(2):179–186. doi:10.1016/j.cyto.2013.04.029
42. Hou H, Cao R, Quan M, et al. Rapamycin and fingolimod modulate Treg/Th17 cells in experimental autoimmune encephalomyelitis by regulating the Akt-mTOR and MAPK/ERK pathways. J Neuroimmunol. 2018;324:26–34. doi:10.1016/j.jneuroim.2018.08.012
43. Alves CC, Castro SB, Costa CF, et al. Anthraquinone derivative O, O’-bis-(3ʹ-iodopropyl)-1,4-dihydroxyanthraquinone modulates immune response and improves experimental autoimmune encephalomyelitis. Int Immunopharmacol. 2012;14(2):127–132. doi:10.1016/j.intimp.2012.06.013
44. Bitounis D, Fanciullino R, Iliadis A, Ciccolini J. Optimizing druggability through liposomal formulations: new approaches to an old concept. ISRN Pharm. 2012;2012:738432.
45. Hosta-Rigau L, Zhang Y, Teo BM, Postma A, Stadler B. Cholesterol–a biological compound as a building block in bionanotechnology. Nanoscale. 2013;5(1):89–109. doi:10.1039/C2NR32923A
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.