")
Back to Journals » Clinical Ophthalmology » Volume 10
Development of lifitegrast: a novel T-cell inhibitor for the treatment of dry eye disease
Received 14 April 2016
Accepted for publication 12 May 2016
Published 10 June 2016 Volume 2016:10 Pages 1083—1094
DOI https://doi.org/10.2147/OPTH.S110557
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Charles P Semba,1 Thomas R Gadek2
1Vascular and Interventional Radiology, Stanford University School of Medicine, Stanford, CA, USA; 2Ophthalma Logic Consulting, Park City, UT, USA
Abstract: Dry eye disease (DED) is a multifactorial disorder of the ocular surface characterized by symptoms of discomfort, decreased tear quality, and chronic inflammation that affects an estimated 20 million patients in the US alone. DED is associated with localized inflammation of the ocular surface and periocular tissues leading to homing and activation of T cells, cytokine release, and development of hyperosmolar tears. This inflammatory milieu results in symptoms of eye dryness and discomfort. Homing of T cells to the ocular surface is influenced by the binding of lymphocyte function-associated antigen-1 (LFA-1; CD11a/CD18; αLβ2), a cell surface adhesion protein, to its cognate ligand, intercellular adhesion molecule-1 (ICAM-1; CD54), which is expressed on inflamed ocular/periocular epithelium and vascular endothelium. LFA-1/ICAM-1 binding within the immunologic synapse enables both T-cell activation and cytokine release. Lifitegrast is a novel T-cell integrin antagonist that is designed to mimic the binding epitope of ICAM-1. It serves as a molecular decoy to block the binding of LFA-1/ICAM-1 and inhibits the downstream inflammatory process. In vitro studies have demonstrated that lifitegrast inhibits T-cell adhesion to ICAM-1-expressing cells and inhibits secretion of pro-inflammatory cytokines including interferon gamma, tumor necrosis factor alpha, macrophage inflammatory protein 1 alpha, interleukin (IL)-1α, IL-1β, IL-2, IL-4, and IL-6, all of which are known to be associated with DED. Lifitegrast has the potential to be the first pharmaceutical product approved in the US indicated for the treatment of both symptoms and signs of DED. Clinical trials involving over 2,500 adult DED patients have demonstrated that topically administered lifitegrast 5.0% ophthalmic solution can rapidly reduce the symptoms of eye dryness and decrease ocular surface staining with an acceptable long-term safety profile. The purpose of this review is to highlight the developmental story – from bench top to bedside – behind the scientific rationale, engineering, and clinical experience of lifitegrast for the treatment of DED.
Keywords: LFA-1, ICAM-1, ocular surface disease, inflammation, drug development
Introduction
Why develop a novel pharmaceutical agent for dry eye disease (DED)? In the annals of ophthalmic drug discovery and development, dry eye has a vast history of clinical trial failures leading to collective disappointment for patients and clinicians who are seeking new therapeutic options. The general approach for managing DED has not changed dramatically over the past 50 years; lubricating artificial tears and punctal plugs represent the mainstay of therapy to alleviate disease symptoms and enhance ocular surface tear film volume. For patients, the symptoms of chronic ocular discomfort, dryness, and irritation are associated with significant impairment in their visual-related quality of life.1 For eye care specialists, DED remains one of the most common reasons for patient visits and the burden is increasing as the population ages.2 Attempts to advance new drug treatments to the marketplace have not been without significant effort. Of the nearly 30 programs that have endeavored to develop a pharmaceutical agent for DED (and estimated billions of dollars invested in research and development), only cyclosporine-A (CsA) 0.05% ophthalmic emulsion (Restasis®; Allergan, Inc., Irvine, CA, USA) has been approved by the US Food and Drug Administration (FDA).3 However, cyclosporine emulsion has its limitations in the treatment of dry eye. It is indicated solely to increase tear production and not for the treatment of the oft-disabling symptoms associated with DED. It has a long onset of action (eg, up to 6 months) and has failed to show a significant dose response; many patients discontinue its use due to the burning sensation associated with administration.4,5 Even with the availability of cyclosporine, the overwhelming majority of surveyed ophthalmologists (94%) desire additional treatment options.6 Thus, a large unmet need remains for newer agents that can rapidly diminish the symptoms of disease, have a rapid onset of action, protect the ocular surface, and are well tolerated.
In February 2016, a New Drug Application (NDA) was received by the FDA for a novel small molecule T-cell inhibitor – lifitegrast – for the treatment of DED, and a regulatory decision to approve the NDA is pending for July 2016.7 If approved, lifitegrast has the potential to be the first pharmaceutical therapy approved in the US indicated for the treatment of both signs and symptoms of DED with a rapid onset of action in as little as 2 weeks, a statistically significant dose response, and appears safe and well tolerated.8–10 In this review, the scientific rationale and approach toward developing lifitegrast in a notoriously difficult developmental arena will be described. Just as the development program for CsA 0.05% helped set the clinical and regulatory precedent for a US dry eye pharmaceutical approval, it is our hope that this summary will provide learning for future programs in creating the next generation of topically administered ophthalmic agents in managing the complexities of ocular surface disease.
DED: a disease state primer
Extensive literature exists on the epidemiology, diagnosis, and treatment of DED; therefore, only a fundamental primer is provided here from the drug development perspective. As scientific interest in ocular surface disease increased in the early 1990s, there was growing awareness by the medical, scientific, and regulatory communities that standardized definitions and clinical methodology were needed in order to foster innovation for industry partners. The National Eye Institute/Industry Workshop on Clinical Trials in Dry Eyes formed the initial basis of establishing a coordinated framework on the approach toward DED.11 The Tear Film and Ocular Surface Society (TFOS) sponsored subsequent evidence-based critical assessments on the definition, classification, diagnosis, and treatment of ocular surface disease resulting in two landmark publications: the 2007 International Dry Eye Workshop (DEWS) and the 2011 International Workshop on Meibomian Gland Dysfunction (MGD).2,12 A second international TFOS DEWS is planned for September 2016 and reflects the dramatic increase in the knowledge and progress in the clinical understanding of DED over the past decade, particularly advancing dry eye from a syndrome to a disease. It is the DEWS report along with leading clinical experts in the field that helped provide the guiding foundation for the development of lifitegrast.
Definition
DED, as defined by DEWS, is a multifactorial disease of the tears and ocular surface that results in symptoms of discomfort, visual disturbance, and tear film instability. It is accompanied by increased osmolarity of the tear film and inflammation of the ocular surface.2 Epidemiologic evidence shows that the disease is common and increasing in prevalence. In the US, the Women’s Health Study and Physicians’ Health Study estimates that DED affects 7.8% of women and 4.3% of men over the age of 50 years, with increasing prevalence with age.13,14 DED of any severity has been estimated to be as high as 20 million in the US alone.15
Risk factors
DED is associated with female sex, advancing age, various environmental factors (eg, low humidity conditions, office environments), hormonal imbalance (eg, menopause), lid margin disorders (eg, ptosis, droopy lids, ectropion), certain medications, ocular allergies, autoimmune diseases (eg, Sjögren’s disease), LASIK surgery, contact lenses, and use of computer displays (decreased blink rate due to gazing).2
Diagnosis
There is no single “gold standard” diagnostic test for DED, and the clinician must evaluate the patient’s symptomatic history, risk factors, and focused physical exam to make the diagnosis based on the collective evidence. In attempts to codify an approach toward diagnosis, the DEWS report classified DED into two general pathoetiologic groups: aqueous-deficient and evaporative tear-loss dry eye.2 Aqueous-deficient dry eye is generally characterized as insufficient lacrimal tear secretion and tear volume while evaporative tear-loss dry eye is characterized by excessive water loss from the exposed ocular surface in presence of normal lacrimal function due to diminished protection from the outer lipid layer of the tear film. Despite the proposed classifications, in clinical practice the patient presentation is often mixed with features of both aqueous-deficient and evaporative tear-loss components.
Subjective evaluation (symptoms)
Symptoms of ocular discomfort remain one of the leading complaints to eye care practitioners in patients with DED.16 The range of reported perceptions of symptoms is highly variable from patient-to-patient and includes sensations of dryness, discomfort, irritation, burning/stinging, foreign body sensation, eye pain, and/or grittiness. Quantifying the magnitude of ocular symptoms for the purposes of measuring outcomes for investigational drugs or devices has been difficult because of the waxing and waning course of DED, the nonspecific nature of patient-reported symptoms (my eyes feel uncomfortable), and the lack of uniformity of how patients perceive and report their symptoms – one patient’s “burning” may be another patient’s “grittiness” sensation. Furthermore, patients may describe their worse symptom as “foreign body sensation” on one day and “dryness” on another day. Several psychometric inventories have been proposed to help quantify and characterize the subjective component of DED (eg, Ocular Surface Disease Index [OSDI], Symptom Assessment in Dry Eye [SANDE] questionnaire, Visual Function Questionnaire-25 [VFQ-25]), but there remains no consensus on a standardized tool.17–19 These inventories remain largely reserved for academic research or drug trials rather than the harried pace of daily routine ophthalmic practice.
Objective evaluation (signs)
Slit-lamp evaluation for DED often consists of evaluating one or more of the following: measuring the presence and severity of epithelial damage to the cornea/conjunctiva based on staining with vital dyes (eg, fluorescein, rose bengal, lissamine green), determining tear production (eg, Schirmer tear test evaluation), assessing the degree of conjunctival hyperemia, and/or measuring tear-film breakup time. Even with these quantitative tests, there remains no consensus on a uniform grading system or scale (eg, Oxford scale, National Eye Institute scale) or standardized methodology on how these individual tests should be conducted.20 In this age of increasing sophistication with digital imaging and medical informatics, the diagnosis of DED remains highly reliant on analog technology that lacks precision and specificity and often times defaults to clinician gestalt.21 The procedures used to evaluate DED have not fundamentally changed over the past century. Vital dyes to evaluate ocular surface epithelial integrity were first reported by Pflüger in 1882 and Schirmer described his method of tear volume assessment in 1903.22 Further confounding the diagnosis is the well-documented observation that signs and symptoms of disease have no strong correlation;23 early stages of DED can manifest simply as symptoms of ocular discomfort and dryness without any supportive objective findings, whereas severe advanced stages of the disease may be associated with afferent nerve fiber damage with minimal reported patient symptoms.24 In attempts to devise more objective, non-operator-dependent tests, progress has been made with use of point-of-care digital tear osmolarity,25 tear film inflammatory biomarker testing,26 digital interferometry,27 and high-resolution optical coherence tomography imaging.28 However, these tests are not pathognomonic of DED and the diagnosis still rests on the experienced clinician’s judgment.
Treatment
Options for treatment include modification of risk factors, artificial tears, lid hygiene procedures, punctal plugs, and pharmaceutical treatment with cyclosporine drops.29 There is no universally accepted regimen and the specific treatment regimen must be tailored to the individual patient’s presentation. Examples of risk factor modifications include periodic breaks during prolonged visual-tasking activities, removal of topically administered medications that contain preservatives such as benzalkonium chloride that damage corneal epithelial cells, and surgical correction of lid margin pathology (eg, blepharoplasty, ectropion repair). Over-the-counter lubricating artificial tears are a common first-line treatment and may contain vasoconstrictive agents (eg, tetrahydrozoline) to reduce the often cosmetically displeasing conjunctival hyperemia associated with DED. Lid hygiene procedures are focused toward patients with meibomitis and/or blepharitis and involve gentle lid scrubs and warm compresses to soften inspissated meibum, concomitant antibiotic therapy, and oral fish oil supplements. Punctal occlusion with short-term collagen or long-term silicone plugs is used to increase thickness of the tear film by blockade of the normal nasolacrimal drainage. Finally, CsA 0.05% ophthalmic emulsion is often added to the treatment regimen to increase tear production.
Novel treatment approaches for DED by drug and device developers have commonly focused on enhancing one of the three key components of the tear film: the inner mucin layer (eg, goblet cell stimulators/secretagogues30), the middle aqueous tear layer (eg, lacrimators to produce more tear volume either using drugs4 or nasal neurostimulation devices31), or the outer lipid layer (eg, enhancement of normal lipid production from the meibomian glands32). It was our observation that, regardless of the etiology of DED, a common pathophysiologic condition was establishment of ocular surface inflammation and that a core element of the treatment paradigm should focus on addressing the immunologic basis of disease.
Immunologic basis of DED
Despite the multifactorial nature and complex presentation of dry eye, the development of chronic inflammation on the ocular surface and periocular tissues appears to be a central pathoetiologic theme and is characterized by the activation and migration of T lymphocytes to the inflamed tissues.33 It remains unclear as to whether chronic inflammation leads to the clinical manifestations of DED or, conversely, if DED leads to chronic inflammation. Regardless, several lines of evidence support the major role of T-cell-mediated inflammation in DED. Lymphocytic infiltration has been observed in the conjunctiva and lacrimal glands in animal models of Sjögren’s disease,34 canine dry eye,35 and conjunctival epithelium of dry eye patients.36,37 Elevated levels of inflammatory cytokines expressed by T lymphocytes have been profiled in the tear film of patients with aqueous-deficient38 or evaporative-loss DED.39
Activation and homing of lymphocytes to the site of inflammation are influenced by the expression of two cell surface proteins, lymphocyte function-associated antigen-1 (LFA-1; CD11a/CD18; αLβ2) and intercellular adhesion molecule-1 (ICAM-1; CD54) (Figure 1).40 LFA-1 is a 275 kD molecular weight (MW) heterodimeric protein of the integrin family bound to the membrane surface of CD4 lymphocytes (T cells).41 ICAM-1 is the cognate ligand to LFA-1 and is a 95 kD transmembrane protein found on the surface of inflamed endothelial and epithelial cells as well as immune function cells including T cells, B cells, and antigen-presenting cells (APCs).42 The LFA-1/ICAM-1 interaction is critical to the firm adhesion of T cells to the vascular endothelium of inflamed tissues and influences the diapedesis and migration of these adhered lymphocytes out of the vasculature and directly into the adjacent tissues on the ocular surface.43,44 Once present in tissues, the LFA-1/ICAM-1 interaction allows for the formation of the immunological synapse between T cells and APCs by enabling engagement of the T-cell receptor (TCR) to the major histocompatibility complex (MHC) present in the APC membrane.45 The immunological synapse is comprised of concentric rings of segregated protein rafts with a central core of a cluster of TCR/MHC molecules surrounded by a ring of LFA-1/ICAM-1 molecules.46 Simultaneous engagement of both LFA-1/ICAM-1 and TCR/MHC between the T lymphocytes and APCs leads to amplification of the chronic inflammatory process by stimulating intracellular signals that cause activation and proliferation of T cells, the release of inflammatory cytokines, and the subsequent recruitment of additional T cells at the site of inflammation.47
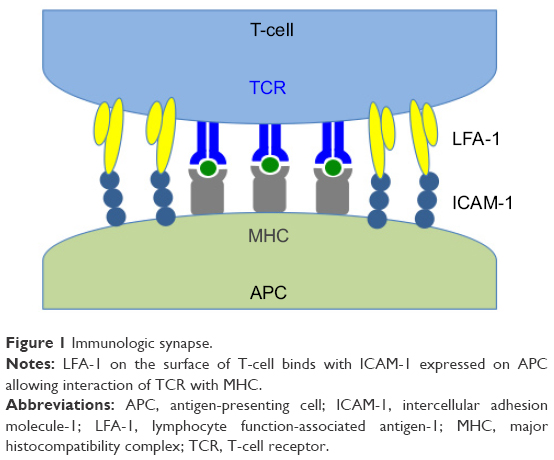 | Figure 1 Immunologic synapse. |
Our approach toward developing lifitegrast for DED was inspired, in large part, by the groundbreaking immunologic research conducted by the Stern Lab at Allergan that culminated in the approval of CsA ophthalmic emulsion.34–36 CsA is an immunosuppressant drug that interferes with the activity of T-cell-mediated immune responses by suppressing calcineurin that normally controls the transcription of interleukin-2 (IL-2).48 IL-2 mediates tolerance and immunity by influencing the production of both T-regulatory cells and effector T cells during homeostasis and activation of the immune system in dry eye. CsA was originally approved as an oral agent that is widely used in organ transplantation to prevent organ rejection. Early clinical trials with CsA ophthalmic emulsion in moderate-to-severe dry eye patients demonstrated a reduction in total number of activated lymphocytes in the conjunctiva accompanied by a significant reduction in LFA-1 expression, suggesting that topical CsA was exerting an immunomodulatory effect by blocking the migration of lymphocytes into the ocular surface.49 Though CsA did not block the LFA-1/ICAM-1 interaction directly, the investigators proposed that indirect immunomodulation of this interaction by CsA helped reduce the overall inflammation in dry eye by reducing the expression of both LFA-1 and ICAM-1 by cells on the ocular surface. This proposal captured our attention toward developing a direct antagonist of the LFA-1/ICAM-1 interaction as a novel pathway for the treatment of dry eye.
Discovery of lifitegrast
Lifitegrast, previously referred to as SAR 1118, is a member of the class of direct competitive LFA-1 antagonists that mimic the binding epitope of LFA-1’s cognate ligand ICAM-1.50,51 As such, lifitegrast is “purpose built” for the treatment of DED to bind LFA-1 on leukocytes and block their binding to ICAM-1 in the adhesion, extravasation, migration, activation, cytokine secretion, and proliferation of these leukocytes in inflammatory diseases.52
At the inception of this program, the prospect for the discovery of a small molecule (MW <1 kD) capable of disrupting the substantially larger LFA-1/ICAM-1 complex (MW ~370 kD) was daunting and controversial.50,53 However, with the expectation that a high-affinity mimic of the ICAM-1 epitope would bind to LFA-1 and outcompete ICAM-1, the quest began.
Lifitegrast was discovered in a rational design process that began with alanine point mutagenesis of the ICAM-1 protein to identify the amino acid side chains that were critical for LFA-1 epitope binding (Figure 2). Six key binding residues from the first immunoglobulin domain of ICAM-1 were identified: glutamic acid 34 (E34), lysine 39 (K39), methionine 64 (M64), tyrosine 66 (Y66), asparagine 68 (N68), and glutamine 73 (Q73).50 Although these residues are not directly linked together in the primary amino acid sequence of ICAM-1, they are presented in a spatially contiguous manner within the folded tertiary structure of native ICAM-1 that falls within the dimensions suitable for a novel small molecule. Using the dimensional coordinates obtained through X-ray crystallography and other structural techniques, work was initiated to create small molecule scaffolds that could mimic aspects of this epitope.
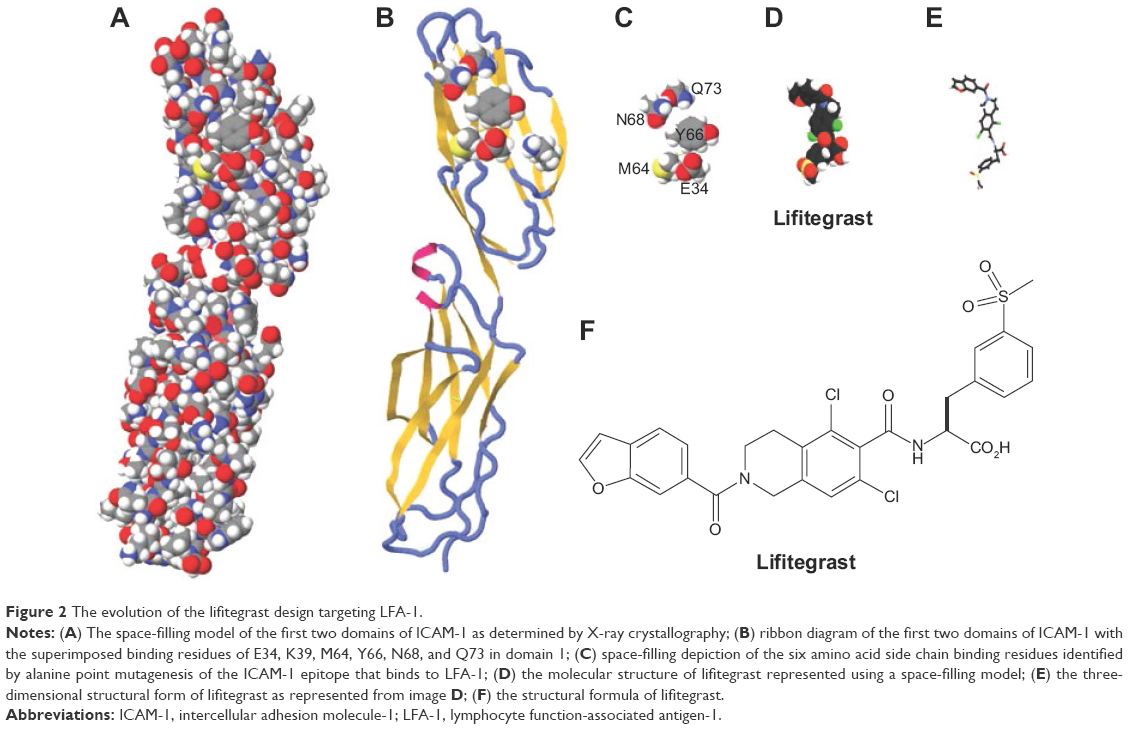 | Figure 2 The evolution of the lifitegrast design targeting LFA-1. |
The E34, K39, M64, Y66, N68, and Q73 amino acid side chains were presumed to be the actual atoms in contact with LFA-1 and were stripped away from the rest of the ICAM-1 structure in silico. An initial active compound mimicking E34 and Y66 was isolated with modest LFA-1/ICAM-1 antagonist activity using relatively simple benzoyl amino acid moieties.54,55 Further enhancements were elaborated off the benzoyl moiety to incorporate mimicry of the N68 and Q73 side chains resulting in a ~30-fold improvement in potency and specificity for LFA-1. Eventually, a small set of non-peptide lead compounds was created that satisfied the criteria for LFA-1 binding, but additional optimization was necessary to ultimately identify a tailor-made molecule that could be a commercially viable pharmaceutical for specifically treating DED.
Based on the philosophy that the properties of a molecule can be seen as the sum of its parts, the optimization process was initiated by dividing the lead compound into five sub-fragments or modules. Each individual module was modified – while holding the remaining modules constant – resulting in the creation of a laborious combinatorial series of over 3,000 analogs.52 These analogs were further assessed based initially on the potency of LFA-1/ICAM-1 inhibition using an enzyme-linked immunosorbent assay followed by secondary testing using ex vivo cellular immune assays to assess potency of human T-cell inhibition (eg, Jurkat cell and mixed lymphocyte reaction assays).50 This exhaustive process led to the identification of SAR 1118 – lifitegrast (MW, 615 g/mol) – a new chemical entity whose three-dimensional structure activity relationship could effectively compete with the key binding residues from ICAM-1 and serve as a novel decoy to block the LFA-1/ICAM-1 interaction. Although lifitegrast was designed as an ICAM-1 mimic, one report has questioned the validity of the design effort already discussed in this document.56 Additional studies have conclusively disproved this assertion and demonstrated that lifitegrast is indeed a mimic of ICAM-1 as designed.50–52,57
Characterization of lifitegrast
The selection process that ultimately resulted in lifitegrast was multifactorial since it had to satisfy not only the onslaught of critical bench-top scientific validations but also meet the more pragmatic aspects of a viable commercial formulation for patients. The desired features of a custom-engineered small molecule we were seeking had to possess the following characteristics: high affinity and specificity for LFA-1; potent inhibition of LFA-1/ICAM-1 binding; high water solubility with sufficient stability to be compatible with the tonicity, osmolarity, and pH necessary for a topical ophthalmic formulation; no systemic or local drug accumulation with rapid systemic clearance; and an efficient manufacturing process.
In vitro assays
Lifitegrast potently inhibits human T-cell binding to human ICAM-1, T-cell activation and cytokine release, and formation of the immunologic synapse.46,58 The concentration of lifitegrast that inhibits 50% binding of T cells (IC50) is 3 nM as assessed with a Jurkat cell assay.58 Lifitegrast inhibits the release of cytokines interferon gamma, tumor necrosis factor alpha, macrophage inflammatory protein 1 alpha, IL-1α, IL-1β, IL-2, IL-4, and IL-6 from activated lymphocytes at levels as low as ~2 nM.58 In vitro cellular imaging studies demonstrated that lifitegrast inhibits formation of the immunologic synapse at concentrations >100 nM.46
In vivo studies
Dose-escalation tolerability studies in dogs demonstrated the safety of lifitegrast solution when dosed topically up to three times a day at a concentration up to 10% solution for 1 month.58 Clinical efficacy of lifitegrast 1.0% solution administered three times a day for 28 days was evaluated in a study of dogs diagnosed with keratoconjunctivitis sicca. At the end of the course of treatment, clinical signs of dry eye were markedly improved and histological evaluation of conjunctival biopsies demonstrated reduction of periocular T-cell inflammation compared to the study baseline samples.58
In rats treated with a single administered drop of 6.5% 14C-labeled lifitegrast, the drug was rapidly distributed (less than 30 minutes) into ocular and periocular tissues and cleared from the eye by normal drainage of the tear through the nasolacrimal duct into the nasopharynx. There was no evidence of absorption from the gastrointestinal tract as evaluated by whole body autoradiography,59 and the drug absorbed into the systemic circulation after ophthalmic drop administration was rapidly cleared by the hepatic circulation into the bile and feces.58 In a similar radiolabeled study in dogs, there was high distribution to the ocular surface and periocular tissues, lesser distribution to the aqueous humor, and undetectable levels in posterior ocular tissues (retina/choroid) and systemic plasma across a 24-hour time period.52,58
Most drugs administered to the ocular surface are rapidly cleared from the tear film within the first 30–60 minutes following topical administration due to the normal tear turnover. Lifitegrast has excellent aqueous solubility (>100 mg/mL). The high solubility allows a relatively large concentration to be administered into the tear film and theoretically allows significant residual concentration despite the normal tear turnover. In dogs given a single dose of lifitegrast 1.0% ophthalmic solution, concentrations of lifitegrast in excess of 1 μM is detected in the tear film for 12–24 hours; this residual concentration is 2–3 log orders higher than the IC50 for blocking T-cell adhesion and inhibition of immunologic synapse formation and cytokine secretion in vitro46,58 and supportive of a dosing rationale of once to twice a day.
Formulation
The final step toward enabling human investigations of lifitegrast was to formulate the drug and package it in appropriate dosing ampules for clinical trials. Simply putting the drug into a bottle is no trivial feat and represents, arguably, the most complex and underappreciated step toward developing a viable pharmaceutical. At the start of this effort, we could not find a “road map” in the literature for the rational development of a new chemical entity as an ophthalmic drop. Numerous examples of the reformulation of systemic drugs to repurpose them for ophthalmic indications exist (eg, cyclosporine administered orally for the prevention of transplant rejection repurposed as an ophthalmic drop for the treatment of dry eye). We reasoned that for a topical drop targeting LFA-1 on the extracellular surface of T cells in the ocular surface and periocular tissues, the desired properties for lifitegrast should include solubility and stability in water enabling an aqueous formulation isotonic with tear, metabolic stability in biologic matrices, and long retention time in tear at therapeutic levels and periocular tissues supporting once or twice daily dosing. For clinical investigations, lifitegrast was prepared in phosphate buffered saline in concentrations up to 5.0% at a pH, tonicity, and osmolarity range consistent with currently approved topical ophthalmic products while maintaining potency for LFA-1/ICAM-1 inhibition.52 Because of the harm to the ocular surface epithelium associated with chronic exposure to preservatives, lifitegrast was formulated preservative-free in single-unit dose ampules.
By 2008, nearly two decades after the initial descriptions of LFA-1, lifitegrast was being prepared for dosing in the first human subject. The clinical development of lifitegrast as the first ophthalmic drop specifically engineered to target T-cell-mediated inflammation in dry eye was ready to begin.
Regulatory perspectives
What is the FDA’s expectation for the demonstration of efficacy? At least two adequate and well-controlled pivotal trials are recommended that demonstrate statistical significance in a sign and a symptom of DED.60 Since no sign or symptom has been shown to be more important than another, the FDA has stated that a statistically significant change in a sign associated with a statistically significant change in a symptom will be considered to cross-validate each endpoint. Because signs and symptoms are not highly correlative, two to four adequate and well-controlled trials can be submitted – two trials replicating the sign and two trials replicating symptoms; these can be conducted in different study populations.61 In addition, efficacy trials with a single endpoint (sign) have been allowed where the goal is to evaluate the proportions of subjects decreasing their risk of infection (eg, whole corneal staining score =0) or increasing the proportion of subjects with clearly increased tear production (eg, Schirmer tear test increased by at least 10 mm). There has not been allowance for pivotal programs based solely on a symptom endpoint due to the potential for formulations to incorporate anesthetics that mask the perception of DED but are not biologically active in treating the disease itself.61 To provide sufficient evidence to support a labeled claim of treating both a sign and a symptom of the disease, a four-trial strategy may be the preferred approach given the historically difficult ability to demonstrate meeting the a priori sign and symptom endpoints in a single trial.
The clinical safety database for a chronically administered new chemical entity topical ophthalmic drug should have safety data on at least 100 subjects exposed for ≥1 year and at least 300 subjects exposed for ≥6 months to support the New Drug Application. Endothelial cell counts assessed by specular microscopy should also be included to verify the absence of long-term toxicity to the cornea (baseline and 1 year).61
Clinical development of lifitegrast
Lifitegrast has been evaluated in over 2,500 adult DED patients with twice-daily dosing of a 5.0% ophthalmic solution for up to 1 year. A summary of the six clinical trials are provided in Table 1. All the trials were randomized, double-masked, placebo-controlled, parallel arm designs including the Phase 1 first-in-human and the 1-year safety (SONATA) trials.8–10,62–64 In the efficacy studies (Phase 2, OPUS-1, -2, -3), eligible patients were treated in an identical manner according to the study protocol, which called for a 2-week open-label vehicle run-in period followed by randomized treatment to either lifitegrast (twice daily) or vehicle (twice daily) for 84 days and assessed for signs and symptoms of DED on days 14, 42, and 84 in the normal ambient (natural) environment; baseline and clinical endpoint evaluations were pre-specified to be conducted in the natural environment. The Phase 2 and OPUS-1 trials incorporated the use of the controlled adverse environment for selection of DED patients with acute, active disease. The OPUS-2 and OPUS-3 trials required patients to have a recent history of artificial tear use within 30 days of the first screening visit as an empiric proxy for acutely active disease.
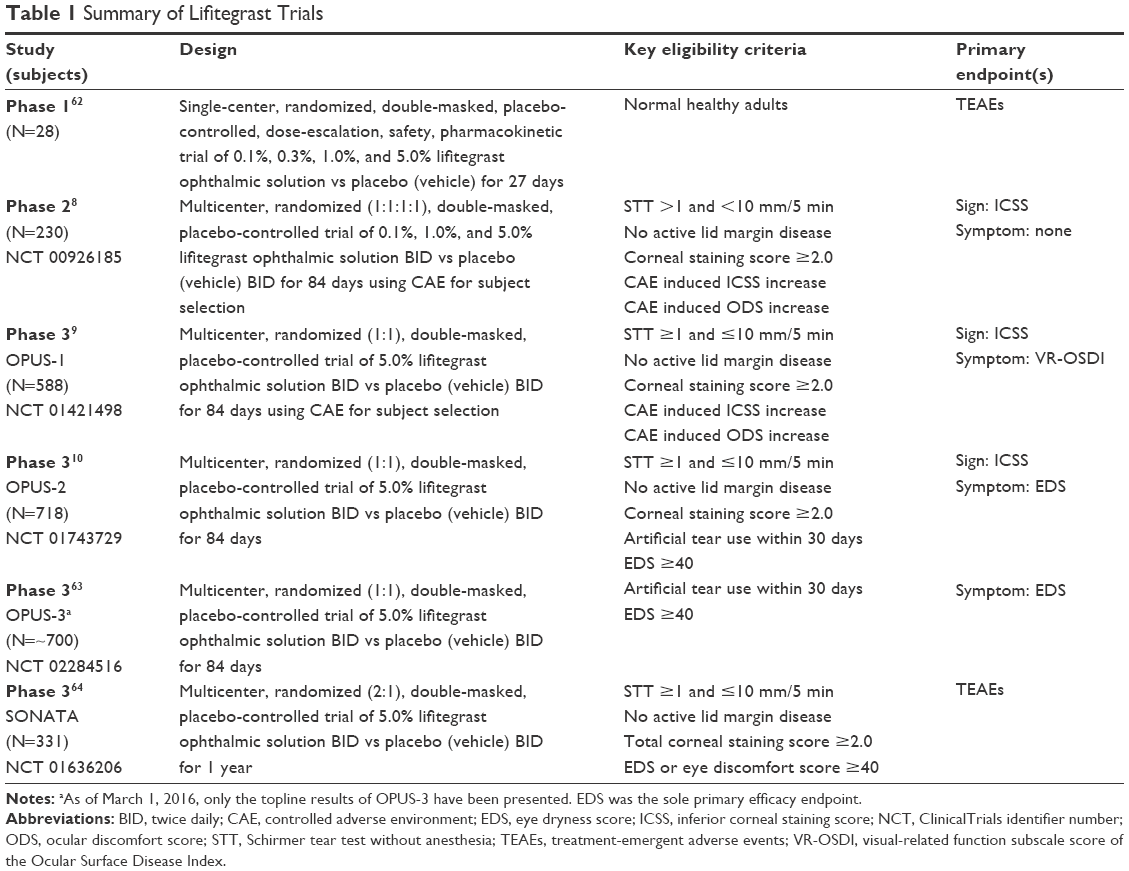 | Table 1 Summary of Lifitegrast Trials |
A summary of key findings from each study is provided in Table 2. The objective efficacy endpoint (sign) was inferior corneal staining score (ICSS) (0–4 points, 0= no staining). The subjective efficacy endpoint (symptom) was either the visual-related function subscale score of the OSDI (Questions 6–9; 0–4 points, 0= no symptoms) or the eye dryness score (EDS; 0–100 points, 0= no discomfort). The primary statistical analysis involved evaluating the difference in the endpoint score using mean change from baseline to day 84 comparing the lifitegrast group to the placebo (vehicle) group. Long-term safety was supported by SONATA, a double-masked, placebo-controlled study that compared 5.0% lifitegrast to vehicle in DED patients treated for 1 year.
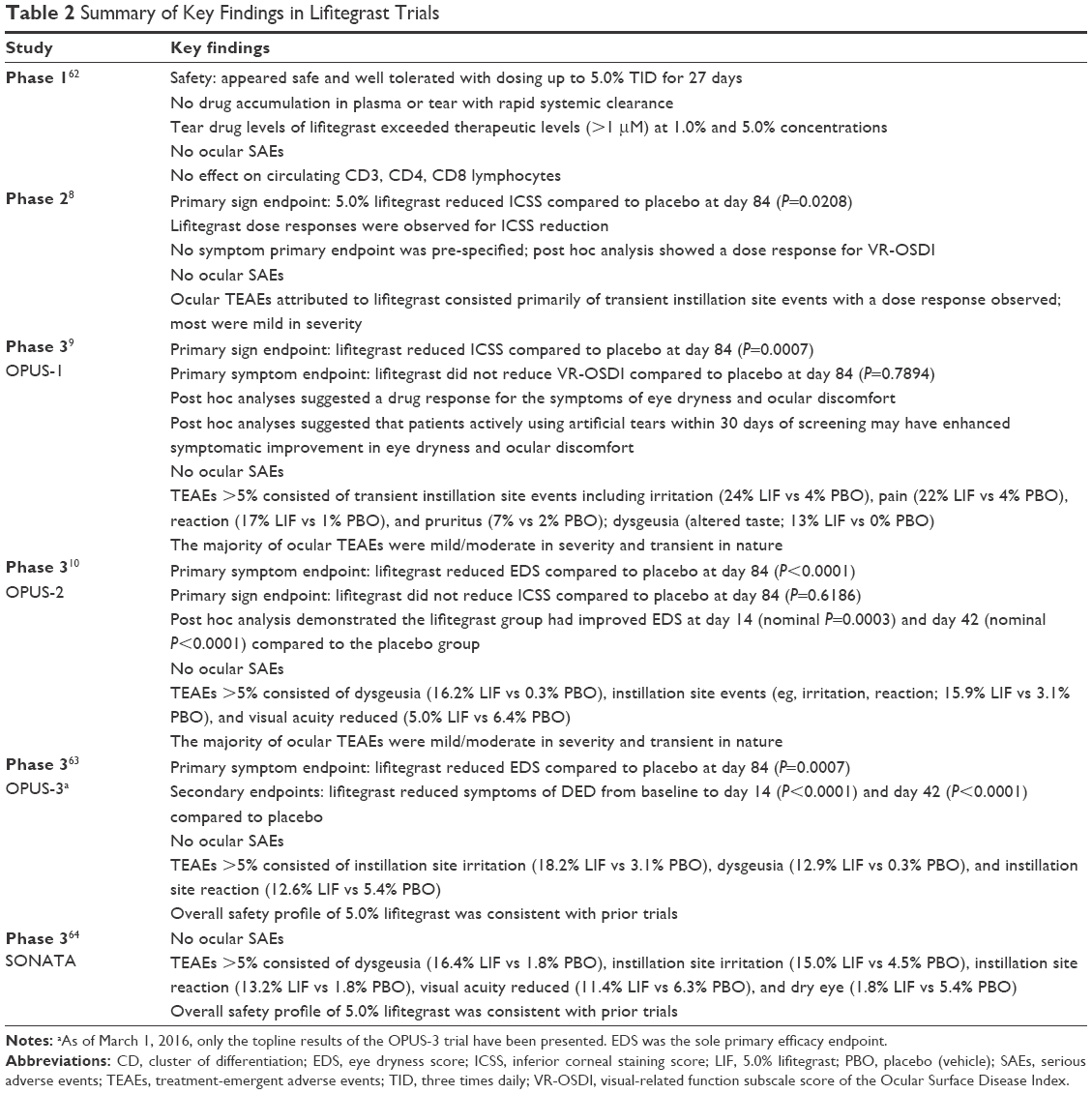 | Table 2 Summary of Key Findings in Lifitegrast Trials |
Lifitegrast demonstrated reduction in ICSS at day 84 compared to placebo in two consecutive trials in accordance with the overall lifitegrast study protocol following the use of controlled adverse environment for patient selection (Phase 2, P=0.0208; OPUS-1, P<0.0001).8,9 Consequently, we determined that the replication of the sign was met and consistent with regulatory guidelines. Though OPUS-1 did not meet the symptom endpoint (visual-related function subscale score of the OSDI, P=0.7894), post hoc analyses suggested a beneficial drug response using the eye dryness score (EDS; 7-item Visual Analog Scale). The apparent drug response appeared particularly enhanced in patients who were actively using artificial tears within 30 days of screening and had a baseline EDS ≥40 points. Therefore, this hypothesis was evaluated prospectively in OPUS-2 in accordance with the overall lifitegrast study protocol where these two parameters (artificial tear use and baseline EDS ≥40 points) were incorporated into the eligibility criteria with EDS as the pre-specified primary symptom endpoint.10 The symptom results from OPUS-2 were highly robust and confirmed that lifitegrast markedly decreased EDS by day 84 compared to placebo (P<0.0001); OPUS-2 did not demonstrate a simultaneous reduction in ICSS (P=0.6186). Given the highly statistically significant symptom outcome from OPUS-2 as a single trial, it was reasonable to conclude that the totality of the evidence from three consecutive adequate and well-controlled trials were statistically persuasive in demonstration of efficacy of the sign (Phase 2 and OPUS-1) and symptom (OPUS-2) endpoints consistent with regulatory guidelines.65 However, the FDA’s reliance on only a single trial (eg, OPUS-2) is generally limited to situations in which a “second trial would be practically or ethically impossible” and that “a conclusion based on two persuasive studies will always be more secure than a conclusion based on a single, comparably persuasive study”.66 OPUS-3, conducted in identical manner to OPUS-2 and specifying EDS as the sole efficacy endpoint, provided the confirmatory evidence that lifitegrast rapidly and robustly decreased EDS compared to placebo (P=0.0007).63 Secondary outcomes from OPUS-3 were consistent with OPUS-2 in demonstrating that the onset of symptom reduction was as early as day 14 (P<0.0001) and day 42 (P<0.0001) and continued through the duration of the treatment.10,63 Given the known discordance between signs and symptoms, it is not wholly unreasonable to observe that different experimental conditions were required to elicit a drug response in the sign (inferior corneal staining score, Phase 2 and OPUS-1 trials) and symptom (eye dryness score, OPUS-2 and OPUS-3 trials). This outcome provides clinical validation that statistically significant treatment effects of one variable (sign) can behave independent of the other variable (symptom).
Lifitegrast appears safe and well tolerated when administered twice daily up to 1 year.64 The most commonly reported treatment-emergent adverse events associated with lifitegrast were dysgeusia (altered taste sensation) and instillation site events (eg, irritation); these adverse events were generally transient and mild to moderate in severity. There have been no drug-related ocular serious adverse events, and neither localized nor systemic immunosuppressive events were reported.
Conclusion
We have described the design, discovery, and development of lifitegrast, a novel T-cell inhibitor, as the first anti-inflammatory ophthalmic drop to demonstrate a dose response in the moderation of both signs and symptoms of DED. It began with the selection of LFA-1 as a target, ICAM-1 as a lead molecular mimic, and pharmaceutical refinement of lifitegrast as a purpose-built treatment. Preclinical safety and efficacy in treating canine keratoconjunctivitis sicca led to initiating the challenging and complex human clinical trials and navigating regulatory expectations. Along the way, all major aspects of the story – from bench top to bedside – have been published in the peer-reviewed scientific literature in order to provide one potential “road map” for those who will follow our footsteps in developing future therapeutics for ocular surface diseases.
Disclosure
Charles P Semba is a former employee of SARcode Bioscience, Inc. (Brisbane, CA, USA; now a wholly owned subsidiary of Shire Plc) and Shire (Lexington, MA, USA) during the development of lifitegrast for the treatment of dry eye disease.
Thomas R Gadek is a former employee of SARcode Bioscience, Inc. (Brisbane, CA, USA; now a wholly owned subsidiary of Shire Plc) during the development of lifitegrast for the treatment of dry eye disease.
References
Miljanovic B, Dana R, Sullivan DA, Schaumberg DA. Impact of dry eye syndrome on vision-related quality of life. Am J Ophthalmol. 2007;143:409–415. | ||
[No authors listed]. The definition and classification of dry eye disease: report of the Definition and Classification Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007;5(2):75–92. | ||
New Drug Application 21-023. Cyclosporine 0.05% ophthalmic emulsion. Available from: https://www.accessdata.fda.gov/scripts/cder/drugsatfda/. Accessed March 12, 2016. | ||
Full Prescribing Information. Restasis® 0.05% Ophthalmic emulsion, Allergan, Inc. Available from: http://www.allergan.com/assets/pdf/restasis_pi.pdf. Accessed March 12, 2016. | ||
Sheppard JD, Scoper SV, Samudre S. Topical loteprednol pretreatment reduces cyclosporine stinging in chronic dry eye disease. J Ocul Pharmacol Ther. 2011;27(1):23–27. | ||
Asbell PA, Spiegel S. Ophthalmologist perceptions regarding treatment of moderate to severe dry eye: results of a physician survey. Trans Am Ophthalmol Soc. 2009;107:205–210. | ||
US FDA acknowledges receipt of resubmission of Shire’s New Drug Application for lifitegrast for dry eye disease in adults [press release]. Shire Plc; 2016 [February 4]. Available from: https://www.shire.com/newsroom/2016/february/lifitegrast-resubmission-press-release-fda-acknowledgement-2-4-16. Accessed March 12, 2016. | ||
Semba CP, Torkildsen GL, Lonsdale JD, et al. A phase 2 randomized, double-masked, placebo-controlled study of a novel integrin antagonist (SAR 1118) for the treatment of dry eye. Am J Ophthalmol. 2012;153(6):1050–1060. | ||
Sheppard JD, Torkildsen GL, Lonsdale JD, et al. Lifitegrast ophthalmic solution 5.0% for treatment of dry eye disease: results of the OPUS-1 phase 3 study. Ophthalmology. 2014;121(2):475–483. | ||
Tauber J, Karpecki P, Latkany R, et al. Lifitegrast ophthalmic solution 5.0% versus placebo for treatment of dry eye disease: results of the randomized phase III OPUS-2 study. Ophthalmology. 2015;122(12):2423–2431. | ||
Lemp MA. Report of the National Eye Institute/Industry Workshop on clinical trials in dry eyes. CLAO J. 1995;21(4):221–232. | ||
Nichols KK, Foulks GN, Bron AJ, et al. The international workshop on meibomian gland dysfunction: executive summary. Invest Ophthamol Vis Sci. 2011;52(4):1922–1929. | ||
Schaumberg DA, Sullivan DA, Buring JE, Dana MR. Prevalence of dry eye syndrome among US women. Am J Ophthalmol. 2003;136(2):318–326. | ||
Schaumberg DA, Dana R, Buring JE, Sullivan DA. Prevalence of dry eye disease among US men: estimates from the Physicians’ Health Studies. Arch Ophthalmol. 2009;127(6):763–768. | ||
Gayton JL. Etiology, prevalence, and treatment of dry eye disease. Clin Ophthalmol. 2009;3:405–412. | ||
Pflugfelder SC. Dry eye: the problem. In: Pflugfelder SC, Beuerman RW, Stern ME, editors. Dry Eye and Ocular Surface Disorders. New York: Marcel Dekker; 2004:1–10. | ||
Schiffman RM, Christianson MD, Jacobsen G, Hirsch JD, Reis BL. Reliability and validity of the Ocular Surface Disease Index. Arch Ophthalmol. 2000;118(5):615–621. | ||
Schaumberg DA, Gulati A, Mathers WD, et al. Development and validation of a short global dry eye symptom index. Ocul Surf. 2007;5(1):50–57. | ||
Nichols KK, Mitchell GL, Zadnik K. Performance and repeatability of the NEI-VFQ-25 in patients with dry eye. Cornea. 2002;21(6):578–583. | ||
[No authors listed]. Methodologies to diagnose and monitor dry eye disease: report of the Diagnostic Methodology Subcommittee on the International Dry Eye WorkShop (2007). Ocul Surf. 2007;5(2):108–152. | ||
Savini G, Prabhawasat P, Kojima T, Grueterich M, Espana E, Goto E. The challenge of dry eye diagnosis. Clin Ophthalmol. 2008;2(1):31–55. | ||
Kim J. The use of vital dyes in corneal disease. Curr Opin Ophthalmol. 2000;11(4):241–247. | ||
Nichols KK, Nichols JJ, Mitchell GL. The lack of association between signs and symptoms in patients with dry eye disease. Cornea. 2004;23(8):762–770. | ||
Adatia, Michaeli-Cohen A, Naor J, Caffery B, Bookman A, Slomovic A. Correlation between corneal sensitivity, subjective dry eye symptoms and corneal staining in Sjögren’s syndrome. Can J Ophthalmol. 2004;39(7):767–771. | ||
Pepose JS, Sullivan BD, Foulks GN, Lemp MA. The value of tear osmolarity as a metric in evaluating the response to dry eye therapy in the clinic and in clinical trials. Am J Ophthalmol. 2014;157(1):4–6. | ||
Schargus M, Ivanova S, Kakkassery V, Dick HB, Joachim S. Correlation of tear film osmolarity and 2 different MMP-9 tests with common dry eye tests in a cohort of non-dry eye patients. Cornea. 2015;34(7):739–744. | ||
Finis D, Pischel N, Schrader S, Geerling G. Evaluation of lipid layer thickness measurement of the tear film as a diagnostic tool for Meibomian gland dysfunction. Cornea. 2013;32(12):1549–1553. | ||
Czajkowski G, Kaluzny BJ, Laudencka A, Malukiewicz G, Kaluzny JJ. Tear meniscus measurement by spectral optical coherence tomography. Optom Vis Sci. 2012;89(3):336–342. | ||
[No authors listed]. Management and therapy of dry eye disease: report of the Management and Therapy Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007;5(2):163–178. | ||
Tauber J, Davitt WF, Bokosky JE, et al. Double-masked, placebo-controlled safety and efficacy trial of diquafosol tetrasodium (INS365) ophthalmic solution for the treatment of dry eye. Cornea. 2004;23(8):784–792. | ||
Kossler AL, Wang J, Feuer W, Tse DT. Neurostimulation of the lacrimal nerve for enhanced tear production. Ophthal Plast Reconstr Surg. 2015;31(2):145–151. | ||
Qiao J, Yan X. Emerging treatment options for meibomian gland dysfunction. Clin Ophthalmol. 2013;7:1797–1803. | ||
Stevenson W, Chauhan SK, Dana R. Dry eye disease: an immune-mediated ocular surface disorder. Arch Ophthalmol. 2012;130(1):90–100. | ||
Gao J, Morgan G, Tieu D, et al. ICAM-1 expression predisposes ocular tissues to immune-based inflammation in dry eye patients and Sjögren’s syndrome-like MRL/lpr mice. Exp Eye Res. 2004;78:823–835. | ||
Gao J, Schwalb TA, Addeo JV, Ghosn CR, Stern ME. The role of apoptosis in the pathogenesis of canine keratoconjunctivitis sicca: the effect of topical cyclosporin A therapy. Cornea. 1998;17:654–663. | ||
Stern ME, Gao J, Schwalb TA, et al. Conjunctival T-cell subpopulations in Sjögren’s and non-Sjögren’s patients with dry eye. Invest Ophthalmol Vis Sci. 2002;43:2609–2614. | ||
Barabino S, Montaldo E, Solignani F, Valente C, Mingari MC, Rolando M. Immune response in the conjunctival epithelium of patients with dry eye. Exp Eye Res. 2010;91(4):524–529. | ||
Lam H, Bleiden L, de Paiva CS, Farley W, Stern ME, Pflugfelder SC. Tear cytokine profiles in dysfunctional tear syndrome. Am J Ophthalmol. 2009;147(2):198–205. | ||
Enríquez-de-Salamanca A, Castellanos E, Stern ME, et al. Tear cytokine and chemokine analysis and clinical correlations in evaporative-type dry eye disease. Mol Vis. 2010;16:862–873. | ||
Smith A, Stanley P, Jones K, Svensson L, McDowall A, Hogg N. The role of the integrin LFA-1 in T-lymphocyte migration. Immunol Rev. 2007;218:135–146. | ||
Kürzinger K, Springer TA. Purification and structural characterization of LFA-1, a lymphocyte function-associated antigen, and Mac-1, a related macrophage differentiation antigen associated with the type three complement receptor. J Biol Chem. 1982;257(20):12412–12418. | ||
Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell. 1987;51(5):813–819. | ||
Springer TA, Dustin ML, Kishimoto TK, Marlin SD. The lymphocyte function-associated LFA-1, CD2, and LFA-3 molecules: cell adhesion receptors of the immune system. Annu Rev Immunol. 1987;5:223–252. | ||
Kishimoto TK, Hollander N, Roberts TM, Anderson DC, Springer TA. Heterogeneous mutations in the beta subunit common to the LFA-1, Mac-1, and p150,95 glycoproteins cause leukocyte adhesion deficiency. Cell. 1987;50(2):193–202. | ||
Grakoui A, Bromley SK, Sumen C, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285(5425):221–227. | ||
Dustin ML, Groves JT. Receptor signaling clusters in the immune synapse. Annu Rev Biophys. 2012;41:543–556. | ||
Hogg N, Laschinger M, Giles K, McDowall A. T-cell integrins: more than just sticking points. J Cell Sci. 2003;116(Pt 23):4695–4705. | ||
Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci U S A. 1992;89(9):3686–3690. | ||
Kunert KS, Tisdale AS, Stern ME, Smith JA, Gipson IK. Analysis of topical cyclosporine treatment of patients with dry eye syndrome. Effect on conjunctival lymphocytes. Arch Ophthalmol. 2000;118:1489–1496. | ||
Gadek TR, Burdick DJ, McDowell RS, et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science. 2002;295:1086–1090. | ||
Keating SM, Clark KR, Stefanich LD, et al. Competition between intercellular adhesion molecule-1 and a small-molecule antagonist for a common binding site on the αL subunit of lymphocyte function-associated antigen-1. Protein Sci. 2006;15(2):290–303. | ||
Zhong M, Gadek TR, Bui M, et al. Discovery and development of the potent LFA-1/ICAM-1 antagonist SAR 1118 as an ophthalmic solution for treating dry eye. ACS Med Chem Lett. 2012;3(3):203–206. | ||
Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2014;21(9):1102–1114. | ||
Burdick DJ, Paris K, Weese K, et al. N-Benzoyl amino acids as LFA-1/ICAM inhibitors 1: amino acid structure-activity relationship. Bioorg Med Chem Lett. 2003;13(6):1015–1018. | ||
Burdick DJ, Marsters JC Jr, Aliagas-Martin I, et al. N-Benzoyl amino acids as ICAM/LFA-1 inhibitors. Part 2: structure-activity relationship of the benzoyl moiety. Bioorg Med Chem Lett. 2004;14(9):2055–2059. | ||
Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Small molecule integrin antagonists that bind to the beta-2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity. 2003;19(3):391–402. | ||
Chigaev A, Skalr LA. Aspects of VLA-4 and LFA-1 regulation that may contribute to rolling and firm adhesion. Front Immunol. 2012;3:242. | ||
Murphy CJ, Bentley E, Miller PE, et al.The pharmacologic assessment of a novel lymphocyte function-associated antigen-1 antagonist (SAR 1118) for the treatment of keratoconjunctivitis sicca in dogs. Invest Ophthalmol Vis Sci. 2011;52(6):3174–3180. | ||
Rao VR, Prescott E, Shelke NB, et al. Delivery of SAR 1118 to the retina via ophthalmic drops and its effectiveness in a rat streptozotocin (STZ) model of diabetic retinopathy (DR). Invest Ophthalmol Vis Sci. 2010;51(10):5198–5204. | ||
Chambers W. Proceedings from the Dry Eye Summit, Panel Discussion. Fort Lauderdale, FL; 2010. Available from: http://www.dryeyesummit.org. Accessed March 12, 2016. | ||
Chambers W. Regulatory perspectives. Lecture, Dry Eye Summit, Ft. Lauderdale, FL; 2010. | ||
Semba CP, Swearingen D, Smith VL, et al. Safety and pharmacokinetics of a novel lymphocyte function-associated antigen-1 antagonist ophthalmic solution (SAR 1118) in healthy adults. J Ocul Pharm Ther. 2011;27(1):99–104. | ||
Press Release. Shire’s OPUS-3 phase 3 trial with lifitegrast meets primary and key secondary endpoints, significantly reducing patient-reported symptoms for dry eye disease [press release]. Shire Plc; 2015 [October 27]. Available from: https://www.shire.com/newsroom/2015/october/shires-opus-3-phase-3-trial-with-lifitegrast-meets-primary-and-key-secondary-endpoints. Accessed March 12, 2016. | ||
Donnenfeld ED, Karpecki PM, Majmudar PA, et al. Safety of lifitegrast ophthalmic solution 5.0% in patients with dry eye disease: a 1-year, multicenter, randomized, placebo-controlled study. Cornea. 2016;35(6):741–748. | ||
Shire submits application to the US FDA for approval of lifitegrast for treatment of dry eye disease in adults [press release]. Shire Plc; 2015 [March 2]. Available from: https://www.shire.com/newsroom/2015/march/shire-submits-application-to-the-us-fda-for-approval-of-lifitegrast-for-treatment-of. Accessed March 12, 2016. | ||
FDA Guidance for Industry. Providing clinical evidence of effectiveness for human drug and biological products; 1998. Available from: http://www.fda.gov/downloads/Drugs/GuidanceCompliance%20RegulatoryInformation/Guidances/UCM078749.pdf+Providing+clinical+evidence+of+effectiveness+for+human+and+bio&client=FDAgov&site=FDAgov&lr=&proxystylesheet=FDAgov&output=xml_no_dtd&ie=UTF-8&access=p&oe=UTF-8. Accessed March 12, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.