")
Back to Journals » International Journal of Nanomedicine » Volume 13
Development of biocompatible and VEGF-targeted paclitaxel nanodrugs on albumin and graphene oxide dual-carrier for photothermal-triggered drug delivery in vitro and in vivo
Authors Deng W, Qiu J, Wang S, Yuan Z, Jia Y, Tan H, Lu J, Zheng R
Received 6 September 2017
Accepted for publication 24 October 2017
Published 17 January 2018 Volume 2018:13 Pages 439—453
DOI https://doi.org/10.2147/IJN.S150977
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Wentao Deng,1,* Juhui Qiu,1,* Shaoting Wang,1 Zhi Yuan,1 Yuefeng Jia,2 Hailin Tan,2 Jiru Lu,1 Ruqiang Zheng1
1Department of Urinary Surgery, Dongying People’s Hospital, Dongying, 2Department of Urinary Surgery, The Affiliated Hospital of Qingdao University, Qingdao, Shandong, China
*These authors contributed equally to this work
Abstract: In this study, we performed the characterization and synthesis of biocompatible and targeted albumin and graphene oxide (GO) dual-carrier paclitaxel (PTX) nanoparticles for photothermal-triggered tumor therapy. PTX absorbed on GO nanosheets as cores were coated with human serum albumin (HSA), following surface conjugation with monoclonal antibodies (mAb) against vascular endothelial growth factor (VEGF; denoted as mAbVEGF) via polyethylene glycol linker to form targeted nanoparticles (PTX-GHP-VEGF). The spherical nanoparticles were 191±5 nm in size with good stability and biocompatibility. GO functioned as the first carrier and a near infrared absorber that can generate photothermal effects under 5-minute 808-nm laser irradiation to thermal trigger the release of PTX from the second carrier HSA nanoparticles. The mechanism of thermal-triggered drug release was also investigated preliminarily, in which the heat generated by GO induced swelling of PTX-GHP-VEGF nanoparticles which released the drugs. In vitro studies found that PTX-GHP-VEGF can efficiently target human SW-13 adrenocortical carcinoma cells as evaluated by confocal fluorescence microscopy as well as transmission electron microscopy, and showed an obvious thermal-triggered antitumor effect, mediated by apoptosis. Moreover, PTX-GHP-VEGF combined with near infrared irradiation showed specific tumor suppression effects with high survival rate after 100 days of treatment. PTX-GHP-VEGF also demonstrated high biosafety with no adverse effects on normal tissues and organs. These results highlight the remarkable potential of PTX-GHP-VEGF in photothermal controllable tumor treatment.
Keywords: paclitaxel, graphene oxide, human serum albumin, tumor targeting, photothermal-triggered tumor therapy
Introduction
Cancer remains a serious threat to human health worldwide. Adrenocortical carcinoma (ACC), derived from the adrenal cortex, is a rare heterogeneous neoplasm where the pathogenesis is not well characterized and has a poor prognosis, resulting in the lack of effective treatment.1,2 Some attempts such as the combination of drugs and mitotane have been recommended as first-line therapy in advanced ACC, but the overall prognosis remains very limited.3,4 In addition, the low target effects (from local low-dose drugs) and high side effects have also limited the utility of chemotherapeutic drugs. Therefore, development of new chemotherapeutic agent formulations is ongoing, such as tumor target nanoparticles-loaded anticancer drugs.5–8
In recent decades, natural antitumor drugs have been found to be effective against certain cancers such as resveratrol, curcumin, and artemisinin.9–13 Paclitaxel (PTX) is a Food and Drug Administration-approved clinical tumor chemotherapeutic agent, which is a natural secondary metabolite purified from the bark of the gymnoside yew.14,15 PTX is mostly used as treatment for ovarian and breast cancer. It has also been reported to have high toxicity against other cancers such as non-small cell lung cancer and ACC.6,16–18 However, since PTX has poor aqueous solubility in its natural form, it is formulated in a mixture of Cremophor EL/absolute ethanol (1:1 v/v, Taxol®) for clinical use.19,20 However, Cremophor EL in the media has serious side effects such as hypersensitivity and acute renal function impairment.21,22 To solve these limitations of PTX, various alternative strategies have been developed, such as loading PTX by carbon nanomaterials, protein, and liposomes.23–25
Graphene oxide (GO), a two-dimensional carbon nanomaterial, is usually used as a drug carrier due to its near infrared (NIR) light absorbance property, large surface area, and modifiable surface structure.26,27 Some aromatic and insoluble antitumor drugs such as SN38 and camptothecin as well as small molecule fluorescent dyes have been shown to be loaded onto nanosized GO surfaces through π–π stacking interactions and hydrogen bonds.28–30 Moreover, GO has remarkable NIR photothermal properties that can control drug release on its surface.31–33 For example, Kim et al have reported that the GO derivative reduced GO-loaded anticancer drugs doxorubicin showed photothermally triggered tumor chemotherapy.34 However, GO has low biocompatibility and further surface modification must be conducted for biomedical application. Furthermore, high concentrations of GO may have toxicity issues.35
Apart from GO, other organic materials such as liposomes, hydrogels, and serum albumin can also be used as drug carriers.36–39 Compared with these carriers, serum albumin as an endogenous protein with good biocompatibility, non-toxicity, and non-immunogenicity has become one of the most exciting carriers to deliver insoluble anticancer drugs.40–42 Moreover, the presence of functional carboxylic and amino groups on the surface facilitates surface functionalization for albumin nanoparticles such as conjugation of targeting moieties. However, the release of drugs in albumin nanoparticles cannot be controlled, which is unfavorable for efficient tumor therapy.
Based on the demand for novel formulations with greater biocompatibility, tumor targeting abilities, and controlled drug release for efficient tumor therapy, this study utilizes GO nanosheets to carry lipophilic PTX (PTX-GO). Human serum albumin (HSA) as the second carrier was used as a shell to encapsulate the PTX-GO forming a nanodrug, which is surface functionalized with a tumor targeting molecule, monoclonal antibodies against vascular endothelial growth factor (VEGF; denoted as mAbVEGF) via a polyethylene glycol (PEG) “bridge” (PTX-GHP-VEGF).1,2 In addition to in vitro and in vivo biocompatibility, PTX-GHP-VEGF showed thermal-controlled drug release due to the photothermal effect of GO and further thermal-induced expansion of HSA shells. The in vitro and in vivo tumor target potential of PTX-GHP-VEGF was validated via cell uptake and NIR thermal imaging experiments. At last, the anticancer effect of PTX-GHP-VEGF combined NIR laser irradiation on ACC tumor bearing mice was evaluated. The obtained results demonstrate that PTX-GHP-VEGF promises to be a kind of novel therapeutic agent for clinical translation medicine.
Materials and methods
Materials
GO platelet (PLT) was obtained from Nanjing XFNANO Materials Tech Co, Ltd (Nanjing, China). N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich Co (St Louis, MO, USA). Amino-polyethylene glycol-7500-maleimide (NH2-PEG-MAL) was obtained from Creative PEGWorks (Durham, NC, USA). mAbVEGF produced in mouse was obtained from Sigma-Aldrich. PTX (≥99%, Aladdin, Shanghai, China) was provided by Shanghai Jingchun Biotech Corporation (Shanghai, China). 6-Diamidino-2-phenylindole (DAPI) was obtained from Aladdin. L-15 media and phosphate-buffered saline (PBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Cell counting kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan).
Synthesis of PTX-GO
GO PLT was added into distilled water (2 mg/mL) for GO dispersion, following ultrasonication in an ice bath for 5 hours. Centrifugation at 12,000 rpm was used to remove the bulk GO, and the supernatant was collected to load PTX. In detail, PTX powder was dissolved in dimethyl sulfoxide (DMSO) and then added into GO suspension with stirring at 4°C for 6 hours. Repeated dialyzing (MW cutoff =10 kDa) against distilled water was then carried out for 2 days to eliminate the unbound PTX and free DMSO to obtain a purified PTX-GO solution. The concentration of bound PTX was measured by ultraviolet–visible (UV–vis) spectroscopy (PerkinElmer Inc, Waltham, MA, USA) with the absorption intensity at 227 nm.
Synthesis of PTX-GO-HSA
HSA powder (10 mg) was added into 20 mL PTX-GO solution with slight stirring for 6 hours at 25°C. This was then processed by cross-linking with 0.5% glutaraldehyde (200 μL). Afterwards, the obtained mixture was dialyzed against distilled water (MW cut off =10 kDa) for 2 days to give purified PTX-GO-HSA solution. The amount of loaded PTX was detected by an UV–vis spectrophotometer by monitoring the absorption peak at 230 nm.
Synthesis of PTX-GHP-VEGF
The mAbVEGF solution (0.3 mL, 500 μg/mL) in 0.1 M carbonate buffer at pH 8.6 was incubated with 10 μL 14 mM Traut’s reagent for 1 hour at room temperature. Excess Traut’s reagent was eliminated by gel-filtration on Sephadex G-25. In addition, NH2-PEG-MAL (50 μL, 20 mg/mL) was added into the thiolated antibodies solution, following incubation for 4 hours at 4°C. Next, EDC solution (8 μL, 5 mg/mL) and NHS solution (10 μL, 20 mg/mL) were mixed with GO-PTX-HSA solution (5 mL, 4.5 mg/mL) to activate the carboxylic groups of HSA, and then the mixture was allowed to react with PEGylated mAbVEGF for 2 hours at 4°C. Purified mAbVEGF-modified nanoparticles (PTX-GO-HSA-PEG-VEGF, PTX-GHP-VEGF) were obtained by gel-filtration on Sepharose CL-6B (PBS, column 50×2.5 cm, 0.5 mL/min).
Characterizations
The morphology of the nanoparticles was observed by a transmission electron microscope (H-600; Hitachi Ltd, Tokyo, Japan) and scanning electron microscopy (Phenom G2 pro; Phenom-World, Eindhoven, the Netherlands). The size and zeta potential of the nanoparticles were measured by a Zetasizer (Nano ZS; Malvern Instruments, Malvern, UK). The absorbance spectra were recorded by a UV–vis spectrophotometer (UV-2550; Shimadzu, Kyoto, Japan). A laser source at 808 nm (diode-pumped solid-state laser system; Beijing Laserwave OptoElectronics Technology Co Ltd, China) was used to conduct photothermal experiments. The temperature variation was recorded every 30 seconds using a thermocouple thermometer (Fluke Corporation, Everett, WA, USA).
Photothermal properties of PTX-GHP-VEGF
GO, PTX-GHP-VEGF without GO (PTX-HP-VEGF), and PTX-GHP-VEGF dispersions (with the same concentration of GO) in a 96-well plate were irradiated under 808-nm laser irradiation for 5 minutes (1 W/cm2). Real-time temperature and thermal images were recorded once every 30 seconds by a thermocouple thermometer and infrared thermal camera (Fluke TI10; Fluke Corporation), respectively.
Cell culture and cellular uptake study
Human ACC SW-13 cells were purchased from American Type Culture Collection (Manassas, VA, USA) and cultured in L-15 media supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37°C with 5% CO2.
To observe cellular uptake, a fluorescence dye, fluorescein isothiocyanate (FITC), was used to label PTX-GHP and PTX-GHP-VEGF. SW-13 cells were incubated with FITC-labeled PTX-GHP, and PTX-GHP-VEGF at the same concentration of FITC (0.05 mg/mL) for 3 hours, and the free FITC-treated cells were set as the control group. Afterwards, the treated cells were washed by PBS thrice slightly, fixed with 0.2 mL glutaraldehyde, and stained with DAPI for 10 minutes. A confocal laser scanning microscope (FV1200; Olympus Corporation, Tokyo, Japan) was used to detect the fluorescence signals of cells.
A flow cytometer (FCM) was further used to evaluate cellular uptake. As described above, PTX-GHP and PTX-GHP-VEGF were labeled with FITC (0.05 mg/mL) and incubated with cells for 3 hours. Thereafter, the treated cells were washed three times with PBS and digested by trypsin-EDTA. The suspended cells were directly introduced into an FCM (Beckman Coulter, Miami, FL, USA). Furthermore, Bio-TEM was used to observe the location of the nanoparticles intracellularly as well as extracellularly.
In vitro biocompatibility
Whole blood was collected from healthy mice via orbit and the red blood cells (RBCs) were separated for hemolysis test. PTX-GHP-VEGF (0.8 mL, in PBS) at predetermined concentrations (25, 50, 100, 200, and 400 μg/mL) was first mixed with 0.2 mL RBCs. Then, the mixture was incubated at 37°C for 1 hour. As the positive and negative controls, purified RBCs were incubated with water and PBS, respectively. Afterwards, the suspensions above were ultracentrifuged (10,000 rpm, 1 minute). Finally, the absorbance of the supernatants was monitored at 541 nm by a UV–vis spectrometer. The following equation represented the hemolytic percentage (HP).
|
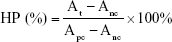
where At, Apc, and Anc are the absorbance of the supernatant at 541 nm of the test sample, positive, and negative controls, respectively.
Next, a standard CCK-8 assay (BestBio, Shanghai, China) was used to evaluate the cytotoxicity of the nanoparticles. SW-13 cells (100 μL, 1×105 cells/mL) were seeded in a 96-well plate. After 24 hours cultivation, the old media were discarded and GHP-VEGF at predetermined concentrations (0.025, 0.05, 0.1, 0.2, and 0.4 mg/mL) in fresh media was incubated with SW-13 cells. After 24 hours treatment in a cell incubator, CCK-8 reagent (10% CCK-8 +90% L-15 media) was added into each well for further 30 minutes incubation at 37°C. The absorbance value at 450 nm was used to characterize the cell viability, which was measured by a Multilabel Plate Reader (EnVision; PerkinElmer Inc).
In vitro anticancer efficacy
SW-13 cells were seeded in 96-well plates (2×104 cells/well) for 24 hours incubation. Free PTX, GHP-VEGF, PTX-GHP, and PTX-GHP-VEGF were serially diluted in cell culture media and dissolved directly into cell media. After 6 hours incubation, the old media were discarded. After addition of fresh media, cells were irradiated with or without an 808 nm laser (1 W/cm2, 5 minutes) at 0, 24, and 48 hours posttreatment with samples. Afterward, cell viability was measured by a CCK-8 assay as mentioned in the ‘In vitro biocompatibility’ section.
Next, live and dead cells were co-stained by calcein-AM/propidium iodide (PI). SW-13 cells were pre-seeded in 35 mm plates at a density of 1×106 cells/plate and were treated with control, control + NIR, PTX-GHP-VEGF, or PTX-GHP-VEGF + NIR. After 6 hours incubation, cells were irradiated under the 808 nm laser (1 W/cm2) for 5 minutes. Cells were stained with calcein-AM/PI for 30 minutes, washed with PBS to remove excess dye solution, and then imaged using a confocal laser scanning microscope (calcein-AM lex =488 nm, lem =515 nm; PI lex =535 nm, lem =617 nm).
Lastly, cell death type was detected using an Annexin V-FITC/PI apoptosis detection kit (BestBio), which was analyzed by a FCM. SW-13 cells were incubated in 6-well plates (1×104 cells/well) for 24 hours. Cells were treated with control, control + NIR, free PTX, PTX-GHP-VEGF, and PTX-GHP-VEGF + NIR at the same PTX concentration of 50 μg/mL with continuous 48 hours incubation. According to the operating instruction, cells were collected, washed, and stained by the Annexin V-FITC/PI kit and analyzed by the FCM, successively.
Animal model and in vivo NIR thermal imaging
BALB/c nude mice were obtained from Charles River Laboratories (Beijing, China). All welfare and experimental procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of Qingdao University Laboratory, and approved by the Ethics Committee of Qingdao University Laboratory. SW-13 subcutaneous tumors were established by subcutaneous injections of 1×106 SW-13 cells in 100 μL PBS onto the back of BALB/c nude mice.
Tumor bearing mice (n=5) were imaged by an NIR thermal imaging system before and at 12, 24, 36, 48, and 60 hours post-intravenous injection of saline, PTX-GHP, or PTX-GHP-VEGF under 808 nm laser irradiation (1 W/cm2) for 5 minutes.
In vivo anticancer efficacy
Tumor bearing mice in the different groups (n=5) were treated by saline, free PTX, GHP-VEGF + NIR, PTX-GHP + NIR, PTX-GHP-VEGF, or PTX-GHP-VEGF + NIR (dose equal to free PTX). NIR laser (808 nm, 1 W/cm2) was used for 5 minutes on the tumor region at 0 and 24 hours post-intravenous injection of these samples. During the treatment, tumor size of the mice was observed every 4 days and calculated as volume = (tumor length) × (tumor width)2/2. The relative tumor volume was equal to the tumor volume at a given time point divided by the tumor volume prior to the initial treatment.
Biosafety evaluation
Healthy BALB/c nude mice (n=5) were intravenously injected with PTX-GHP-VEGF at a dose of 15 mg/kg, which was two-fold of the treatment and imaging dose. At 1 and 30 days postinjection, the PTX-GHP-VEGF-treated mice and untreated mice were sacrificed and blood was collected for serum biochemistry assay and complete blood panel test. In addition, major organs including the heart, liver, spleen, lung, and kidney of those mice were harvested, fixed in 4% formalin, embedded in paraffin, stained with hematoxylin and eosin (H&E), and examined under a digital microscope.
Results and discussion
Preparation and characterization of PTX-GHP-VEGF
The synthesis scheme of PTX-GHP-VEGF is shown in Figure 1. GO nanosheets absorptive PTX were coated with HSA. The surface was then conjugated with mAbVEGF via PEG linker to form targeted nanoparticles (PTX-GHP-VEGF). Transmission electron microscopy (TEM) images were also taken of GO nanosheets and PTX-GO nanosheets (Figure S1). After coating with HSA, PTX-GO-HSA as well as PTX-GHP-VEGF displayed uniform spherical shape on representative scanning electron microscopy and TEM images (Figures 2A, B and S1). The high-resolution magnification TEM image obviously showed the core-shell structure of the nanoparticles. These results indicate that the HSA can be a shell of PTX-GO core, forming a spherical particle. Particle size and zeta potential analysis obtained from the Nano ZS Zetasizer showed that most PTX-GHP-VEGF particles had a diameter of 191±5 nm and zeta potential of −26.8±2.3 mV (Figure 2C and D). The UV–vis spectra of PTX, GO, and PTX-GHP-VEGF showed that PTX-GHP-VEGF had the same feature peak of PTX and GO at 233 and 230 nm, respectively (Figure 3A), demonstrating that PTX-GO had been encapsulated within PTX-GHP-VEGF and that encapsulation did not influence the absorbance intensity of PTX and GO. After calculation, the encapsulation efficiency of PTX on GO was 85.6%±1.1%, and on GO-HSA-PEG was 55.02%±3.1%.
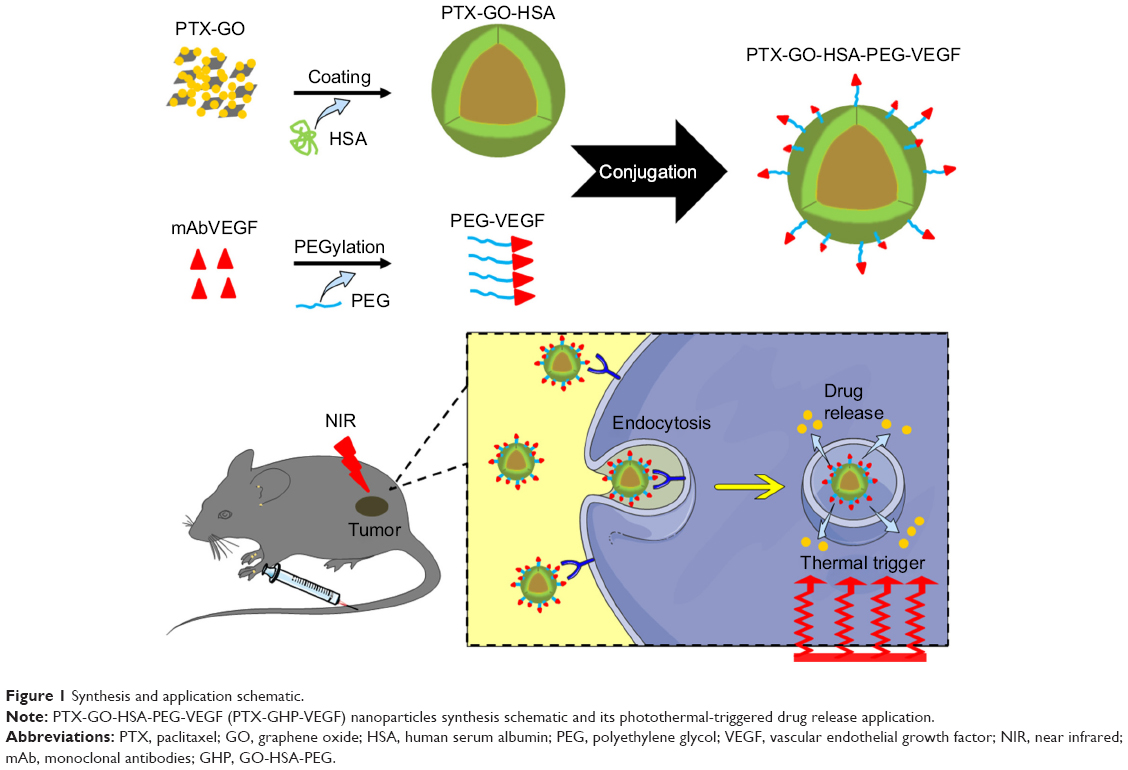 | Figure 1 Synthesis and application schematic. |
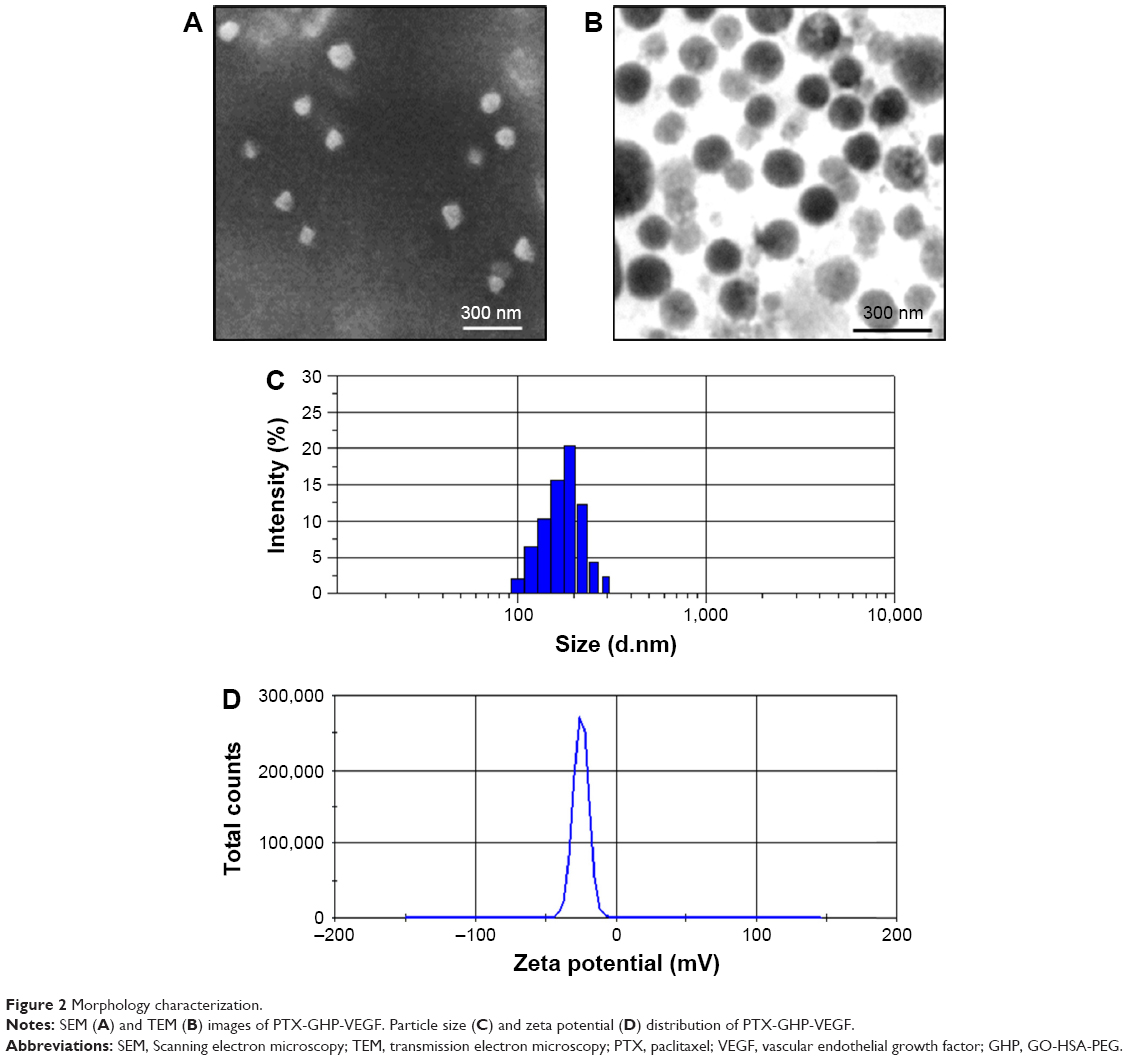 | Figure 2 Morphology characterization. |
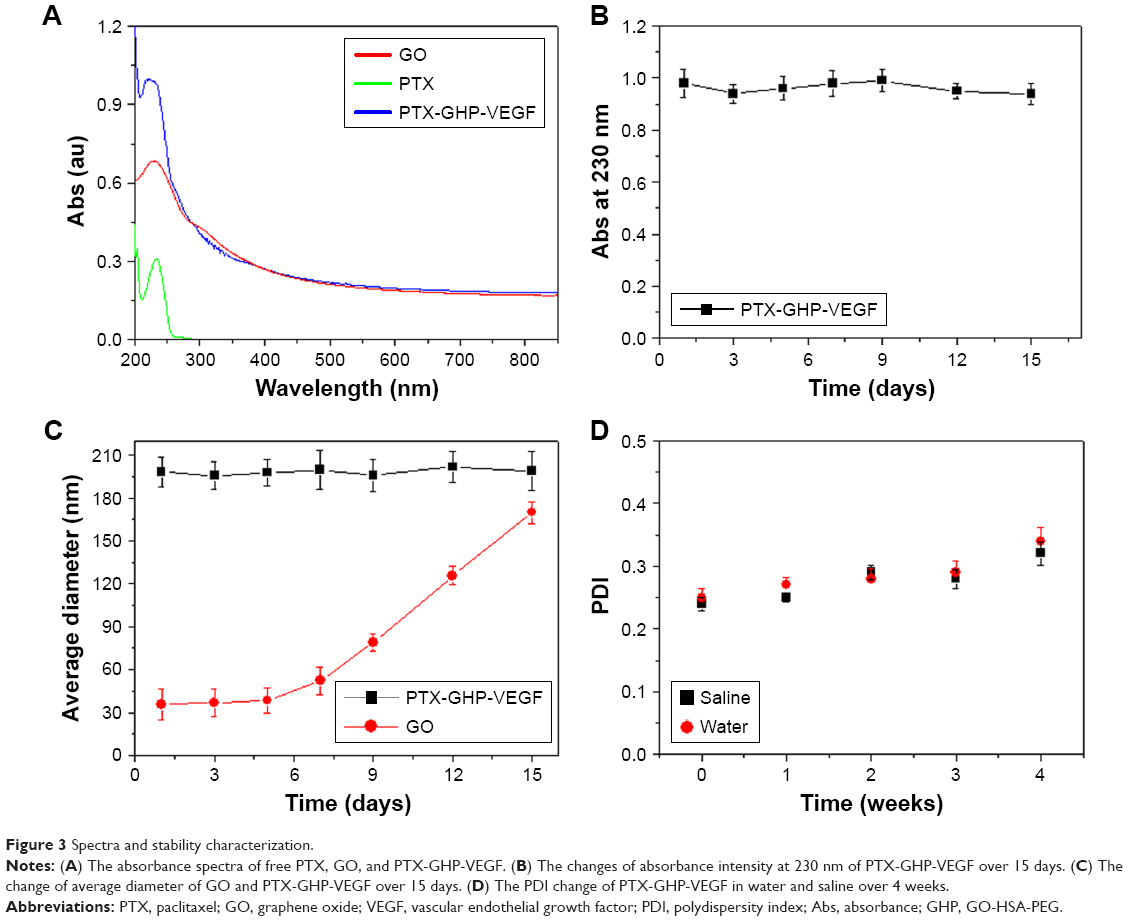 | Figure 3 Spectra and stability characterization. |
After 2 weeks in storage, PTX-GHP-VEGF dissolved in water displayed no obvious changes in absorbance at 230 nm, average size, and polydispersity index (Figure 3B–D); yet, the average diameter of GO increased with time, indicating greater stability as well as dispersity of PTX-GHP-VEGF.
Photothermal properties of PTX-GHP-VEGF
Temperature changes were measured and the corresponding thermal images were obtained for water, PTX-GHP-VEGF without GO (PTX-HP-VEGF), PTX-GHP-VEGF and GO aqueous solution at the same concentrations of GO (10 μg/mL) under 5 minutes NIR laser irradiation (808 nm, 1 W/cm2). After 5 minutes of NIR irradiation, the maximum rising temperature reached about 22°C for PTX-GHP-VEGF and GO solution. As a control, the temperature of water and PTX-HP-VEGF without GO increased by about 2°C under the same conditions. The results indicate the excellent photothermal effects with low concentration of GO (Figure 4A and B). The UV–vis spectra of PTX-GHP-VEGF before and after 5 minutes of NIR irradiation indicated that irradiation did not influence the absorbance intensity (Figure 4C). Moreover, the TEM images of PTX-GHP-VEGF before and after irradiation showed complete and dispersed spherical shape (Figure 4D), demonstrating remarkable photostability.
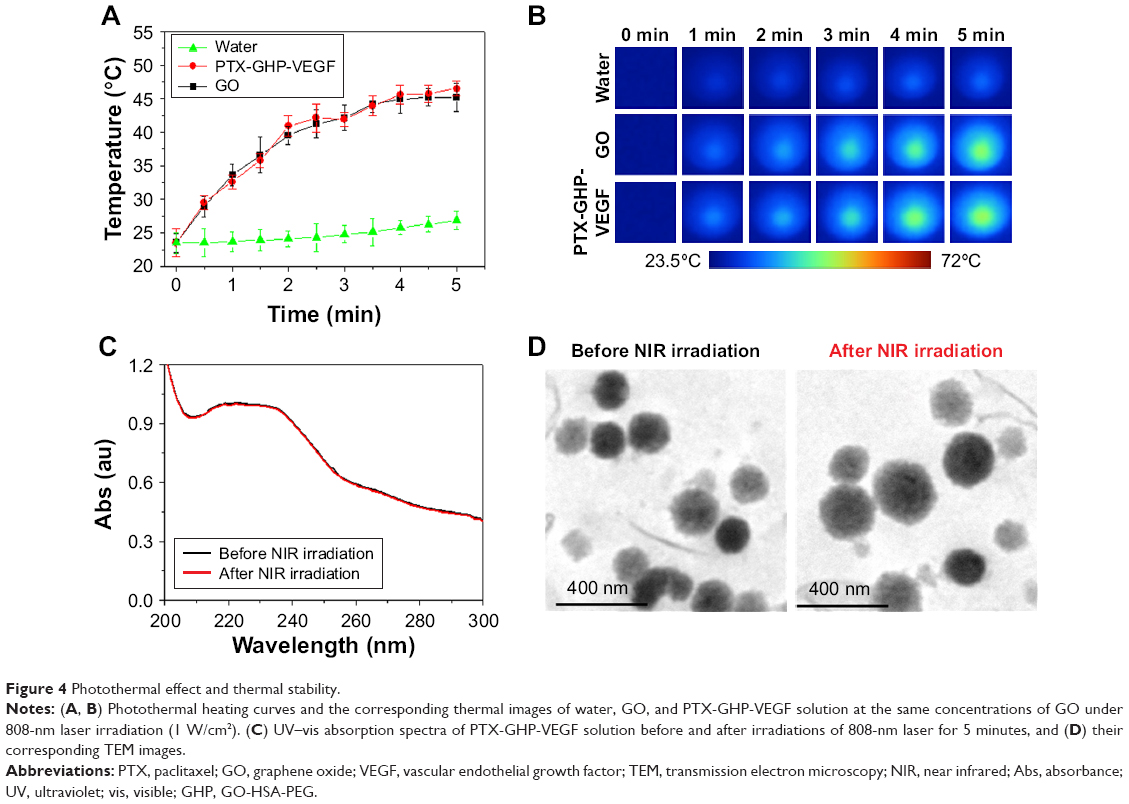 | Figure 4 Photothermal effect and thermal stability. |
Photothermal-triggered drug release
The release of PTX was 6.2% from PTX-GHP-VEGF for 6 hours at pH 7.4 in physiological solution, while at the same condition, PTX release was significantly increased to 20.1% when PTX-GHP-VEGF was irradiated for 5 minutes (808 nm, 1 W/cm2) (Figure 5A). After additional three cycles of 5-minute irradiation at 12, 24, and 36 hours, the accumulated release of PTX approached about 81.2% after 48 hours, much higher than that of PTX released without NIR irradiation (11.2%). In addition, without NIR irradiation, PTX-GHP-VEGF showed 15.1% PTX release after 48 hours at pH 6.5, which was slightly higher than that at pH values of 7.4 and 9.0 (Figure S2). These results indicate that apart from the acidic environment and tumor endogenous factors, NIR light-induced local hyperthermia can function as a stimulus to on–off control the PTX release from PTX-GHP-VEGF. Furthermore, PTX-GHP-VEGF incubated in different temperatures, from 25°C to 55°C, showed an increase in the diameter of PTX-GHP-VEGF from 35°C to 55°C (Figure 5B). The release process could have two potential steps: 1) PTX was released from the GO surface due to the heat generated by GO when absorbing NIR light, which weakens the non-covalent adsorption interactions between PTX and the GO surface, and 2) the released PTX then leaked from HSA nanoparticles, which can be attributed to the heat-induced expansion of HSA nanoparticles.32,34,43
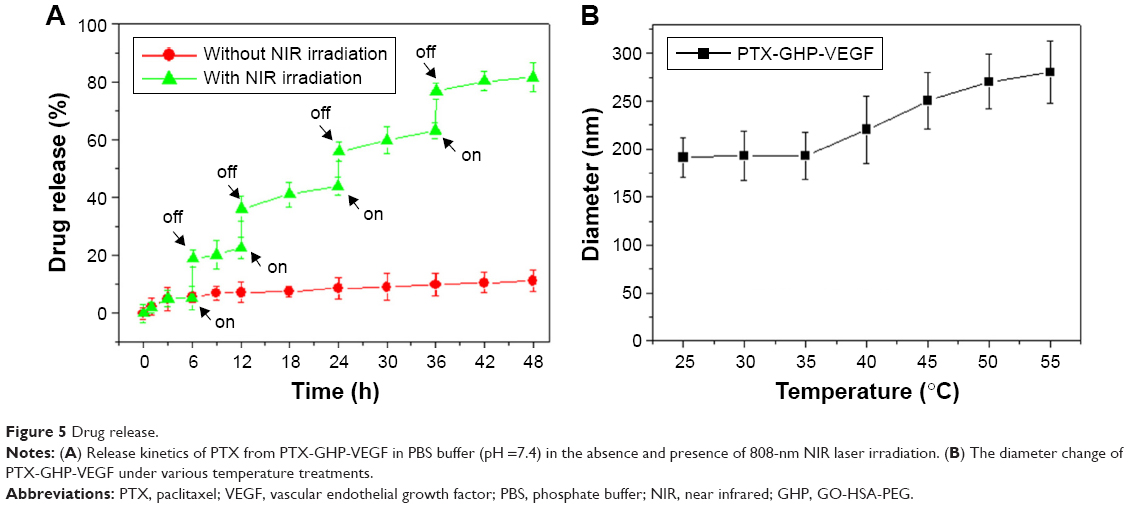 | Figure 5 Drug release. |
In vitro biocompatibility
The cytotoxicity of the nanoparticles on SW-13 cells was detected by the CCK-8 assay (Figure 6A). GHP-VEGF as the target carrier without PTX below 400 μg/mL exhibited negligible toxicity to SW-13 cells. Moreover, no obvious hemolysis phenomenon was observed for PTX-GHP-VEGF-treated RBCs (<400 μg/mL), similar to that of the PBS-treated group (Figure 6B), suggesting excellent hemocompatibility. These results indicate that PTX-GHP-VEGF has remarkable in vitro biocompatibility.
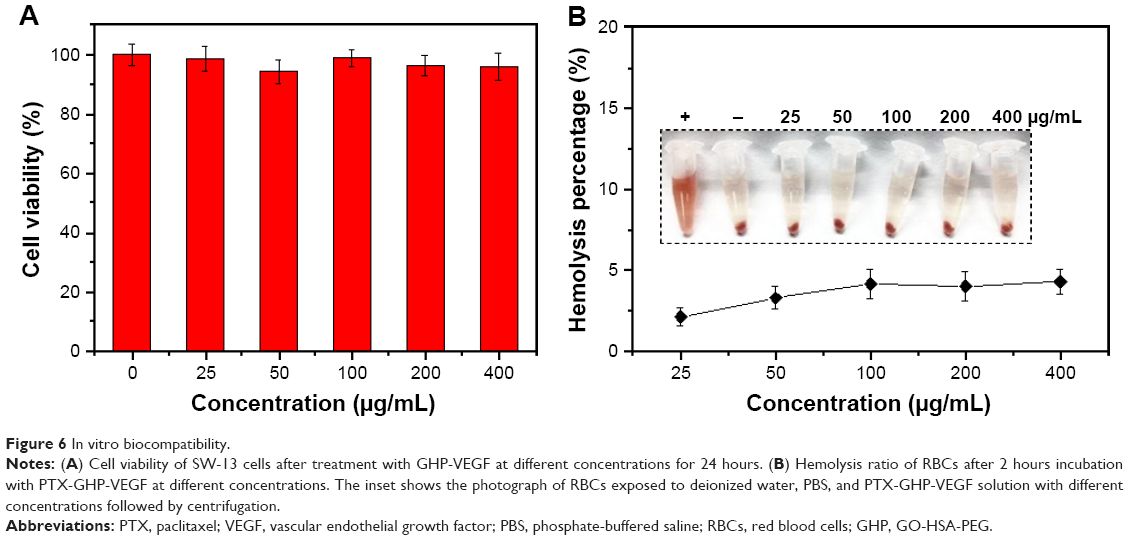 | Figure 6 In vitro biocompatibility. |
Cellular uptake
To evaluate cellular uptake, PTX-GHP and PTX-GHP-VEGF were labeled with FITC, a fluorescence dye. After incubation with PTX-GHP and PTX-GHP-VEGF for 3 hours, FITC fluorescence was much stronger inside the cytoplasm in PTX-GHP-VEGF-treated cells than that of PTX-GHP- and free FITC-treated cells (Figure 7A). The flow cytometry results further showed the cellular uptake ratio of PTX-GHP-VEGF was 49.6%±4.1%, which was higher than that of PTX-GHP (13.6%±3.9%) (Figure 7B–E). According to the Bio-TEM images, the cytoplasm of SW-13 cells showed more PTX-GHP-VEGF nanoparticles (Figure 7F), as confirmed from high-magnification images (Figure 7G). These results indicate that mAbVEGF facilitated the uptake of PTX-GHP-VEGF into the cells, likely due to the receptor-mediated endocytosis pathway.
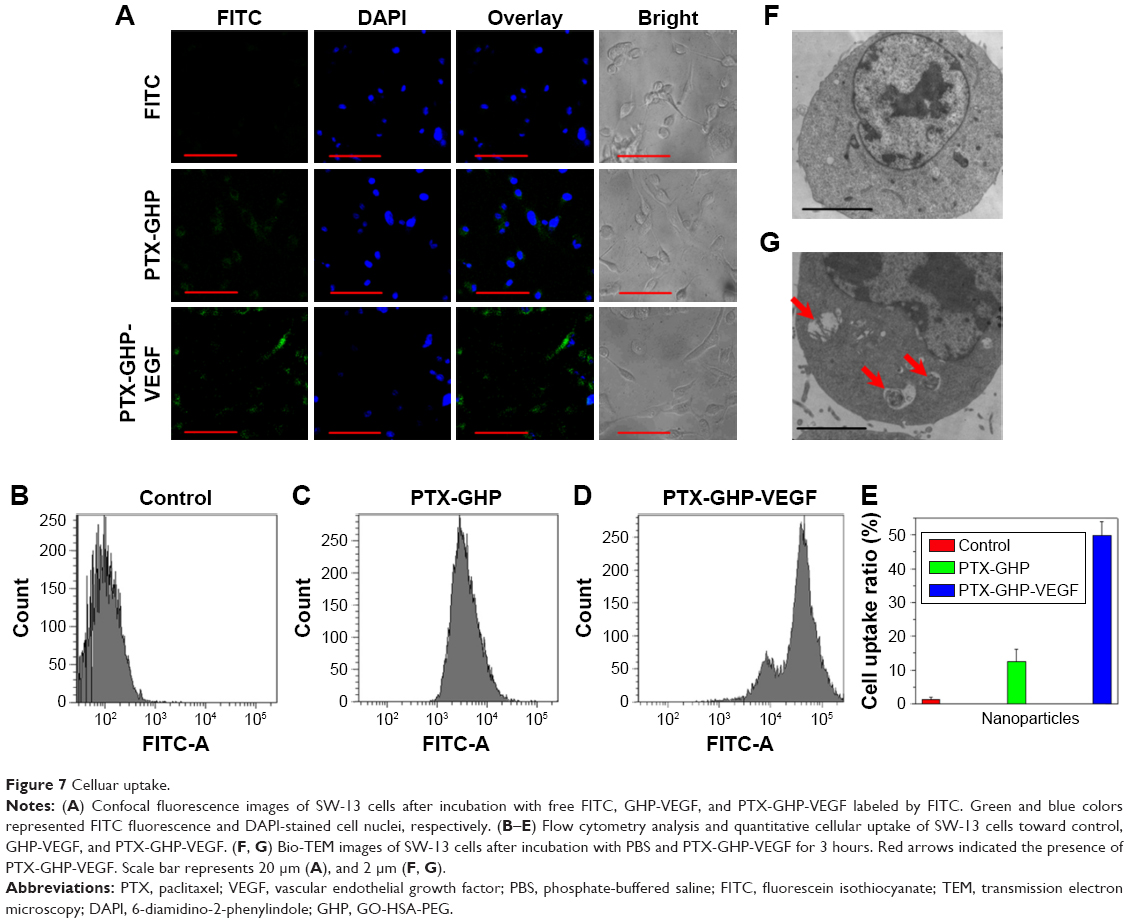 | Figure 7 Celluar uptake. |
In vitro cytotoxicity and detection of apoptosis
Treatment with free PTX, PTX-GHP, and PTX-GHP-VEGF at PTX concentrations ranging from 0 to 100 μg/mL decreased cell viability in a dose- and time-dependent manner from 0 to 48 hours (Figure 8A–C). However, under the same PTX concentration, PTX-GHP-VEGF induced a higher cell death rate due to the more efficient mAbVEGF-targeted killing effect of PTX-GHP-VEGF in SW-13 cells. No obvious cytotoxicity was observed from GHP-VEGF on SW-13 cells after 48 hours of treatment (Figure 8A–C), while, after irradiating with NIR laser for 5 minutes, cell viability showed a dose-independent decrease (Figure 8D), indicating slight photothermal cytotoxicity of GO-based nanoparticles at initial treatment (0 hours). After two cycles of 5-minute NIR irradiation at subsequent 24 and 48 hours, PTX-GHP-VEGF-treated cells showed a considerably greater cell viability decrease compared to PTX-GHP and PTX in a dose- and time-dependent manner (Figure 8E and F).
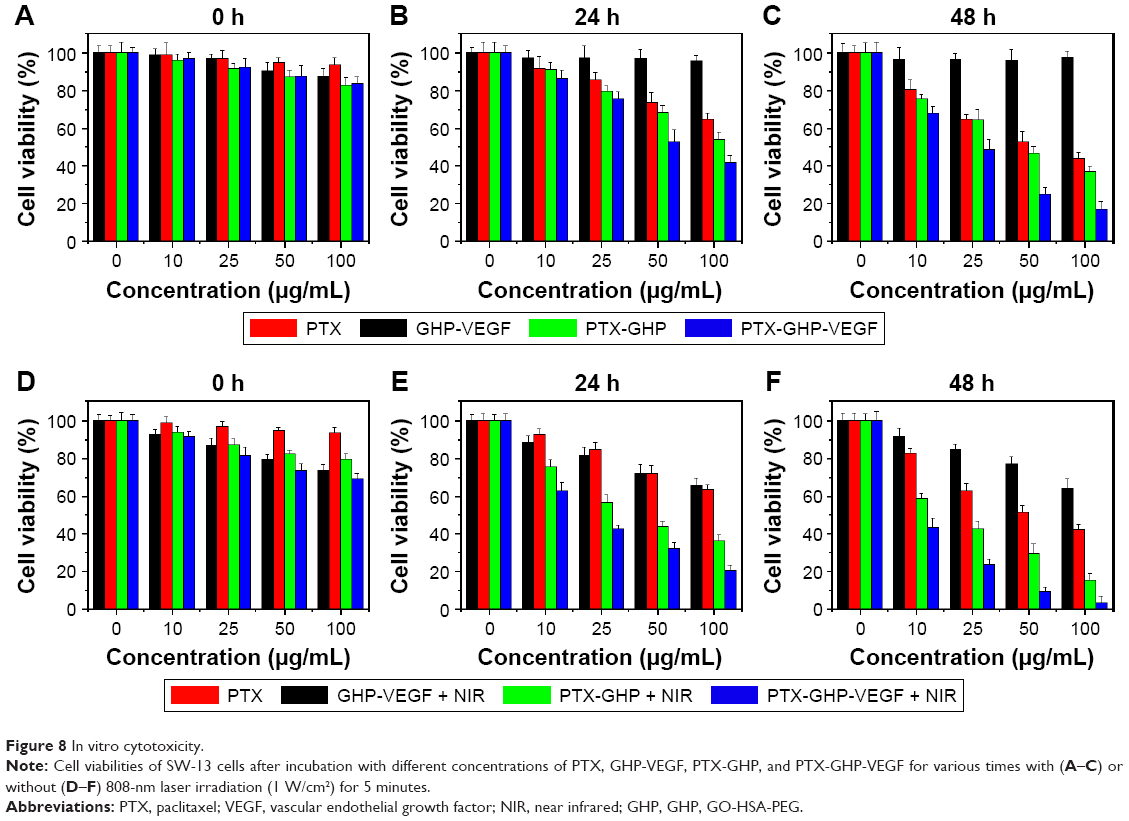 | Figure 8 In vitro cytotoxicity. |
Calcein-AM/PI co-staining was further used to investigate the cell killing effect of PTX-GHP-VEGF in vitro. Cells displayed green fluorescence in the control and control + NIR groups, suggesting no effect of pure laser irradiation on cells (Figure 9A and B). In the PTX-GHP-VEGF-treated group, some cells were dead and showed red fluorescence, while with laser irradiation, almost all cells were killed (Figure 9C and D), which corresponded with the results from the CCK-8 assay (Figure 8F).
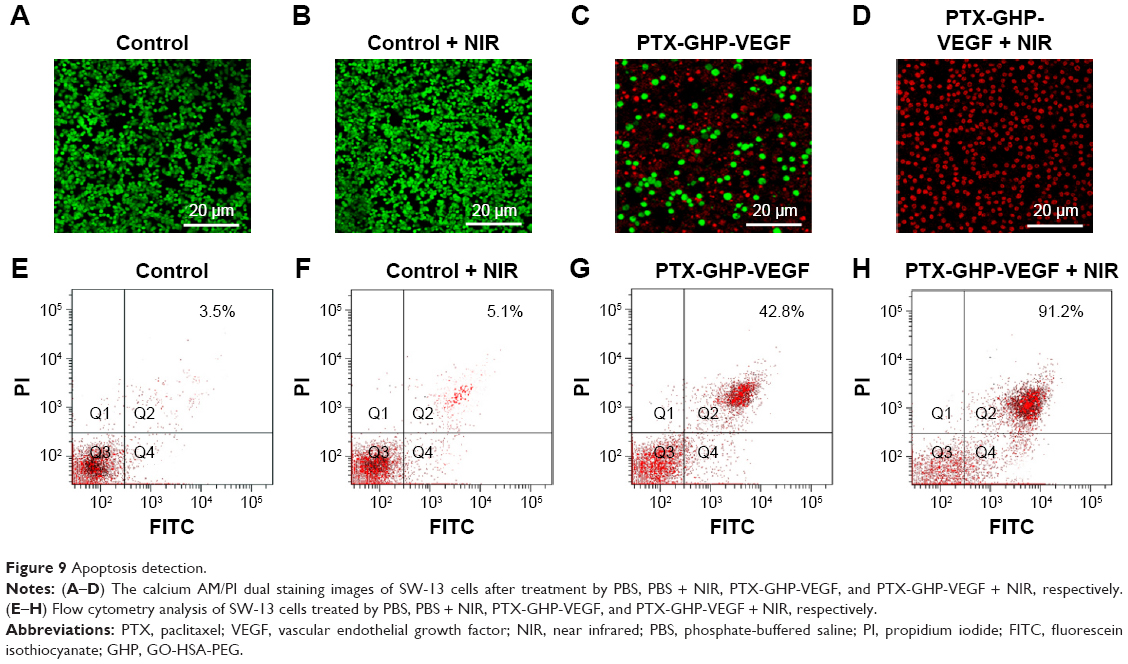 | Figure 9 Apoptosis detection. |
In addition, the type of cell death induced by PTX-GHP-VEGF was further detected using an Annexin V-FITC/PI double staining kit and analyzed by FCM. On the basis of literature, the signal in Q2 + Q4 region in the representative dot plots are defined as apoptotic cells.9 Cells treated with control and control + NIR showed few dead cells (Figure 9E and F), while PTX-GHP-VEGF + NIR-treated cells showed a 91.2% apoptosis rate (Figure 9H), which was higher than PTX-GHP-VEGF (42.8%) and free PTX (32.6%)-treated groups (Figures 9G and S3). These findings suggest a significant synergistic cell killing effect of PTX-GHP-VEGF with NIR laser irradiation, which could be mainly ascribed to hyperthermia triggering more PTX release for enhanced chemotherapy.
NIR thermal imaging guided tumor therapy
The accumulation of nanoparticles in tumors at various time points following an intravenous injection of PTX-GHP and PTX-GHP-VEGF was then assessed. After an intravenous injection of saline, PTX-GHP, or PTX-GHP-VEGF, the tumor regions of the mice in different groups were irradiated for 5 minutes by NIR laser at various time points. The temperature of the tumor was monitored using a thermal imager (Figure 10A). The thermal images and statistical results showed that the maximum temperature of tumors post-PTX-GHP-VEGF injection was reached at 24 hours, which was higher than that of the control and PTX-GHP-treated groups (Figure 10A and B). These results indicated that PTX-GHP-VEGF can reach the maximum accumulation in tumor regions at 24 hours postinjection. These may be caused by the PEG coating of the nanoparticles that can improve blood circulation duration and effectively decrease macrophage clearance of nanoparticles by the reticuloendothelial system, the enhanced permeability and retention (EPR) effect of solid tumor, as well as the mAbVEGF tumor targeting effect enhancing tumor accumulation.44–47
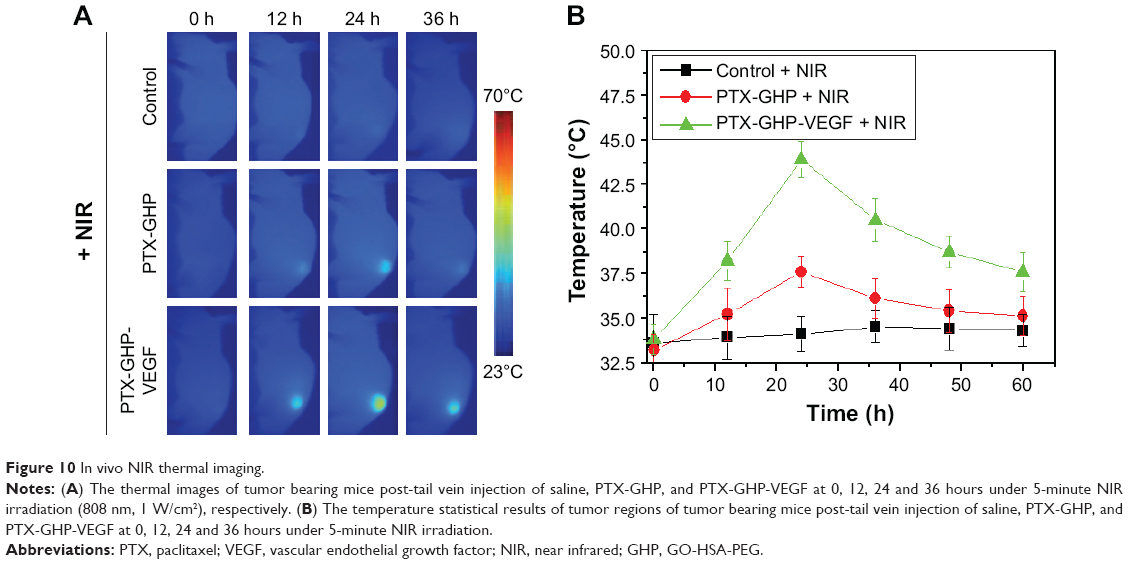 | Figure 10 In vivo NIR thermal imaging. |
According to the imaging diagnosis result, NIR irradiation treatment was conducted at 24 hours post-intravenous injection of samples. Intravenous injection of PTX-GHP-VEGF with or without 5-minute NIR irradiation exhibited a significant difference (Figure 11A). In the former group, it showed noticeable tumor growth inhibition and no tumor relapse after about 1 month of treatment. However, in the latter group, no tumor growth inhibition was observed compared to the control group. Because of the EPR effect, PTX-GHP without targeting moiety under NIR irradiation displayed in vivo tumor inhibition but not completely, indicating that active targeting of nanoparticles plays an important role in tumor therapy. In addition, GHP-VEGF + NIR and free PTX showed little tumor inhibition effect. Furthermore, after 100 days of treatment, the survival rate of mice in the PTX-GHP-VEGF + NIR-treated group was 100%, which was higher than that of the other groups (Figure 11B). These results indicate that PTX-GHP-VEGF combined with NIR irradiation features an excellent in vivo anticancer efficacy likely due to active targeting thermal-triggered drug release for chemotherapy.
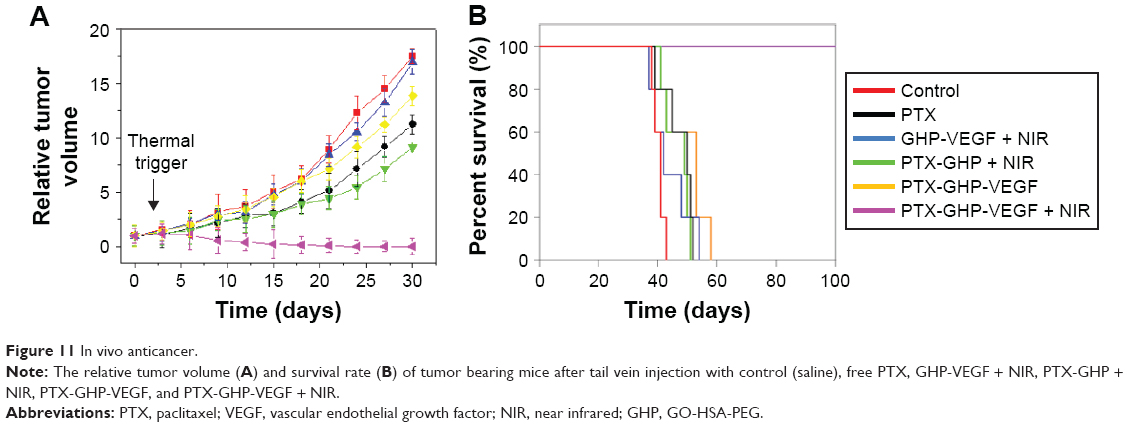 | Figure 11 In vivo anticancer. |
Blood examination and histology
After about 1 month of treatment, no significant body weight loss was observed in all groups (Figure 12A). Furthermore, blood chemistry analysis results and complete blood panel tests were acquired from PTX-GHP-VEGF-injected (15 mg/kg) healthy BALB/c mice 1 and 30 days postinjection. All test indexes, including aspartic transaminase, alanine transaminase, white blood cells, RBCs, hemoglobin, mean corpuscular hemoglobin, mean PLT volume, hematocrit, mean corpuscular volume, PLTs, and mean corpuscular hemoglobin concentration were within reference ranges for healthy BALB/c mice (Figure 12B–D). Major organs (including heart, liver, spleen, lungs, and kidneys) in the mice of each group were stained by H&E to further evaluate the tissue toxicity of PTX-GHP-VEGF. None of the tested organs showed significant histological lesions (Figure 12E). Taken together, PTX-GHP-VEGF exhibited remarkable in vivo biocompatibility characteristics, which will be beneficial for its future nanomedicine applications.
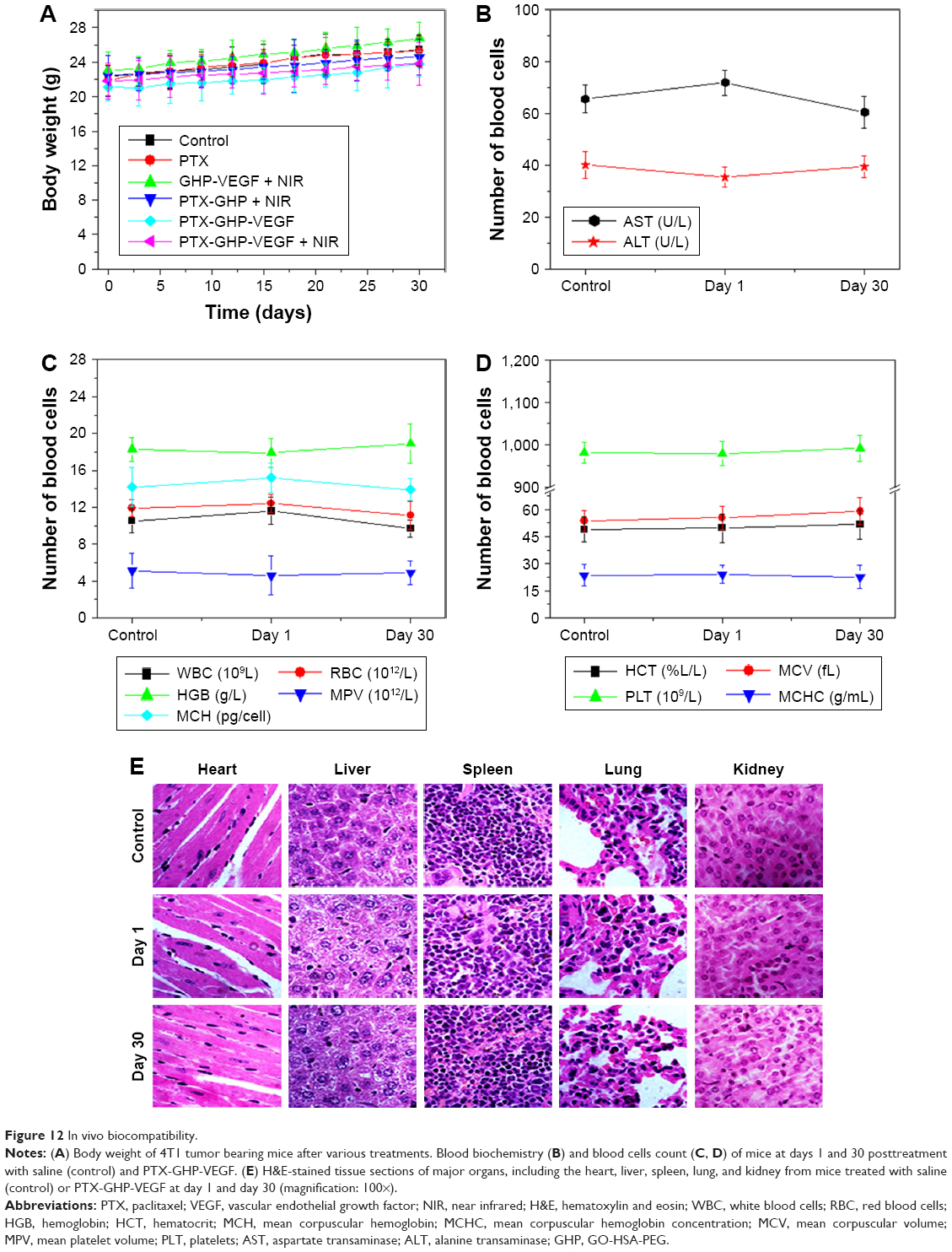 | Figure 12 In vivo biocompatibility. |
Conclusion
Novel mAbVEGF-conjugated and photothermal-triggered drug release HSA nanoparticles containing PTX-GO (PTX-GHP-VEGF) were designed and prepared. The dual-carriers, GO and HSA, respectively, functioned as a photothermal agent and biocompatible shell for the nanocomposite. Under NIR irradiation, the heat generated by GO triggered PTX molecules to be released from GO nanosheets and then released from the HSA shell. The targeting effect of the PTX-GHP-VEGF was confirmed by fluorescence and TEM images in vitro. The targeted photothermal-triggered tumor chemotherapy effect was evaluated and PTX-GHP-VEGF showed significant cell killing ability in vitro and tumor suppression ability in vivo with 100% survival rate over 100 days and no relapse. Moreover, the PTX-GHP-VEGF showed high biosafety with remarkable biocompatibility as confirmed by hematology and histological analyses. Considering all these findings, the development of PTX-GHP-VEGF is an attractive strategy for chemotherapeutic drug delivery to tumors for efficient photothermal controllable tumor therapy.
Acknowledgment
The authors acknowledge Dr Xu from the Department of Pathology, the Affiliated Hospital of Qingdao University for his kind help in H&E staining experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
Xu Y, Zhu Y, Shen Z, et al. Significance of heparanase-1 and vascular endothelial growth factor in adrenocortical carcinoma angiogenesis: potential for therapy. Endocrine. 2011;40(3):445–451. | ||
O’Sullivan C, Edgerly M, Velarde M, et al. The VEGF inhibitor axitinib has limited effectiveness as a therapy for adrenocortical cancer. J Clin Endocrinol Metab. 2014;99(4):1291–1297. | ||
Terzolo M, Zaggia B, Allasino B, De Francia S. Practical treatment using mitotane for adrenocortical carcinoma. Curr Opin Endocrinol Diabetes Obes. 2014;21(3):159–165. | ||
Germano A, Rapa I, Volante M, et al. Cytotoxic activity of gemcitabine, alone or in combination with mitotane, in adrenocortical carcinoma cell lines. Mol Cell Endocrinol. 2014;382(1):1–7. | ||
Glover AR, Zhao JT, Gill AJ, et al. MicroRNA-7 as a tumor suppressor and novel therapeutic for adrenocortical carcinoma. Oncotarget. 2015;6(34):36675–36688. | ||
Demeure MJ, Stephan E, Sinari S, et al. Preclinical investigation of nanoparticle albumin-bound paclitaxel as a potential treatment for adrenocortical cancer. Ann Surg. 2012;255(1):140–146. | ||
Subramanian C, Kuai R, Zhu Q, et al. Synthetic high-density lipoprotein nanoparticles: a novel therapeutic strategy for adrenocortical carcinomas. Surgery. 2016;159(1):284–295. | ||
Hantel C, Jung S, Mussack T, Reincke M, Beuschlein F. Liposomal polychemotherapy improves adrenocortical carcinoma treatment in a preclinical rodent model. Endocr Relat Cancer. 2014;21(3):383–394. | ||
Xin Y, Liu T, Yang C. Development of PLGA-lipid nanoparticles with covalently conjugated indocyanine green as a versatile nanoplatform for tumor-targeted imaging and drug delivery. Int J Nanomedicine. 2016;11:5807–5821. | ||
Gwak HR, Kim S, Dhanasekaran DN, Song YS. Resveratrol triggers ER stress-mediated apoptosis by disrupting N-linked glycosylation of proteins in ovarian cancer cells. Cancer Lett. 2016;371(2):347–353. | ||
Gurunathan S, Han JW, Kim ES, Park JH, Kim JH. Reduction of graphene oxide by resveratrol: a novel and simple biological method for the synthesis of an effective anticancer nanotherapeutic molecule. Int J Nanomedicine. 2015;10:2951–2969. | ||
Jayaprakasha GK, Murthy KNC, Patil BS. Enhanced colon cancer chemoprevention of curcumin by nanoencapsulation with whey protein. Eur J Pharmacol. 2016;789:291–300. | ||
Wang D, Zhou J, Chen R, et al. Controllable synthesis of dual-MOFs nanostructures for pH-responsive artemisinin delivery, magnetic resonance and optical dual-model imaging-guided chemo/photothermal combinational cancer therapy. Biomaterials. 2016;100:27–40. | ||
Koudelka Š, Turánek J. Liposomal paclitaxel formulations. J Control Release. 2012;163(3):322–334. | ||
Zhu M, Li W, Lu Y, et al. Alpha fetoprotein antagonizes apoptosis induced by paclitaxel in hepatoma cells in vitro. Sci Rep. 2016;6:26472. | ||
Geller MA, Cooley S, Judson PL, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. 2011;13(1):98–107. | ||
Bradley JD, Paulus R, Komaki R, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): a randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015;16(2):187–199. | ||
Berruti A, Sperone P, Ferrero A, et al. Phase II study of weekly paclitaxel and sorafenib as second/third-line therapy in patients with adrenocortical carcinoma. Eur J Endocrinol. 2012;166(3):451–458. | ||
Xiao K, Luo J, Fowler WL, et al. A self-assembling nanoparticle for paclitaxel delivery in ovarian cancer. Biomaterials. 2009;30(30):6006–6016. | ||
Zhang C, Lu T, Tao J, Wan G, Zhao H. Co-delivery of paclitaxel and indocyanine green by PEGylated graphene oxide: a potential integrated nanoplatform for tumor theranostics. RSC Adv. 2016;6(19):15460–15468. | ||
Gogaté US, Schwartz PA, Agharkar SN. Effect of unpurified Cremophor EL on the solution stability of paclitaxel. Pharm Dev Technol. 2009;14(1):1–8. | ||
Kim SC, Kim DW, Shim YH, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72(1):191–202. | ||
Angelopoulou A, Voulgari E, Diamanti EK, Gournis D, Avgoustakis K. Graphene oxide stabilized by PLA–PEG copolymers for the controlled delivery of paclitaxel. Eur J Pharm Biopharm. 2015;93:18–26. | ||
Kinoshita R, Ishima Y, Chuang VTG, et al. Improved anticancer effects of albumin-bound paclitaxel nanoparticle via augmentation of EPR effect and albumin-protein interactions using S-nitrosated human serum albumin dimer. Biomaterials. 2017;140:162–169. | ||
Ruttala HB, Ko YT. Liposome encapsulated albumin-paclitaxel nanoparticle for enhanced antitumor efficacy. Pharm Res. 2015;32(3):1002–1016. | ||
Zhang W, Guo Z, Huang D, Liu Z, Guo X, Zhong H. Synergistic effect of chemo-photothermal therapy using PEGylated graphene oxide. Biomaterials. 2011;32(33):8555–8561. | ||
Chung C, Kim YK, Shin D, Ryoo S, Hong B, Min D. Biomedical applications of graphene and graphene oxide. Acc Chem Res. 2013;46(10):2211–2224. | ||
Liu Z, Robinson JT, Sun X, Dai H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J Am Chem Soc. 2008;130(33):10876–10877. | ||
Chen J, Liu C, Zeng G, et al. Indocyanine green loaded reduced graphene oxide for in vivo photoacoustic/fluorescence dual-modality tumor imaging. Nanoscale Res Lett. 2016;11(1):85. | ||
Wang YW, Fu YY, Peng Q, et al. Dye-enhanced graphene oxide for photothermal therapy and photoacoustic imaging. J Mater Chem B. 2013;1(42):5762–5767. | ||
Yang X, Zhang X, Ma Y, Huang Y, Wang Y, Chen Y. Superparamagnetic graphene oxide–Fe3O4 nanoparticles hybrid for controlled targeted drug carriers. J Mater Chem. 2009;19(18):2710–2714. | ||
Kurapati R, Raichur AM. Near-infrared light-responsive graphene oxide composite multilayer capsules: a novel route for remote controlled drug delivery. Chem Commun. 2013;49(7):734–736. | ||
Muthoosamy K, Abubakar IB, Bai RG, Loh HS, Manickam S. Exceedingly higher co-loading of curcumin and paclitaxel onto polymer-functionalized reduced graphene oxide for highly potent synergistic anticancer treatment. Sci Rep. 2016;6:32808. | ||
Kim H, Lee D, Kim J, Kim TI, Kim WJ. Photothermally triggered cytosolic drug delivery via endosome disruption using a functionalized reduced graphene oxide. ACS Nano. 2013;7(8):6735–6746. | ||
Hu W, Peng C, Lv M, et al. Protein corona-mediated mitigation of cytotoxicity of graphene oxide. ACS Nano. 2011;5(5):3693–3700. | ||
Sarfraz M, Afzal A, Raza SM, et al. Liposomal co-delivered oleanolic acid attenuates doxorubicin-induced multi-organ toxicity in hepatocellular carcinoma. Oncotarget. 2017;8(29):47136–47153. | ||
Akash MS, Rehman K, Chen S. Polymeric-based particulate systems for delivery of therapeutic proteins. Pharm Dev Technol. 2016;21(3):367–378. | ||
Ma C, Shi Y, Pena DA, Peng L, Yu G. Thermally responsive hydrogel blends: a general drug carrier model for controlled drug release. Angew Chem Int Ed Engl. 2015;127(25):7484–7488. | ||
Yu Z, Yu M, Zhang Z, Hong G, Xiong Q. Bovine serum albumin nanoparticles as controlled release carrier for local drug delivery to the inner ear. Nanoscale Res Lett. 2014;9(1):343. | ||
Geng T, Zhao X, Ma M, Zhu G, Yin L. Resveratrol-loaded albumin nanoparticles with prolonged blood circulation and improved biocompatibility for highly effective targeted pancreatic tumor therapy. Nanoscale Res Lett. 2017;12(1):437. | ||
Zong Y, Wu J, Shen K. Nanoparticle albumin-bound paclitaxel as neoadjuvant chemotherapy of breast cancer: a systematic review and meta-analysis. Oncotarget. 2017;8(10):17360–17372. | ||
Qu N, Lee RJ, Sun Y, et al. Cabazitaxel-loaded human serum albumin nanoparticles as a therapeutic agent against prostate cancer. Int J Nanomedicine. 2016;11:3451–3459. | ||
Han Y, Shchukin D, Yang J, Simon CR, Fuchs H, Möhwald H. Biocompatible protein nanocontainers for controlled drugs release. ACS Nano. 2010;4(5):2838–2844. | ||
Robinson JT, Welsher K, Tabakman SM, et al. High performance in vivo near-IR (>1 μm) imaging and photothermal cancer therapy with carbon nanotubes. Nano Res. 2010;3(11):779–793. | ||
Yang Q, Jones SW, Parker CL, Zamboni WC, Bear JE, Lai SK. Evading immune cell uptake and clearance requires PEG grafting at densities substantially exceeding the minimum for brush conformation. Mol Pharm. 2014;11(4):1250–1258. | ||
Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9(9–10):1521–1532. | ||
Abakumov MA, Nukolova NV, Sokolsky-Papkov M, et al. VEGF-targeted magnetic nanoparticles for MRI visualization of brain tumor. Nanomedicine. 2015;11(4):825–833. |
Supplementary materials
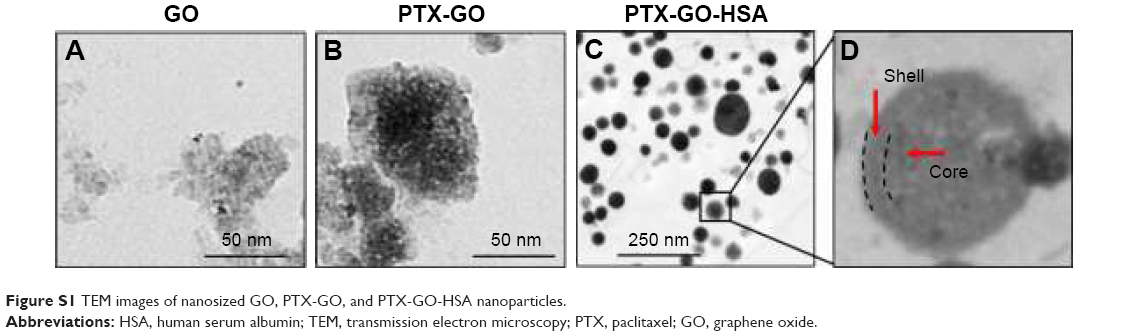 | Figure S1 TEM images of nanosized GO, PTX-GO, and PTX-GO-HSA nanoparticles. |
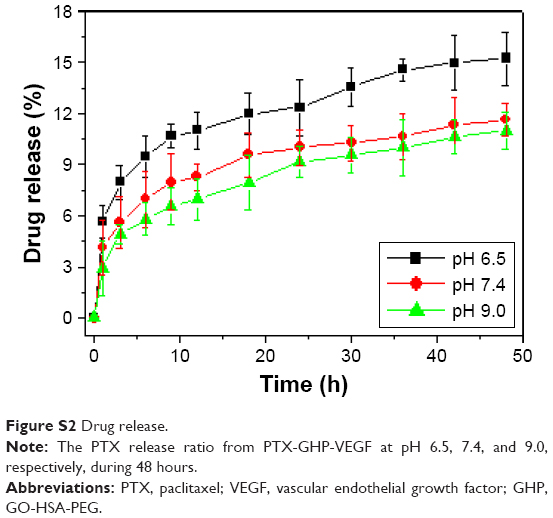 | Figure S2 Drug release. |
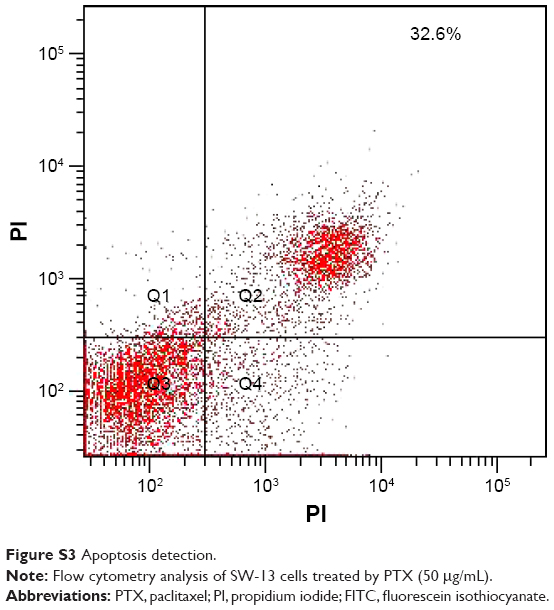 | Figure S3 Apoptosis detection. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.