")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Development, Characterization, and in-vivo Pharmacokinetic Study of Lamotrigine Solid Self-Nanoemulsifying Drug Delivery System
Authors Abdelmonem R , Azer MS , Makky A , Zaghloul A , El-Nabarawi M , Nada A
Received 21 May 2020
Accepted for publication 24 September 2020
Published 19 October 2020 Volume 2020:14 Pages 4343—4362
DOI https://doi.org/10.2147/DDDT.S263898
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Jianbo Sun
Rehab Abdelmonem,1 Marian Sobhy Azer,2 Amna Makky,3 Abdelazim Zaghloul,4 Mohamed El-Nabarawi,3 Aly Nada4
1Department of Industrial Pharmacy, Faculty of Pharmacy, Misr University for Science and Technology, 6th of October City, Giza, Egypt; 2Department of Pharmaceutics, Faculty of Pharmacy, Misr University for Science and Technology, 6th of October City, Giza, Egypt; 3Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 4Department of Pharmaceutics, Faculty of Pharmacy, Kuwait University, Kuwait City, Kuwait
Correspondence: Marian Sobhy Azer
Department of Pharmaceutics, Faculty of Pharmacy, Misr University for Science and Technology, 6th of October City, Giza, Egypt
Tel +2 010 050 529 83
Email [email protected]
Mohamed El-Nabarawi
Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Kasr El-Aini Street, Cairo 11562, Egypt
Tel +2 010 014 244 39
Email [email protected]
Purpose: This study aimed to prepare solid self-nanoemulsified drug delivery system (S-SNEDDS) of lamotrigine (LMG) for enhancing its dissolution and oral bioavailability (BA).
Methods: Nineteen liquid SNEDDS were prepared (R1-R19) using D-optimal design with different ratios of oil, surfactant (S), and cosurfactant (Cos). The formulations were characterized regarding robustness to dilution, droplet size, thermodynamic stability testing, self-emulsification time, in-vitro release in 0.1 N HCl and phosphate buffer (PB; pH 6.8). Design Expert® 11 software was used to select the optimum formulations. Eight S-SNEDDS were prepared (S1-S8) using 23 factorial design, and characterized by differential scanning calorimetry (DSC), powder x-ray diffraction (PXRD), and scanning electron microscopy (SEM). The optimum formulation was chosen regarding in-vitro drug released in 0.1 N HCl and PB, compared to pure LMG and commercial tablet (Lamictal®). The BA of LMG from the optimized S-SNEDDS formulation was evaluated in rabbits compared to pure LMG and Lamictal®.
Results: The optimized S-SNEDDS was S2, consisting of R9 adsorbed on Aeroperl® 300 in a ratio of 1:1, with the best results regarding in-vitro drug released in 0.1 N HCl at 15 min (100%) compared to pure LMG (73.40%) and Lamictal® (79.43%), and in-vitro drug released in PB at 45 min (100%) compared to pure LMG (30.46%) and Lamictal® (92.08%). DSC, PXRD, and SEM indicated that LMG was molecularly dispersed within the solid nano-system. The BA of S2 was increased 2.03 and 1.605 folds compared to pure LMG, and Lamictal®, respectively.
Conclusion: S2 is a promising S-SNEDDS formulation. It can be a potential carrier for improving dissolution, and BA of LMG.
Keywords: antiepileptic drug, lipid-based formulation, porous carriers, Aeroperl® 300, oral bioavailability
Introduction
Lamotrigine is a novel antiepileptic drug of the phenyltriazine class which is used widely in epilepsy, simple and complex seizures, generalized tonic-clonic seizures, and bipolar disorder.1,2 Its chemical name is 3, 5-diamino-6-(2,3-dichlorophenyl)-1,2,4-triazine.3 It acts by regeneration and modification of neurons function and Schwann cells by reducing glutamate release from nerve terminals in addition to a protective effect against cytotoxicity resulting from inhibition of mitochondrial respiratory complex I.4–6 It exerts a neuroprotective effect in experimental ischemia and cerebroprotective effect after focal ischemia.7 LMG is very slightly soluble in water (0.17 mg/mL at 25 °C) and slightly soluble in 0.1 N HCl (4.1 mg/mL at 25 °C).8 It is a weak base with a pka of 5.7, melting point of 218°C, and classified as a BCS class II-b drug. It exhibits high solubility and dissolution rate at the stomach acidic pH and may precipitate in the small intestine in a complex poorly understood manner.9–11
To ensure the presence of the drug in the dissolved form in the gastrointestinal tract, lipid microemulsion formulations, especially SNEDDS, are one of the promising formulations. SNEDDS is isotropic mixture of oil, surfactants, and cosurfactants that produce oil in water nanoemulsion in aqueous media upon mild agitation. SNEDDS spreads rapidly in the gastrointestinal tract with the aid of peristaltic movement producing droplets <100 nm containing the drug in a soluble form thus enhancing drug dissolution and bioavailability.12–15 The improvement of drug absorption by SNEDDS may be due to facilitating transcellular absorption by increasing membrane fluidity, allowing paracellular transport by opening tight junction, and bypassing the hepatic first-pass effect, reducing cytochrome P-450 metabolism, and protecting the drug from enzymatic degradation thus promoting highly lipophilic drug absorption by lymphatic transport.16–18 Liquid SNEDDS can be inserted in soft or hard gelatin capsules, but this may lead to some drawbacks as high production cost, excipients incompatibility with the capsule shell leading to either shell shrinkage or swelling, formulation leakage, and drug precipitation if stored at lower temperatures.19 To overcome these problems, converting liquid SNEDDS into solid dosage form will gain both advantages of enhancement of drug solubility and bioavailability properties of liquid SNEDDS with enhanced stability of solid dosage forms.20 Many techniques are used for converting liquid SNEDDS into S-SNEDDS such as spray drying, adsorption onto solid carriers, melt granulation, melt extrusion, and melt granulation.21 The simplest way is the physical adsorption onto solid carriers as it involves the adsorption of liquid SNEDDS onto solid carriers by physical mixing to form free-flowing powders which can be filled into hard gelatin capsule or compressed into tablets after the addition of suitable excipients.12,15,22 In recent years, porous carriers with large surface area as Aerosil® 200 Pharma and Aeroperl® 300 Pharma are used as solid carriers for the solidification of SNEDDS.23 Aerosil® 200 Pharma is a high-purity amorphous anhydrous colloidal silicon dioxide with average particle size of 12 nm (primary particles) and a specific surface area of 200 m2/g.24 Aeroperl® 300 Pharma is a granulated colloidal silicon dioxide with particle size ranging from 30 to 40 μm, has a pore volume of 1.6 mL/g and a specific surface area of 300 m2/g.25
Therefore, this study aimed to improve the solubility and oral BA of LMG through the preparation of SNEDDS by using a D-optimal factorial design for the optimization of the prepared LMG-loaded SNEDDS, then converting the optimized liquid SNEDDS to S-SNEDDS by solid adsorption using Aerosil® 200 and Aeroperl® 300 as solid carriers.
Materials and Methods
Materials
Lamotrigine was a gift sample from Global Napi pharmaceutical Company (6th October city, Giza, Egypt). Rose oil (RO) was purchased from alpha chemika™ Company (Mumbai, India). Almond oil, soybean oil, corn oil, linseed oil, isopropyl myristate (IPM), olive oil, castor oil, peanut oil, Cremophor® El (CR-EL), propylene glycol (PG), and polyethylene glycol 400 (PEG 400) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Lauroglycol™ FCC, Lauroglycol™ 90, Capryol™ PGMC, Caprol® ET, Labrasol®, Acconon® CC-6, Acconon® MC8-2, Labrafil® M 1944 CS, Labrafil® M 2125 CS, and Transcutol® P (Trans P) were generously donated by Gattefosse (Saint-Priest, Lyon, France). Tween® 80 (T80) and methanol were purchased from Merck (Darmstadt, Germany). Potassium dihydrogen phosphate, hydrochloric acid, and sodium hydroxide were purchased from El-Nasr Pharmaceutical Chemicals Co. (Cairo, Egypt). Aerosil® 200 and Aeroperl® 300 Pharma were kindly gifted by Evonik Industries (Essen, Germany). Lamictal® tablets containing 25 mg LMG (GlaxoSmithKline pharmaceuticals S.A. Poznan, Poland, Batch No. SX6X). All other reagents and chemicals used were of analytical reagent grade and used without further purification.
Methods
Solubility Studies of Lamotrigine
The solubility of LMG in different oils, surfactants, and cosurfactants were performed by adding an excess amount of LMG powder into a screw capped glass vial containing 2 mL of each vehicle, i.e., oil, surfactant, or cosurfactant. Oils employed were almond oil, soybean oil, corn oil, linseed oil, rose oil, IPM, olive oil, castor oil, peanut oil, Lauroglycol™ FCC, Lauroglycol™ 90, Capryol™ PGMC, and Caprol® ET. Surfactants and cosurfactants used were Labrasol®, Cremophor® EL, Acconon® CC-6, Acconon® MC8-2, Labrafil® M 1944 CS, T80, PEG 400, Trans P, and PG. The mixtures were vortexed for 5 min to aid the proper mixing of LMG with the vehicles, shaken for 72 h at 25 ± 1°C in an incubated shaker (Lab Companion, Jeio Tech, Korea) to reach equilibrium, followed by centrifugation at 10,000 rpm for 10 min by Eppendorf Centrifuge 5415 R (Eppendorf AG, 22,331 Hamburg, Germany). The supernatant was then diluted appropriately with methanol or with benzene: methanol (7:3) for oils immiscible with methanol. The concentration of LMG in each solvent was determined by UV spectroscopy (Shimadzu UV1601, Shimadzu Corp, Japan) at λ max 307 nm. The experiment was performed in triplicate.26,27
Preliminary Screening of Surfactants
The surfactants were screened for their ability to emulsify RO. RO was mixed in 1:1 w/w ratio with each surfactant and vortexed for 5 min for proper mixing. Aliquots equivalent to 25 µL from each mixture was diluted to 25 mL with double-distilled water to obtain an emulsion. The emulsions were observed for physical appearance, clarity, and phase separation. The emulsions were left undisturbed for 2 hours and analyzed for transmittance at 638.2 nm on UV-VIS spectrophotometer using ultrapure water as a blank. The experiment was performed in triplicate.17,28
Preliminary Screening of Cosurfactants
The turbidimetric method described under preliminary screening of surfactants was used to screen the cosurfactants. Selected oil/selected surfactant/cosurfactant ratio was kept constant at 3:2:1 w/w during screening. The experiment was performed in triplicate.
Construction of Pseudo-Ternary Phase Diagrams
Based on the data of solubility studies and preliminary screening of surfactants and cosurfactants, rose oil was selected as oil phase. CR-EL as a surfactant, PEG 400 and T80 as cosurfactants. Pseudo-ternary phase diagrams were constructed using water titration method. Water is added dropwise with agitation (by vortex) to the mixture of oil and S/Cos at certain weight ratios (1:1, 2:1, 4:1 w/w) till end point (turbidity).29,30 Six pseudo-ternary phase diagrams were drawn using OriginPro 8.1 program (OriginLab Corporation, One Roundhouse Plaza, Northampton, MA 01060 USA). After identification of microemulsion region, systems of desired component ratios were selected to prepare SNEDDS that forms nanoemulsion upon dilution based on remaining clear even after infinite dilution.31
Preparation of SNEDDS Formulations
To prepare drug loaded SNEDDS, the selected oil, surfactant, and cosurfactant were accurately weighed into glass vials according to their ratios, vortexed, then the amount of LMG (5% w/w) was added. The components were sonicated at 40°C in an ultrasonic bath (Elma Transsonic, 460/H, Germany) until complete dissolution of LMG. The formulation was stored at room temperature for at least 48 hours and examined for any signs of turbidity or phase separation.32
Design of Experiments
D-optimal factorial design was used for the optimization of the preparation of LMG-loaded self-nanoemulsifying formulations. The study involved 3 independent factors namely: concentration of oil (X1), type of Cos (X2), and ratio of S/Cos (X3). Droplet size (DpS; Y1), the percentage of in-vitro drug released after 5 min in 0.1N HCl (Y2), % in-vitro drug released after 15 min in 0.1N HCl (Y3), % in-vitro drug released after 5 min in PB (Y4), and % in-vitro drug released after 15 min in PB (Y5) were selected as dependent variables (Table 1). A total of 19 randomized experimental runs were conducted (R1-R19) as shown in Table 2 using Design Expert® 11 software (Stat-Ease, Inc., Minneapolis, MN, USA). CR-EL was used as a surfactant in all formulations. The Design Expert® was then analyzed the dependent variables by using ANOVA to determine the significance of each factor (p < 0.05).
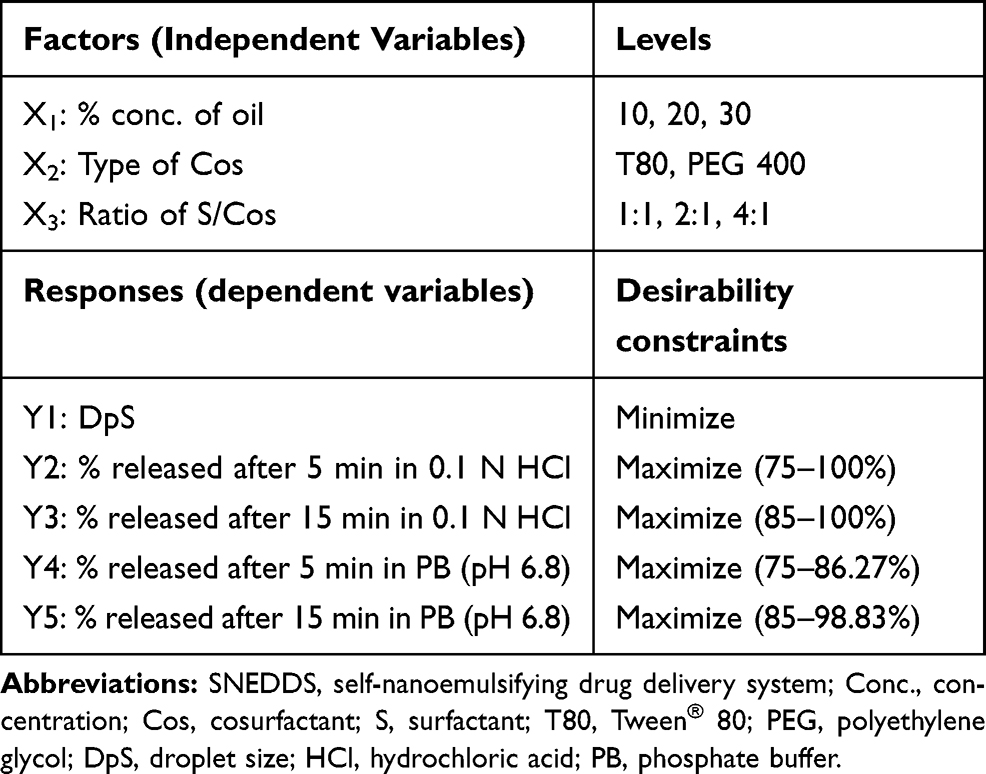 |
Table 1 D-optimal Factorial Design Used for Optimization of SNEDDS Formulations |
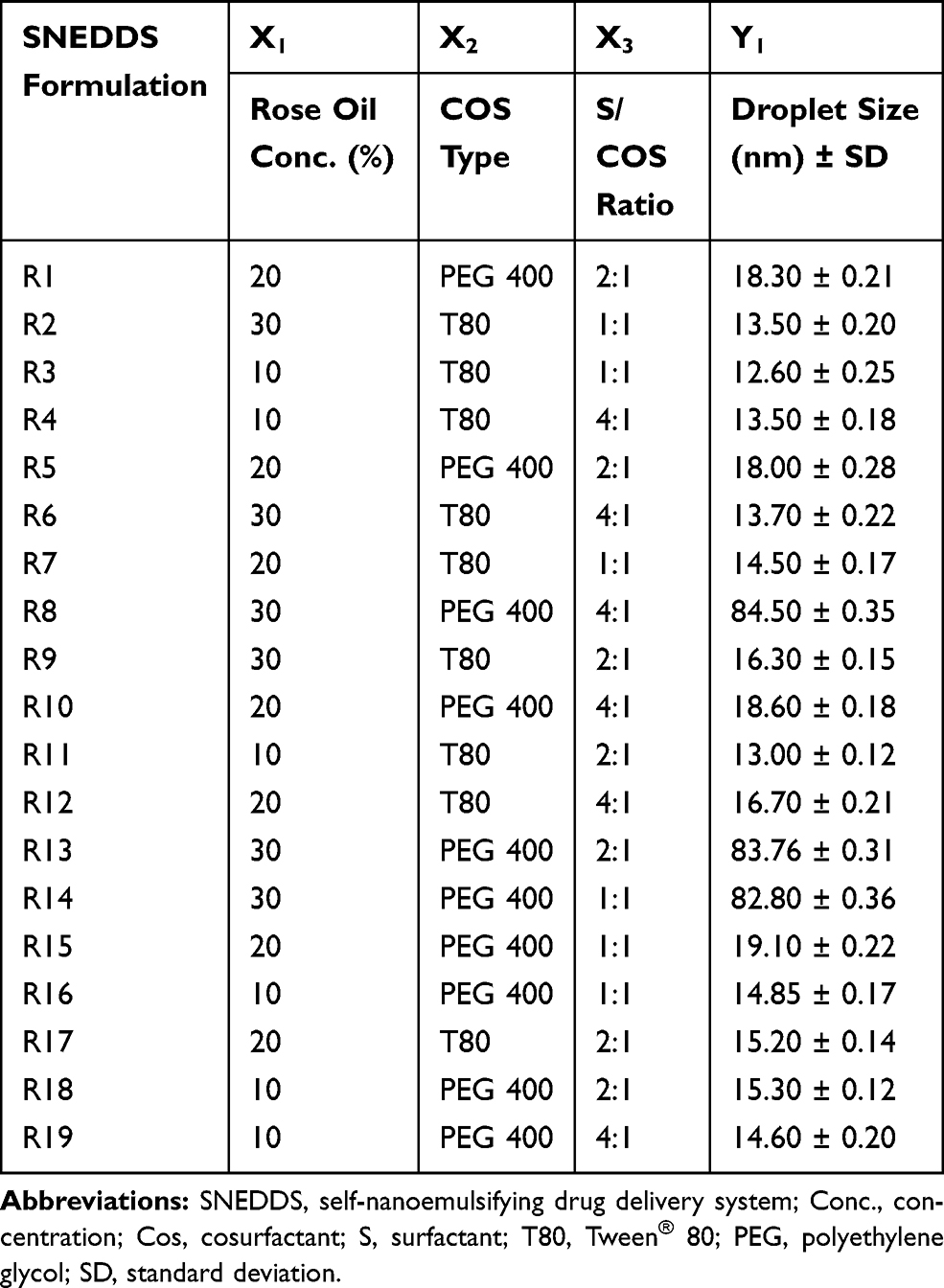 |
Table 2 Experimental Runs, Independent Variables, and Droplet Size Response of D-Optimal Factorial Design of LMG-Loaded SNEDDS Formulations |
Optimization of LMG-Loaded SNEDDS
The optimum formulations, to be used for further investigations, were selected using the desirability function for optimization in Design Expert®. The criteria set for selecting the optimum formulations was achieving the least DpS and the highest % drug released.
In-vitro Characterization of SNEDDS
Liquid SNEDDS formulations were diluted 500 times with ultrapure water and 0.1 N HCl. The diluted nanoemulsions were stored for 24 hours and observed for any signs of phase separation or drug precipitation.33
Thermodynamic Stability Studies
Liquid SNEDDS formulations were subjected to six cycles between cooling (4°C) and heating (45°C) with 24 hours storage at each temperature. The stable formulations were subjected to centrifugation test.15
Liquid SNEDDS that passed heating cooling cycles were centrifuged for 30 min at 3500 rpm. The stable formulations were subjected to freeze thaw stress test.34
Liquid SNEDDS were subjected to three cycles between −21°C and +25°C for 48 hours for each temperature.16
Dispersibility Studies and Self-Emulsification Time
The efficiency of self-emulsification of SNEDDS was evaluated using USP standard dissolution apparatus type II. One mL of each SNEDDS formula was added to 500 mL of distilled water kept at 37°C ± 0.5°C. The paddle was rotating at a speed of 50 rpm. The prepared formulations were examined in terms of rate of emulsification and the final appearance of the nanoemulsion according to the following grading system:35
Rapidly forming emulsion having a clear or bluish appearance (within 1 min).
Rapidly forming, slightly less clear emulsion, having a bluish white appearance (within 1 min).
Fine milky emulsion formed within 2 min.
Dull, grayish white emulsion having slightly oily appearance that is slow to emulsify (longer than 2 min).
Turbidimetric Evaluation
Nephelo-turbidimetric evaluation is performed to monitor the growth of the emulsification. However, because the time required for complete emulsification is too short, it is not possible to monitor the rate of change of turbidity (rate of emulsification). Fifty microliters of SNEDDS formulations were diluted to 25 mL with ultrapure water and then the turbidity was studied using 2100N IS Turbidimeter (Hach Company, Loveland, Colorado, USA). Results were given as nephelometric turbidity unit (NTU).36,37
Optical Clarity Determination
The optical clarity determination was done using % Transmittance. Fifty microliters of SNEDDS were diluted to 25 mL with ultrapure water. The % transmittance of the system was measured at 638.2 nm using UV spectrophotometer using ultrapure water as a blank.38,39
Measurement of Droplet Size
Droplet size of LMG-loaded SNEDDS diluted with water was determined using COULTER® N4 PLUS Submicron Particle Sizer (Coulter Corporation, Miami, Florida, USA), a photon correlation spectrometer which determines particle size by measuring the rate of fluctuations in laser light intensity scattered by particles as they diffuse through a fluid. Fifty microliters of SNEDDS formulations were diluted to 25 mL with ultrapure water and then the droplet size was determined, each determination was done in triplicate.
In-vitro Drug Release Studies
Drug release studies of the plain drug (LMG), liquid SNEDDS formulations, and commercially available tablet (Lamictal®) were performed using USP dissolution apparatus II. Each liquid SNEDDS formulation, equivalent to 25 mg LMG, was accurately weighed, filled into size (0) hard gelatin capsule and introduced into a dissolution apparatus containing 900 mL of 0.1 N HCl, or PB (pH 6.8) at 37 ± 0.5°C. Capsule sinkers were used to avoid the capsule floating in the medium. The speed of the paddle was adjusted to 50 rpm. At predetermined time intervals, aliquots of a 5 mL were withdrawn and replaced with an equal volume of fresh medium to maintain a constant volume. Samples were then properly diluted if needed. The concentration of the drug was measured by UV spectrophotometrically at 264 nm for 0.1 N HCl samples, or at 307 nm for PB samples. All experiments were run in triplicate.40,41 A cumulative correction factor was applied to compensate for the dilution of withdrawn samples in the release studies, according to the following equation.42
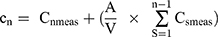
Where:
A: Aliquot volume
V: Release medium volume
Cn meas: The spectrophotometrically measured concentration.
Cn: The concentration of the nth sampling expected in the medium, if the previous sample had not been removed.
n-1: The total number of all samples removed prior to the sample being measured.
Cs meas: The total of all spectrophotometrically measured concentration at n-1 sample.
Preparation of LMG-Loaded S-SNEDDS
The surface adsorption method was used for transforming the optimized SNEDDS formulations into free-flowing S-SNEDDS powder. Eight S-SNEDDS were prepared, according to 23 factorial design, by physical adsorption of the optimized SNEDDS formulations (R9 and R14), using 2 solid carriers (Aerosil® 200 and Aeroperl® 300), and using 2 different SNEDDS: Solid Carrier ratios (1:0.75 and 1:1), by thoroughly mixing in a glass mortar and a pestle. The resultant mass was passed through sieve no. 60 to obtain homogenous free-flowing powder and then stored in tightly closed containers in a desiccator till further use. The 23 factorial design involved 3 independent variables with 2 levels namely: X1: SNEDDS formulation, X2: solid carrier type, X3: SNEDDS: Solid Carrier ratio. The percentage of in-vitro drug released after 5 min in 0.1N HCl (Y1), % in-vitro drug released after 15 min in 0.1N HCl (Y2), % in-vitro drug released after 5 min in PB (Y3), % in-vitro drug released after 15 min in PB (Y4), and % in-vitro drug released after 30 min in PB pH 6.8 (Y5) were selected as dependent variables. Eight formulations were designed (S1-S8) using Design Expert® 11 software as tabulated in Table 3. The Design Expert® then analyzed the dependent variables by using ANOVA to determine the significance of each factor (p < 0.05).
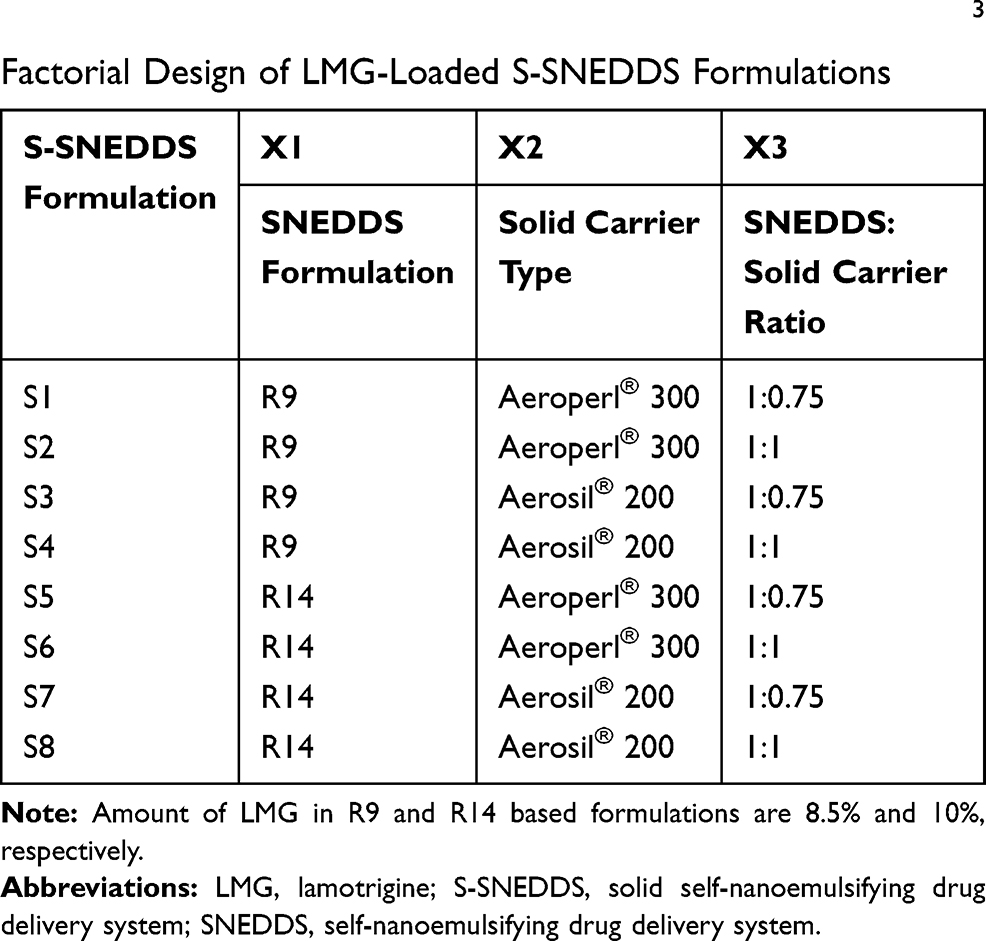 |
Table 3 Experimental Runs, Independent Variables of 23 Factorial Design of LMG-Loaded S-SNEDDS Formulations |
Optimization of LMG-Loaded S-SNEDDS
The optimum formula was selected using the desirability function for optimization in Design Expert®. The criteria set for selecting the optimum formula was achieving the highest % drug released in all responses.
Characterization of LMG-Loaded S-SNEDDS
The prepared S-SNEDDS were diluted, filtered, and examined using COULTER® N4 PLUS Submicron Particle Sizer as described above. The results were compared with their corresponding liquid SNEDDS.
Drug Loading Efficiency
The amount of S-SNEDDS equivalent to 25 mg LMG was dispersed in suitable quantity (50 mL) of methanol, sonicated for 30 min for extraction of the drug, then filtered through a syringe filter (0.2 µm). The filtrate was suitably diluted and analyzed spectrophotometrically at 307 nm.16,43 Each experiment was carried out in triplicate.
Micromeritic Properties of S-SNEDDS
The standard fixed funnel method was used to measure the angle of repose of S-SNEDDS. A funnel was fixed with its tip at a given height, H, above a paper placed on a flat horizontal surface. S-SNEDDS powder was poured through the funnel until the apex of the conical pile just touched the tip of the funnel. The radius, R, of the base of the conical pile was measured.44 The angle of repose (θ) was calculated using the following equation:
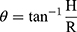
Where θ = Angle of repose, H= Height of pile, and R= Radius of the pile.
A measured 2 g weight (wt) of S-SNEDDS was introduced into a 10 mL dry measuring cylinder. The bulk volume (Vb) was observed before any tapping. The cylinder was then tapped by raising it at a height of 2.5 cm, allowed it to drop under its own weight on a hard surface, and the volume was recorded. The tapping was continued till the change noted in volume was less than 2% between every consecutive tapping then the tapped volume (Vt) was recorded.45 The bulk density (Pb) and tapped density (Pt) were calculated as follows:
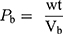
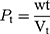
Hausner ratio and Carr’s index are measures of the propensity of S-SNEDDS to be compressed which reflects the importance of interparticulate interactions. These interactions are less significant for free-flowing S-SNEDDS as the bulk and tapped densities will be closer in magnitude, and vice versa for poor flowing materials.46 Hausner ratio and compressibility index reflect these differences, and are calculated by the following equations:
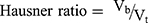
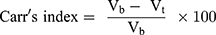
Solid-State Characterization of S-SNEDDS
DSC was used to study the thermal behavior of pure LMG, Aeroperl® 300, their PM, and the optimized S-SNEDDS formulation using DSC (DSC-60 A plus, Shimadzu corp., Kyoto, Japan). Five miligrams were crimped in aluminum pans. The scans were run over temperature range of 35ºC to 250ºC at a heating rate of 10ºC/min under nitrogen purge at a flow rate of 10 mL/min.47
The powder X-ray diffraction patterns of pure LMG, Aeroperl® 300, their PM, and the optimized S-SNEDDS formulation were determined by x-ray diffraction (XRD D8 Discovery, Bruker company, Karlsruhe, Germany) using Cu Kα radiation (λ = 1.54060). The scanning rate was 1º/min over a 2 θ range from 5º to 80º at a voltage conditions of 40 kV and a current of 40 mA.48
The external morphological structure for pure LMG, Aeroperl® 300, their PM, and the optimized S-SNEDDS was done using SEM (SEM, Model Quanta 250 FEG, FEI Company, Eindhoven, Netherlands) attached with EDX unit (Energy Dispersive X-ray Analyses), with accelerating voltage 30 kV.49
In-vitro Drug Release Studies
Drug release studies of the plain drug (LMG), S-SNEDDS formulations, and Lamictal® tablets were performed using USP dissolution apparatus II in 0.1 N HCl and PB (pH 6.8). Each S-SNEDDS formulation, equivalent to 25 mg LMG, was accurately weighed, filled into size (00) hard gelatin capsule and introduced into a dissolution apparatus as illustrated above in SNEDDS release studies. To study the drug release kinetics of each formulation, the in-vitro release data were fitted in various kinetic models: zero order, first order, Higuchi diffusion model, and Hixon-Crowell cube root law.50 To determine the mechanism of drug release, the first 60% drug release data were fitted into Korsmeyer-Peppas equation and the exponent n was calculated from the slope of the straight line.51
Particle Size Analysis and Transmission Electron Microscope (TEM)
Particle size analysis of the SNEDDS used for the preparation of the optimized S-SNEDDS was measured using Malvern Zetasizer (Zetasizer ZS, Malvern, UK). The sample was diluted as illustrated above in droplet size measurement.
The morphology of the optimized SNEDDS was analyzed using transmission electron microscope TEM (JEM-1230, Joel, Tokyo, Japan). Liquid SNEDDS was diluted with water to form nanoemulsion. A drop of nanoemulsion was applied on the surface of copper grid and left for 30 min to allow particles adherence to collodion. After removal of excess nanoemulsion, a drop of 2% uranyl acetate was applied, left to air dried for 1 min, and then examined using TEM.52
Accelerated Stability Studies
The optimized LMG loaded S-SNEDDS was subjected to stability studies for three months at 40 ± 5 ºC and 75 ± 5% RH in order to assess its physical and chemical stability. The stored samples were evaluated every month for appearance, drug content, and in-vitroreleasestudies.53,54
In-vivo Studies
The study was performed for the prepared S-SNEDDS (S2) filled in hard gelatin capsules, Lamictal® tablets (market product), and pure LMG powder filled in hard gelatin capsules. Fifteen healthy New Zealand male rabbits (2.8 ± 0.2 Kg) were randomly divided into three groups, each containing five rabbits. An open-label, randomized parallel design was applied so that each group received a single oral dose of one of the tested formulations. The animals were fasted overnight with free access to water before drug administration. A single oral dose (25 mg) was then given to each rabbit. The study protocol complied with the European community standards for the protection of animals used (Directive 2010/63/EU). The study design was approved by the ethical committee of Faculty of Pharmacy, Cairo University (approval number (PI)=1267). Blood samples were collected in heparinized glass tubes through the marginal ear vein at zero time (pre-dose), 0.5,1,2,4,6,8,10, and 24 h post-dose. Blood samples were centrifuged at 3000 rpm for 10 min then plasma was separated immediately in polyethylene capped tubes and frozen at –20 ºC till assayed.55,56
The analysis was performed using a Shimadzu prominence (Shimadzu, Japan) series LC system equipped with a degasser (DGU-20A3) and solvent delivery unit (LC-20AD) with an auto-sampler (SIL-20A/HT). The system was used to inject 10 µL aliquots of the processed samples on a C18, 100A (50×4.6 mm) (Phenomenex, USA), 5 µm particle size. A sensitive and validated LC-MS/MS method was used for the separation and quantitation of LMG using Ornidazole as an internal standard (IS).57 Isocratic mobile phase consisted of acetonitrile and 0.1% formic acid in water 80:20 (v/v) at a flow rate of 1 mL/min. MS/MS detection in positive ion mode using AB Sciex API 4000 Triple Quadrupole Mass Spectroscopy (Applied Biosystems, Foster city, CA, USA), equipped with a TurboIonSpray® source, was used for quantitation. All data were processed using Analyst Software version 1.6 (Applied Biosystems, Foster city, CA, USA).
To prepare the standard calibration samples, aliquots of 0.5 mL drug-free plasma were spiked with LMG stock solution (10 µg/mL) and an aliquot of 100 µL of Ornidazole (200 ng/mL) solution (IS) to produce calibration standards at the following concentrations: 10, 50, 80, 100, 500, 800, 1000, 3000, 6000 ng/mL. For sample preparation, 0.5 mL rabbit plasma and 100 µL Ornidazole solution were vortex mixed in 10 mL glass volumetric flask for 1 min, followed by the addition of 4 mL of ethyl acetate. After vortex mixing for another 3 min, the samples were centrifuged for 10 min at 3000 rpm. The organic layer was separated, evaporated till dryness using centrifugal vacuum concentrator at 45 ºC, and reconstituted in 0.5 mL mobile phase. Ten microliters of the sample were injected into the LC-MS/MS.
Plasma concentration over time profiles of LMG were analyzed by a non-compartmental method using pharmacokinetic modeling program WinNonlin standard edition Version 1.5 (Scientific Consulting Inc., NC, USA). The following pharmacokinetic parameters were calculated: peak plasma concentration (Cpmax), time to reach Cpmax (Tmax), area under plasma concentration time curve extrapolated to infinity (AUC0-α), and percentage of relative bioavailability. The pharmacokinetic parameters of the tested formulations were compared by using non-parametric Mann–Whitney’s test for independent samples, due to low number of subjects (n=5), using SPSS software version 22 (Statistical Package for the Social Sciences; SPSS Inc., Chicago, IL, USA). The significance of the difference was determined at p-value < 0.05.
Results and Discussion
Solubility Studies of Lamotrigine
The SNEDDS was prepared to enhance the solubility and bioavailability of the drug via oral administration. Thus, each component used in the system should have high solubilizing capacity to obtain the optimum drug loading and to minimize the final SNEDDS volume. The solubility of LMG in various vehicles is presented in Figure 1. It was found that rose oil (RO) exhibited the highest drug solubility among the screened oils (Figure 1A). It was also reported that RO has a beneficial antiepileptic, anticonvulsant, and neuroprotective effects.58,59 Therefore, RO was used as the oily phase. The surfactants and cosurfactants were selected based on two parameters, their solubilization ability to LMG and their emulsification ability for the selected oil.
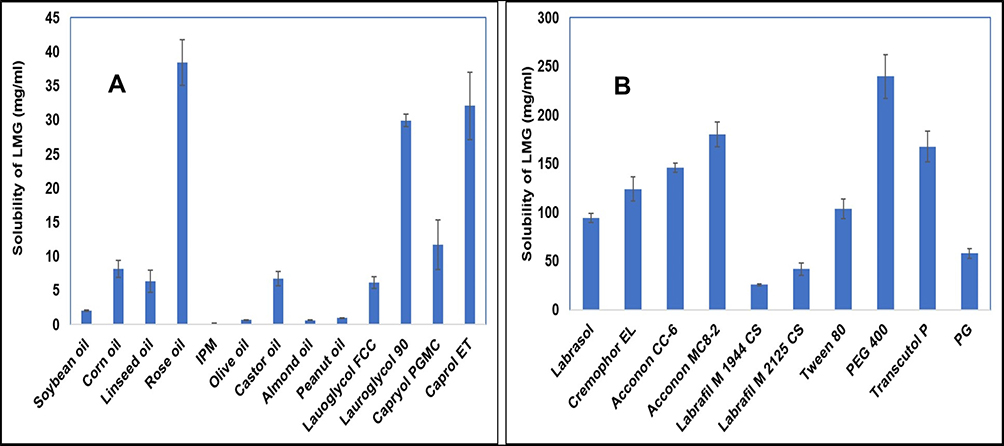 |
Figure 1 Solubility of LMG in (A) oils and (B) surfactants and cosurfactants. |
Preliminary Screening of Surfactants
The evaluation of various surfactants for their emulsification ability to the selected oil phase was done by turbidimetry studies.17 It was essential for oil/surfactant mixture to disperse efficiently and form spontaneous nanoemulsion, as well-formed SNEDDS get dispersed within seconds under gentle stirring conditions.60 It was reported that HLB of surfactants should be in the range of 12–15 to have a good efficiency for self-emulsification.61 Acconon® CC-6, Acconon® MC8-2, CR-EL, T80, and Labrasol® were selected for emulsification study as they showed good solubility for LMG (Figure 1B). The % transmittance values of rose oil/surfactant mixtures are tabulated in Table 4. It was found that the % transmittance values were high in dispersions containing CR-EL, or T80. Therefore, they were selected for further investigations.
 |
Table 4 Emulsification Efficiency of Various Surfactants to Emulsify Rose Oil |
Preliminary Screening of Cosurfactants
Cosurfactants play an important role with surfactants in any formulation for improving its dispersibility and drug absorption.17 As the ratio of surfactant to cosurfactant was kept constant, the turbidity of the resulting microemulsion helped in assessing the relative efficacy of the cosurfactants to improve the emulsification ability of surfactants. T80 and PEG 400 were chosen as T80 gave the highest % Transmittance value with good solubility for LMG, and PEG 400 had the highest solubility for LMG and high transmittance value (Table 5).
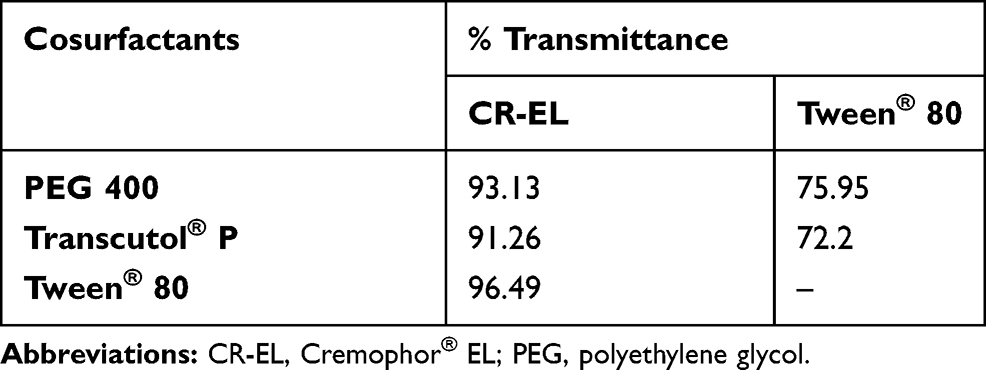 |
Table 5 Emulsification Efficiency of Surfactant/Cosurfactant Combinations to Emulsify Rose Oil |
Based on preliminary screening results, two systems were selected:
System A
RO as an oily phase, CR-EL as a surfactant, T80 as a cosurfactant.
System B
RO as an oily phase, CR-EL as a surfactant, PEG 400 as a cosurfactant.
Construction of Pseudo-Ternary Phase Diagrams
The regions of microemulsion formed are shown in Figure 2. It was found that as the ratio of S/Cos increased, the microemulsion region increased in PEG-based systems. However, increasing S/Cos ratio did not markedly affect microemulsion region in T80-based systems. This might be due to the similar HLB values of both CR-EL (HLB 12–14) and T80 (HLB 15).
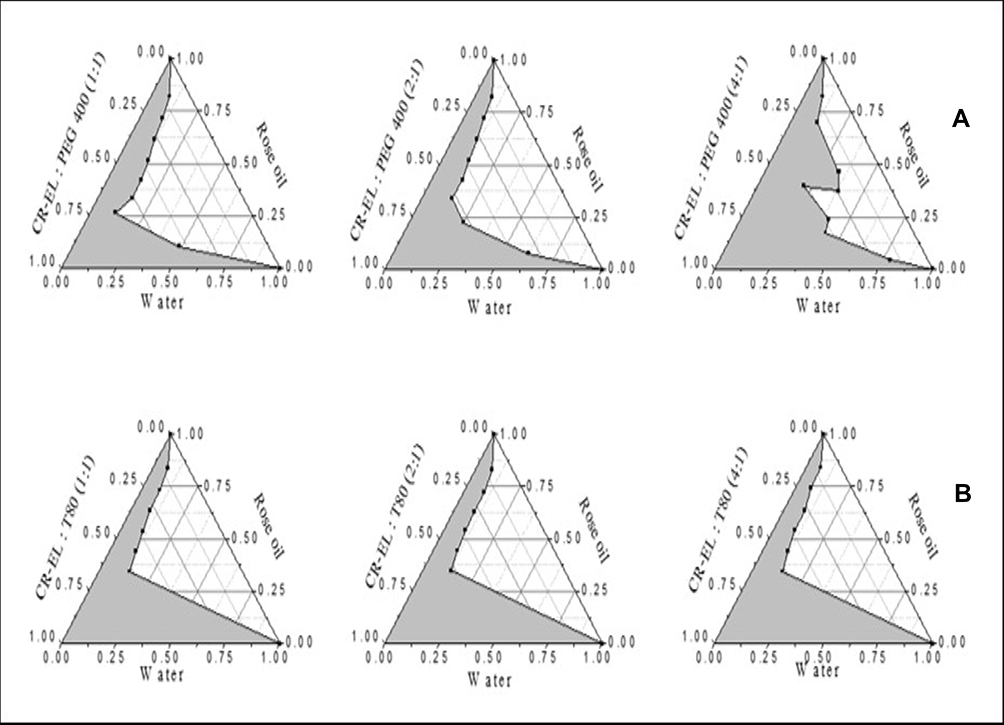 |
Figure 2 Pseudo-ternary phase diagrams of (A) PEG-based systems and (B) T80-based systems. |
In-vitro Characterization of SNEDDS
Robustness to Dilution
All formulations formed nanoemulsion upon dilution with aqueous media, remained clear, transparent, and showed neither precipitation nor phase separation for more than 24 h, giving a good indication for stability of the prepared SNEDDS and the small DpS of the nanoemulsion formed.
Thermodynamic Stability Studies
Thermodynamic stability study was done to evaluate the effect of temperature change on SNEDDS formulations and detect any signs of phase separation. It was found that all formulations passed the three tests, were physically stable and showed no signs of any phase separation nor drug precipitation.
Dispersibility Studies and Self-Emulsification Time
All the liquid SNEDDS formulations prepared as per the experimental design showed good self-emulsification efficiency and formed clear nanoemulsion immediately after dilution with aqueous phase within 1 min. Visual observation showed that all formulations were found to be grade A.
Turbidimetric Evaluation
The turbidity of all SNEDDS formulations was below 10 NTU which confirms good clarity of the formulations.
Optical Clarity Determination
The % transmittance of all SNEDDS formulations was >92 which confirms the formation of clear nanoemulsion with small droplet size. All prepared SNEDDS were clear after 24 h and showed no precipitation after 500 times dilution, confirming the stability of the prepared formulations.
Measurement of Droplet Size (DpS)
The DpS of diluted SNEDDS formulations are tabulated in Table 2. It was found that the DpS of all formulations was smaller than 20 nm except for R8, R13, and R14, prepared with 30% RO using PEG 400 as a cosurfactant, have DpS of 84.5, 83.76, and 82.8 nm, respectively (Figure 3A). But generally, all have mean DpS <100 nm. This in agreement with Porter et al62 who stated that SNEDDS formulations which contain high concentrations of hydrophilic surfactants and cosolvents disperse to give smaller droplets with particle sizes <100 nm, confirming the thermodynamically spontaneous process of the formation of clear nanoemulsion with very low energy required.63
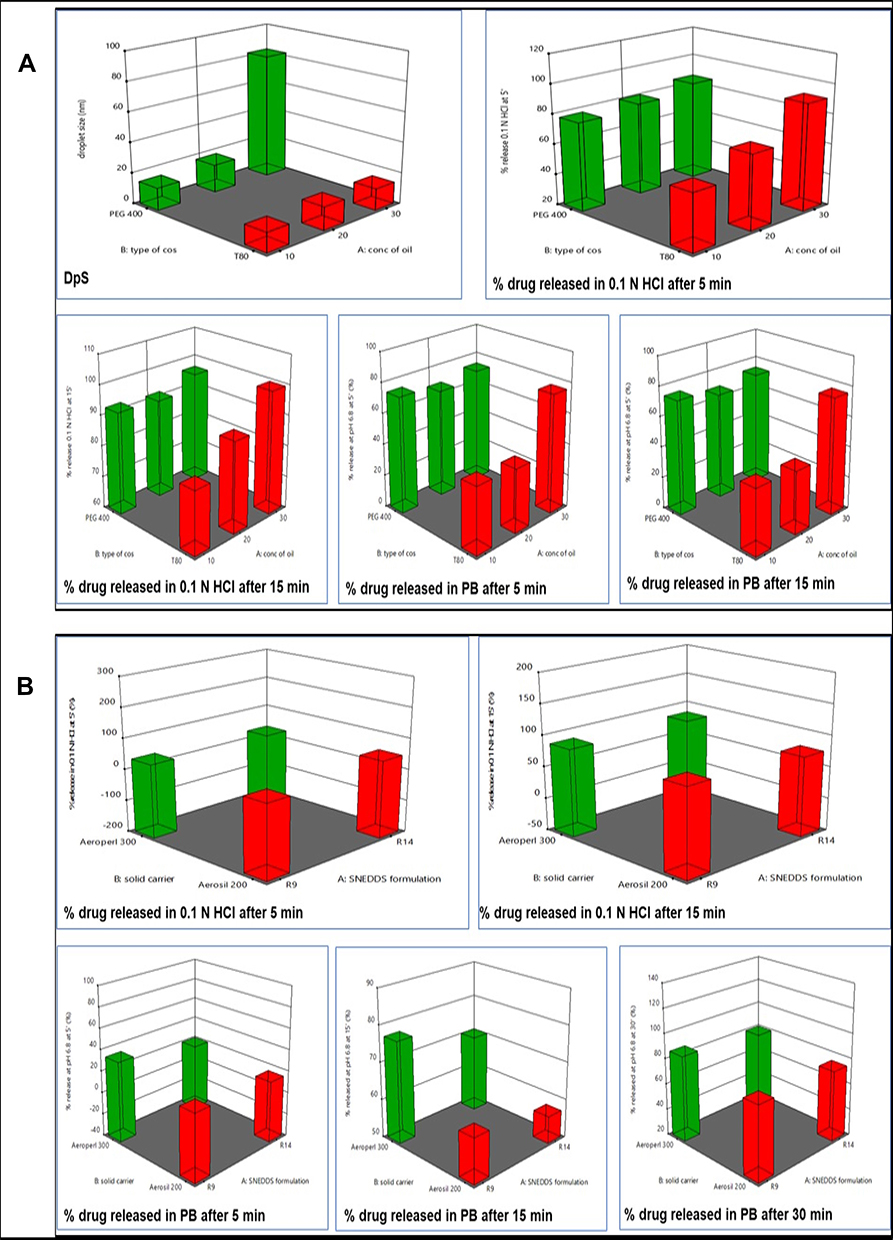 |
Figure 3 Response 3-D plots for (A) the effect of oil concentration and Cos type on SNEDDS responses and (B) the effect of SNEDDS formulation type and solid carrier type on S-SNEDDS responses. |
In-vitro Drug Release Studies
Release studies were performed for liquid SNEDDS containing 25 mg LMG, plain drug, and Lamictal® tablet. The release profile of LMG from different formulations were evaluated and presented in Figure 4A. The data showed that the release profile of LMG from all formulations was faster in 0.1 N HCl than in PB due to the pH-dependent solubility of LMG. After the dissolution of hard gelatin capsules, the drug release was very fast from liquid SNEDDS as a result of fast spontaneous nanoemulsion formation of small droplet size.64 It was found that liquid SNEDDS formulations prepared with 30% oil gave the highest drug release profiles from both T80 and PEG-based formulations in both media. The highest drug released in 0.1 N HCl after 5 min was from R9 (30% RO, CR-EL:T80; 2:1) followed by R14 (30% RO, CR-EL:PEG 400; 1:1), with results of 100% and 96.13% respectively, compared to pure LMG (35.044%), and Lamictal® (77.78%). The % of LMG released in PB after 15 min was higher in R14 (98.82%), followed by R9 (92.7%), followed by R18 (87.06%) compared to pure LMG (7.88%), and Lamictal® (84.29%).
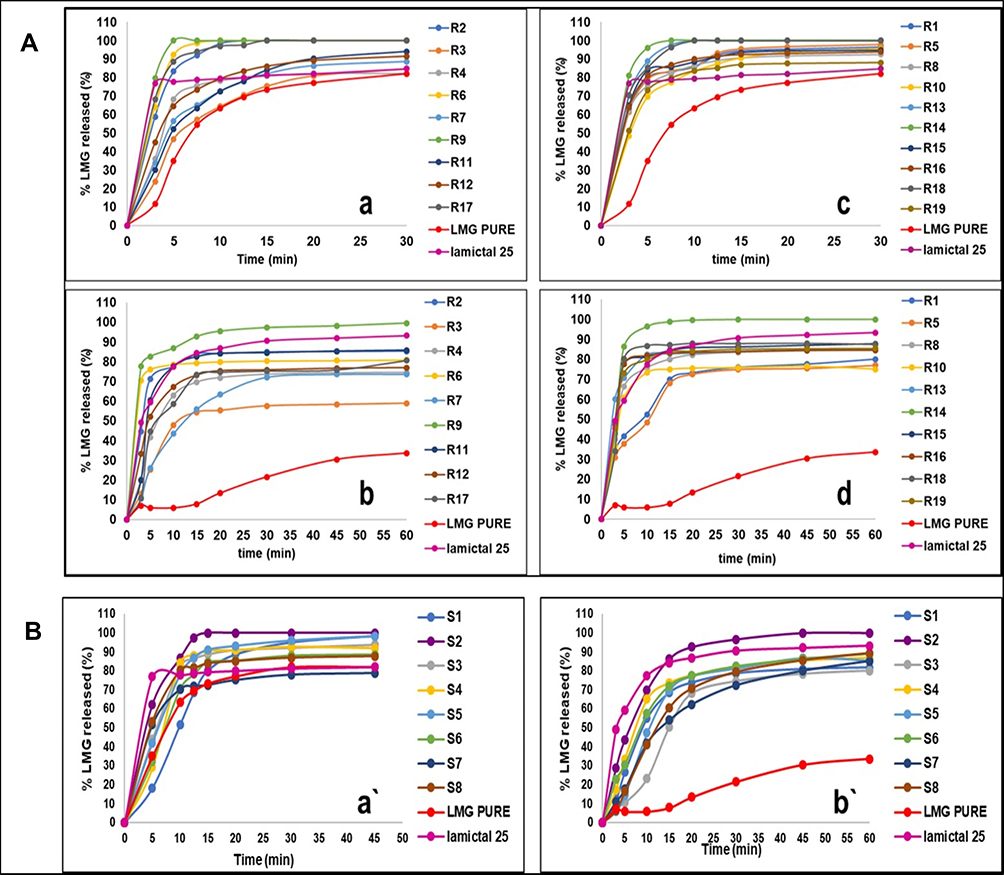 |
Figure 4 In-vitro release profiles of LMG from (A) SNEDDS: (a) T80-based formulations in 0.1 N HCl and PB (b), (c) PEG-based formulations in 0.1N HCl and PB (d), and (B) S-SNEDDS in 0.1 N HCl (a`) and in PB (b`). |
Statistical Analysis and Optimization of LMG-Loaded SNEDDS
For selecting the optimized liquid SNEDDS that achieved maximum acceptable characters, data of evaluation tests were analyzed using the statistical program Design Expert® Software. The mathematical model selected was two-factor interaction and it was noted that the predicted R2 values were in a reasonable agreement with the adjusted R2 values in all responses except for the in-vitro drug release after 5 and 15 min at pH 1.2 (Table 6). The negative predicted R2 values of the in-vitro drug release after 5 and 15 min at pH 1.2 implies that the overall mean may be a better predictor of the responses. This might be due to faster drug release profiles from all formulations due to the pH-dependent solubility of LMG and that the release at pH 1.2 was not affected by the studied factors. The adequate precision with a ratio greater than 4 is desirable which was observed in all responses. Response 3-D plots for the effect of independent factors on the responses are shown in Figure 3A. The solutions obtained from the optimization in Design Expert® software showed that R9 (30% RO, 46.67% CR-EL, and 23.33% T80) exhibited the best results for the criteria set for the optimized formulations with a desirability value of 0.763, followed by R14 (30% RO, 35% CR-EL, 35% PEG 400) with a desirability value of 0.438 as shown in Figure 5A, and R18 (10% RO, 60% CR-EL, 30% PEG 400) with a desirability value of 0.340. In order to validate the design experiment, the predicted and observed responses of the optimized formulations were compared as shown in Table 6. A high correlation was observed between the observed and predicted values confirming the validity of the design. Thus, R9 and R14 were selected for further investigations while R18 was excluded because of its low desirability value.
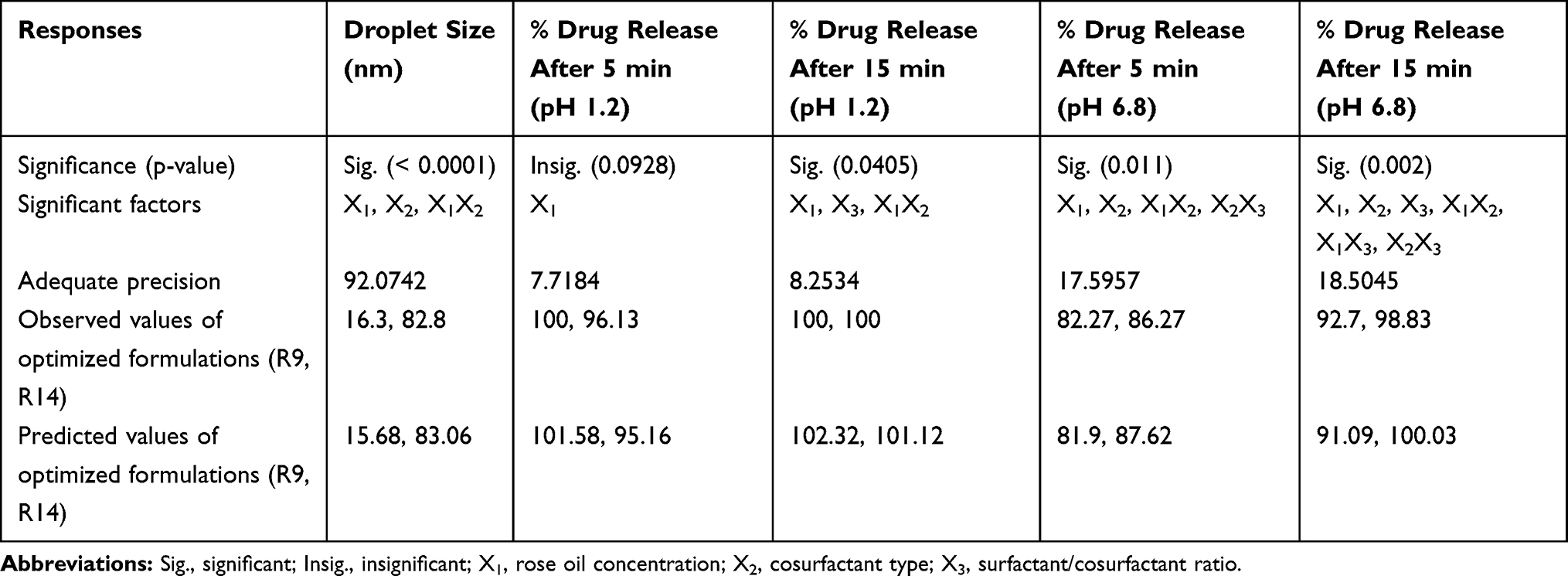 |
Table 6 Output Data of Responses of the D-Optimal Factorial Analysis of SNEDDS Formulations and Observed and Predicted Values of the Optimized Formulations (R9 and R14) |
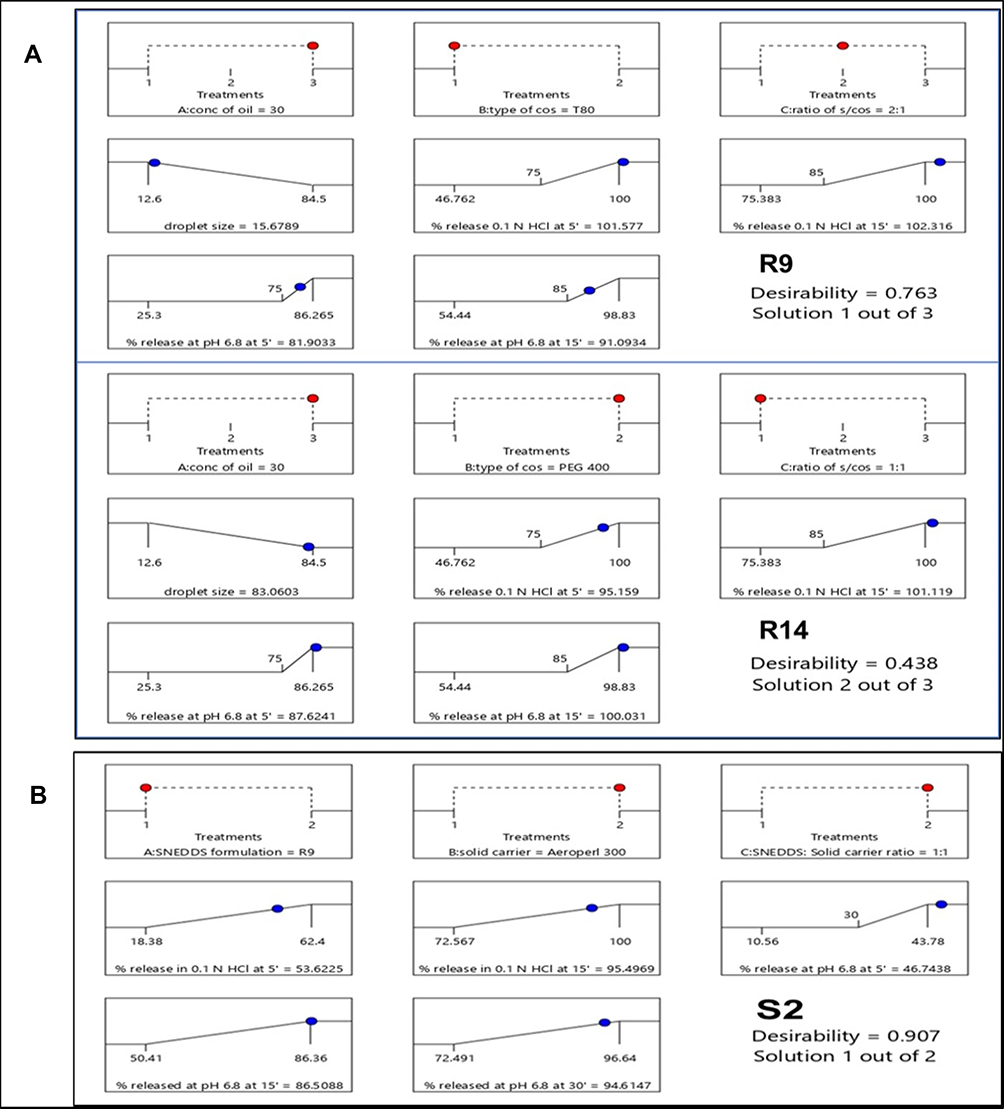 |
Figure 5 Design Expert® solutions according to criteria set for optimization of (A) SNEDDS and (B) S-SNEDDS. |
Preparation of S-SNEDDS
Based on the results of desirability of Design Expert® software for SNEDDS formulation, eight LMG-loaded S-SNEDDS were prepared, according to 23 factorial design by solidification of the optimized SNEDDS (R9 and R14).
Characterization of S-SNEDDS
Droplet Size Measurement
R9 and R14 based S-SNEDDS diluted formulations had droplet size ranging from 18 ± 0.143 to 20 ± 0.116 nm, and from 85 ± 0.201 to 88 ± 0.289 nm respectively, which are close to their corresponding SNEDDS droplet size.
Drug Loading Efficiency
The drug loading efficiency for all LMG-loaded S-SNEDDS was in the range of 96% to 105%, indicating that the drug is uniformly distributed within the solid carrier in the formulation.
Micromeritic Properties
The values obtained for the angle of repose of the optimized S-SNEDDS was 25.43º ± 1.65º indicating the good flowability of the formulation. The bulk and tapped densities were found to be 0.34 ± 0.004 g/mL and 0.392 ± 0.005 g/mL, respectively. Carr’s index and Hausner ratio were 13.45 ± 0.16% and 1.155 ± 0.002, respectively confirming the good flowability of the formulation. The good flowability may be due to good sphericity of particles.
Solid-State Characterization of S-SNEDDS
Figure 6A shows a sharp endothermic peak at 216 ºC corresponding to the melting point of LMG in the DSC thermogram of LMG which confirms its crystalline nature.65 The DSC thermogram of Aeroperl® 300 did not show any peaks over the entire scanned temperature range. The thermogram of the PM of LMG and Aeroperl® 300 showed the endothermic peak of LMG with reduced intensity. However, the DSC thermogram of S-SNEDDS showed complete disappearance of LMG peak indicating that the drug was in its amorphous form and molecularly dispersed in the solid carrier matrix.66
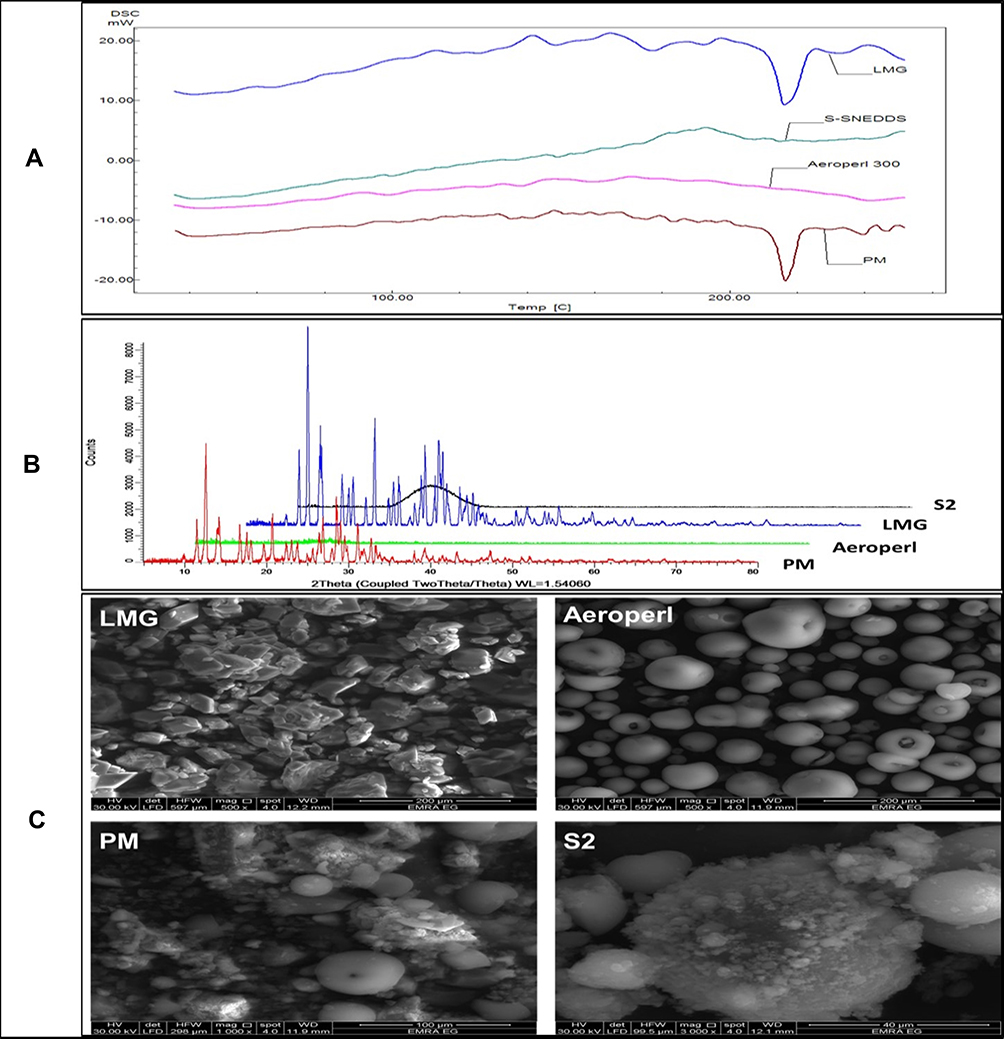 |
Figure 6 Solid-state characterization of LMG, Aeroperl® 300, their PM, and optimized S-SNEDDS (S2): (A) DSC thermograms, (B) PXRD patterns, and (C) SEM micrographs. |
The PXRD patterns of LMG, Aeroperl® 300, PM, and optimized S-SNEDDS are presented in Figure 6B. LMG showed sharp and intense peaks at the diffraction angles, confirming the crystalline nature of the drug. Aeroperl® 300 is amorphous in nature and showed no intense diffraction peaks.67 The same sharp peaks of LMG but with low intensity were observed in the physical mixture of LMG and Aeroperl® 300. The optimized S-SNEDDS (S2) did not show the sharp peaks of LMG, confirming the transformation of LMG from the crystalline state to amorphous or molecularly dispersed state in the S-SNEDDS formulation.68
Figure 6C shows the scanning electron micrographs of LMG, Aeroperl® 300, PM, and the optimized S-SNEDDS. LMG appeared as irregular broken crystals.69 While the mesoporous structure of Aeroperl® 300 appeared as void granules with smooth spherical surface.70 The PM showed both the irregular crystals of LMG and the void spherical shape of Aeroperl® 300. LMG irregular crystals disappeared in the scanning electron micrograph of S-SNEDDS (S2) indicating that the drug is completely dispersed in the S-SNEDDS formulation without any precipitation nor crystallization.31
In-vitro Drug Release Studies
In-vitro drug release studies from the prepared S-SNEDDS, plain LMG, and Lamictal® are shown in Figure 4B. The release profile of LMG from all formulations was faster in 0.1 N HCl than in PB as mentioned in the release of liquid SNEDDS. The drug released from liquid SNEDDS and from Lamictal® in 0.1 N HCl after the first 5 min was faster than from all S-SNEDDS formulations. This delay in drug released for S-SNEDDS may be due to the desorption process from the adsorbent carrier.71 But after 15 min, the highest drug released was from S2 (100%), followed by S5 (91.07%), and S4 (90.45%), compared to LMG (73.40%) and Lamictal® (79.43%). The drug released in PB was also higher at the first 10 min from Lamictal®, then the drug released from S2 after 15 min began to rise till reaching 100% after 45 min, compared to LMG (30.46%) and Lamictal® (92.08%). The drug release kinetics of all S-SNEDDS formulations prepared with 1:1 SNEDDS: solid carrier ratio, S7, pure LMG and Lamictal® tablets obey 1st order kinetics which indicates that the drug release from porous matrix was proportional to the amount of drug remaining in its interior.72 Whereas, S-SNEDDS formulations prepared with 1:0.75 SNEDDS: solid carrier ratio, except S7, obey Higuchi model which indicates that the drug release occurs through the pores in the matrix.73 The drug release mechanism of S2, S6, pure LMG, and Lamictal® tablets showed non-Fickian transport (n <1), while other S-SNEDDS showed super case II transport (n > 1).
Statistical Analysis and Optimization of LMG-Loaded S-SNEDDS
The analyzed data of the evaluation tests using the statistical program Design Expert® Software showed that the mathematical model selected for the S-SNEDDS design was two-factor interaction. The only response that gave significance was in-vitro drug release after 15 min in PB at pH 6.8 (p-value = 0.0258). The significant factors are X1, X2, X3, and X1X3. It was noted that the predicted R2 value (0.9879) was in a reasonable agreement with the adjusted R2 value (0.9987). The adequate precision with a ratio of 91.35 was detected. The insignificance and the negative adjusted and predicted R2 values of the in-vitro drug release at pH 1.2 after 5 and 15 min implies that the overall mean may be a better predictor of the responses. This might be due to faster drug release profile from all formulation due to LMG pH-dependent solubility and that the release at pH 1.2 was not affected by the studied factors. The insignificance and negative predicted R2 values of the in-vitro drug release in PB after 5 min and 30 min may be due to delay in drug release because of the desorption process from the adsorbent for release after 5 min and similar drug release values after 30 min as the majority of the drug was released (>70%) from all formulations. Response 3-D plots for the effect of independent factors on the responses are shown in Figure 3B. The solutions obtained from the optimization showed that S2 gave the best results regarding the criteria set for optimization with a desirability value of 0.907 as shown in Figure 5B, with discarding the second solution (S4) because of its lower desirability value (0.356). A high correlation was observed between the observed (86.36%) and predicted (86.51%) value for % of drug released after 15 min in PB of S2, confirming the validity of the design. Therefore, S2 was selected for further investigations.
Particle Size Analysis and Transmission Electron Microscope (TEM)
R9 used in the preparation of the optimized S-SNEDDS (S2) gave a z-average diameter of 15.013 ± 0.158 nm with PDI of 0.245 ± 0.018, which is similar to R9 droplet size mentioned above by Coulter Counter N4 PLUS Submicron Particle Sizer (16.3 ± 0.148 nm), and a zeta potential of −7.97 mV. The morphology of the formed nanoemulsion of R9 used in the preparation of the optimized S-SNEDDS (S2) is shown in Figure 7. All globules formed were spherical in shape and each oil droplet appeared as a dark spot with a slightly bright surrounding of S/Cos layer. The diameter of the globule size measured was close to the size measured by Malvern zetasizer.
 |
Figure 7 Transmission electron microscope image of optimized SNEDDS (R9). |
Stability Studies
The optimized LMG-loaded S-SNEDDS (S2) samples showed no obvious change in appearance, drug content, nor in-vitro release studies after storage at 40 ± 2 ºC and 75 ± 5% RH for three months. This indicates that LMG is physically and chemically stable in the optimized S-SNEDDS.
In-vivo Studies
The calibration curve of LMG showed a linear response across the concentration used from 10 to 6000 ng/mL and a linear relationship between LMG concentration and its peak ratio to the IS with 0.9993 coefficient of determination (R2). As a result, the method of assay can be used for the quantitative determination of the drug in plasma. A good separation was achieved between LMG and IS as the retention time was 0.36 min for LMG, and 0.586 min for IS. The mean plasma concentration-time curve of LMG following single oral administration of 25 mg of the drug as Lamictal® tablets, pure LMG capsules, and the prepared S2 capsules are shown in Figure 8. The mean pharmacokinetic parameters are tabulated in Table 7. The results showed that Cpmax of S2 (5.25 ± 0.664 µg/mL) was increased by 2.03 folds compared to pure LMG capsule (2.58 ± 0.274 µg/mL), and by 1.45 folds compared to Lamictal® tablet (3.62 ± 0.234 µg/mL), indicating that S2 capsules improved the oral absorption of LMG. The Tmax of LMG was 1 h after administration of S2 capsules and Lamictal® tablets compared to 1.5 h for pure LMG capsules. The AUC0-∞ of S2 (41.74 ± 4.044 µg.h/mL) was increased by 2.03 folds compared to pure LMG (20.53 ± 2.069 µg.h/mL), and 1.605 folds compared to Lamictal® tablets (26.002 ± 3.81 µg.h/mL). Hence, the relative percentage BA of S2 was 203.31% and 160.53%, with respect to pure LMG and Lamictal® respectively, indicating that the amount of drug absorbed by S2 capsules was remarkably higher than both pure LMG capsules and Lamictal® tablets. This was confirmed by the statistical analysis of pharmacokinetic parameters using non-parametric Mann–Whitney’s test which showed that the mean rank of S2 was higher than Lamictal® tablets regarding to Cpmax, AUC0-∞, and elimination t1/2, and higher than pure LMG regarding to Cpmax, AUC0-∞, and Tmax, and showed significant difference (p < 0.05). The enhancement in the BA of LMG loaded S-SNEDDS may be due to the lymphatic absorption of the drug to the systemic circulation.74 Moreover, the presence of bioenhancers as CR-EL and T80 surfactants was reported to further improve the BA of active ingredients by facilitating transcellular and paracellular absorption.75 Thus, it could be concluded that LMG-based S-SNEDDS could be formulated with lower dose due to the enhanced LMG bioavailability.
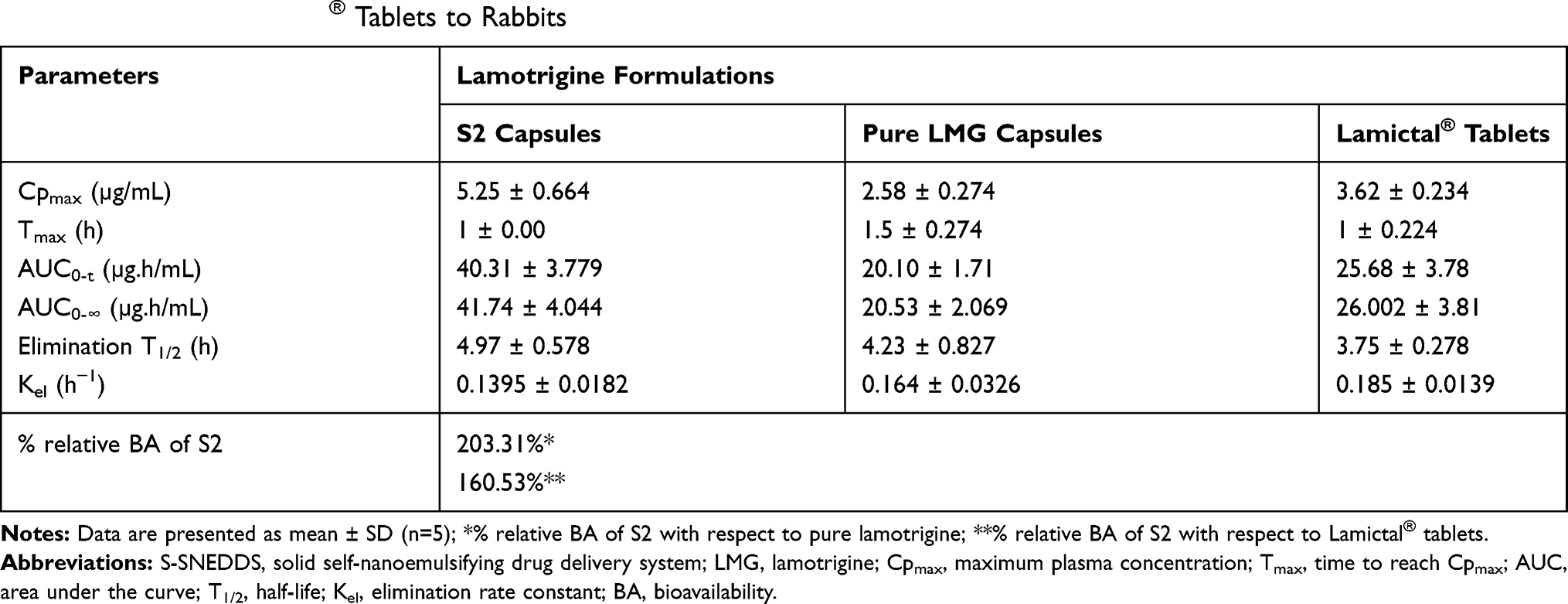 |
Table 7 Pharmacokinetic Parameters of Lamotrigine After Single Oral Administration of Optimized S-SNEDDS (S2) Capsules, Pure LMG Capsules and Lamictal® Tablets to Rabbits |
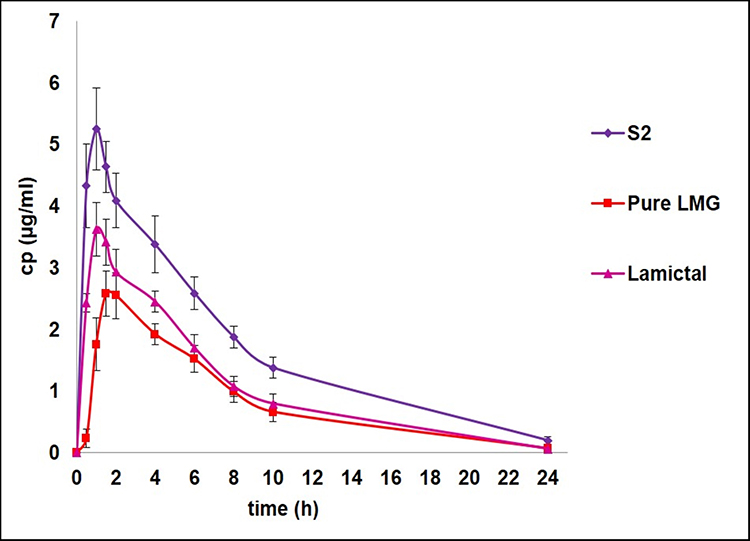 |
Figure 8 Plasma concentration-time profiles of LMG after single oral administration of optimized S-SNEDDS (S2) capsules, pure LMG capsules, and Lamictal® tablets to rabbits. |
Conclusion
The preparation of LMG-loaded SNEDDS formulations enhanced the solubility and dissolution of LMG. All systems prepared were in the nano-size range with short emulsification time, high % transmittance values and acceptable in-vitro drug release profiles compared to both pure LMG and Lamictal® tablet. Moreover, the in-vivo study after oral administration of the optimized S-SNEDDS showed 2.03, and 1.605 folds increase in the bioavailability with respect to pure LMG and Lamictal® respectively. Hence, the developed S-SNEDDS proved to be a promising carrier for LMG for the management and treatment of epilepsy and bipolar disorder.
Acknowledgments
The authors are so grateful to the support staff of Pharmaceutics Department, Faculty of Pharmacy in Health Sciences Center, Kuwait University and Misr University for Science and Technology for their indispensable technical support during conducting the practical work of this research.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gurav YA, Sayyad FJ. Design of regioselective drug delivery system of lamotrigine using curdlan gum. Pharma Innov. 2019;8(4):88–93.
2. Shorvon SD, Bermejo PE, Gibbs AA, Huberfeld G, Kälviäinen R. Antiepileptic drug treatment of generalized tonic–clonic seizures: an evaluation of regulatory data and five criteria for drug selection. Epilepsy Behav. 2018;82:91–103. doi:10.1016/j.yebeh.2018.01.039
3. Shekaari H, Zafarani-Moattar MT, Mokhtarpour M, Faraji S. Exploring cytotoxicity of some choline-based deep eutectic solvents and their effect on the solubility of lamotrigine in aqueous media. J Mol Liq. 2019;283:834–842. doi:10.1016/j.molliq.2019.03.079
4. Kim YJ, Ko HH, Han ES, Lee CS. Lamotrigine inhibition of rotenone-or 1-methyl-4-phenylpyridinium-induced mitochondrial damage and cell death. Brain Res Bull. 2007;71:633–640. doi:10.1016/j.brainresbull.2006.12.006
5. Calabresi P, Picconi B, Saulle E, Centonze D, Hainsworth AH, Bernardi G. Is pharmacological neuroprotection dependent on reduced glutamate release? Stroke. 2000;31(3):766–772. doi:10.1161/01.STR.31.3.766
6. Mohammadi-Samani S, Jalali F, Tavakoli S, Ahmadi F. Solid lipid microparticles of lamotrigine: an injectable controlled release system for local delivery in nerve injuries. J Drug Deliv Sci Technol. 2014;24(4):367–372. doi:10.1016/S1773-2247(14)50075-5
7. Tao S, Sun J, Hao F, et al. Effects of sodium valproate combined with lamotrigine on quality of life and serum inflammatory factors in patients with poststroke secondary epilepsy. J Stroke Cerebrovasc Dis. 2020;29(5):104644. doi:10.1016/j.jstrokecerebrovasdis.2020.104644
8. Kolhe S, Chaudhari P, More D. In-vitro in-vivo studies of lamotrigine tablets prepared by hot melt extrusion technique. Int J Pharm Pharm Sci. 2014;6(11):65–70.
9. Vaithianathan S, Raman S, Jiang W, Ting TY, Kane MA, Polli JE. Biopharmaceutic risk assessment of brand and generic lamotrigine tablets. Mol Pharm. 2015;12(7):2436–2443. doi:10.1021/acs.molpharmaceut.5b00154
10. Tubic-Grozdanis M, Bolger MB, Langguth P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J. 2008;10(1):213–226. doi:10.1208/s12248-008-9023-x
11. Tsume Y, Mudie DM, Langguth P, Amidon GE, Amidon GL. The biopharmaceutics classification system: subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur J Pharm Sci. 2014;57:152–163. doi:10.1016/j.ejps.2014.01.009
12. Soliman KA, Ibrahim HK, Ghorab MM. Formulation of avanafil in a solid self-nanoemulsifying drug delivery system for enhanced oral delivery. Eur J Pharm Sci. 2016;93:447–455. doi:10.1016/j.ejps.2016.08.050
13. Makadia HA, Bhatt AY, Parmar RB, Paun JS, Tank HM. Self-nano emulsifying drug delivery system (SNEDDS): future aspects. Asian J Pharm Res. 2013;3(1):21–27.
14. Fahmy UA, Ahmed OA, Hosny KM. Development and evaluation of avanafil self-nanoemulsifying drug delivery system with rapid onset of action and enhanced bioavailability. AAPS PharmSciTech. 2015;16(1):53–58. doi:10.1208/s12249-014-0199-3
15. Nasr A, Gardouh A, Ghorab M. Novel solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of olmesartan medoxomil: design, formulation, pharmacokinetic and bioavailability evaluation. Pharmaceutics. 2016;8(3):20. doi:10.3390/pharmaceutics8030020
16. Beg S, Swain S, Singh HP, Patra CN, Rao MB. Development, optimization, and characterization of solid self-nanoemulsifying drug delivery systems of valsartan using porous carriers. AAPS PharmSciTech. 2012;13(4):1416–1427. doi:10.1208/s12249-012-9865-5
17. Desai NS, Nagarsenker MS. Design and evaluation of self-nanoemulsifying pellets of repaglinide. AAPS PharmSciTech. 2013;14(3):994–1003. doi:10.1208/s12249-013-9990-9
18. Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13(13–14):606–612. doi:10.1016/j.drudis.2008.04.006
19. Majee SB, Avlani D, Biswas GB. HPMC as capsule shell material: physicochemical, pharmaceutical and biopharmaceutical properties. Int J Pharm Pharm Sci. 2017;9(10):1–6. doi:10.22159/ijpps.2017v9i10.20707
20. Mahmoud EA, Bendas ER, Mohamed MI. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS PharmSciTech. 2009;10(1):183–192. doi:10.1208/s12249-009-9192-7
21. Midha K, Nagpal M, Singh G, Aggarwal G. Prospectives of solid self-microemulsifying systems in novel drug delivery. Curr Drug Del. 2017;14(8):1078–1096. doi:10.2174/1567201813666160824123504
22. Rao BC, Vidyadhara S, Sasidhar RL, Chowdary YA. Formulation and evaluation of liquid loaded tablets containing docetaxel-self nano emulsifying drug delivery systems. Trop J Pharm Res. 2015;14(4):567–573. doi:10.4314/tjpr.v14i4.2
23. Gumaste SG, Serajuddin ATM. Development of solid SEDDS, VII: effect of pore size of silica on drug release from adsorbed self-emulsifying lipid-based formulations. Eur J Pharm Sci. 2017;110:134–147. doi:10.1016/j.ejps.2017.05.014
24. Grumezescu AM. Lipid Nanocarriers for Drug Targeting. William Andrew; 2018.
25. Talekar SD, Dave RH. Solubility enhancement of a BCS class II drug using granulated fumed silica as an adsorbent. J Pharm Res Int. 2017;18(6):1–15. doi:10.9734/JPRI/2017/36872
26. Truong DH, Tran TH, Ramasamy T, et al. Development of solid self-emulsifying formulation for improving the oral bioavailability of erlotinib. AAPS PharmSciTech. 2016;17(2):466–473. doi:10.1208/s12249-015-0370-5
27. Katamreddy JD, Yalavarthi PR, Rao DS, Teja SS, Battu S. In vitro characterization of statistically optimized quetiapine-loaded self-nanoemulsified systems with quality by design. Int J Pharm Investig. 2018;8(1):14–23. doi:10.4103/jphi.JPHI_9_18
28. Mendes C, Buttchevitz A, Kruger JH, et al. Self-nanoemulsified drug delivery system of hydrochlorothiazide for increasing dissolution rate and diuretic activity. AAPS PharmSciTech. 2017;18(7):2494–2504. doi:10.1208/s12249-017-0735-z
29. Meena AK, Sharma K, Kandaswamy M, Rajagopal S, Mullangi R. Formulation development of an albendazole self-emulsifying drug delivery system (SEDDS) with enhanced systemic exposure. Acta Pharm. 2012;62(4):563–580.
30. Ryu K-A, Park PJ, Kim S-B, Bin B-H, Jang D-J, Kim ST. Topical delivery of coenzyme q10-loaded microemulsion for skin regeneration. Pharmaceutics. 2020;12(4):332. doi:10.3390/pharmaceutics12040332
31. Abd-Elhakeem E, Teaima MHM, Abdelbary GA, El Mahrouk GM. Bioavailability enhanced clopidogrel -loaded solid SNEDDS: development and in-vitro/in-vivo characterization. J Drug Deliv Sci Technol. 2019;49:603–614. doi:10.1016/j.jddst.2018.12.027
32. Dixit AR, Rajput SJ, Patel SG. Preparation and bioavailability assessment of SMEDDS containing valsartan. AAPS PharmSciTech. 2010;11(1):314–321. doi:10.1208/s12249-010-9385-0
33. AboulFotouh K, Allam AA, El-Badry M, El-Sayed AM. Development and in vitro / in vivo performance of self-nanoemulsifying drug delivery systems loaded with candesartan cilexetil. Eur J Pharm Sci. 2017;109:503–513. doi:10.1016/j.ejps.2017.09.001
34. Kaur B, Sharma M. Self-microemulsifying drug delivery system-A recent approach in drug delivery system. Curr Pharm Res. 2018;8(4):2504–2513. doi:10.33786/JCPR.2018.v08i04.003
35. Kundarapu S, Srinivas M, Srilalitha G, Sharma JVC. Design and characterization of self emulsifying drug delivery system of repaglinide. Int J Pharm Sci Rev Res. 2014;25(1):41–46.
36. Singh MA, Singh V, Rawat G, Juyal D. Self emulsifying systems: a review. Asian J Pharm. 2015;9(1):13–18. doi:10.4103/0973-8398.150031
37. Czajkowska-Kośnik A, Szekalska M, Amelian A, Szymańska E, Winnicka K. Development and evaluation of liquid and solid self-emulsifying drug delivery systems for atorvastatin. Molecules. 2015;20(12):21010–21022. doi:10.3390/molecules201219745
38. Reddy SM, Reddy MS, Reddy NS, Reddy O. Formulation and evaluation of novel lipid based solid self-nano emulsifying drug delivery system of repaglinide. Int J Pharm Sci. 2014;6:4.
39. Gamal W, Fahmy RH, Mohamed MI. Development of novel amisulpride-loaded liquid self-nanoemulsifying drug delivery systems via dual tackling of its solubility and intestinal permeability. Drug Dev Ind Pharm. 2017;43(9):1530–1538. doi:10.1080/03639045.2017.1322607
40. Suresh G, Yogesh N, Darshana B, Rashmi T, Milind U, Jayshree T. Development and evaluation of lamotrigine soya lecithin solid dispersion: in vitro and pharmacodynamic investigation. Int J App Pharm. 2019;12:1.
41. Gunjal A, Tajane M, Londhe R. Formulation and evaluation of lamotrigine 25 mg immediate release tablet. J Drug Deliv Ther. 2019;9(4–s):295–297.
42. Aronson H. Correction factor for dissolution profile calculations. J Pharm Sci. 1993;82(11):1190. doi:10.1002/jps.2600821126
43. Nawale R, Salunke PB, Jadhav AB. Ketoprofen loaded solid self emulsifying drug delivery system (SEDDS): development and optimization. Int J Pharm Sci Rev Res. 2015;33:102–108.
44. Huang J, Wang Q, Sun R, Li T, Xia N, Xia Q. A novel solid self-emulsifying delivery system (SEDS) for the encapsulation of linseed oil and quercetin: preparation and evaluation. J Food Eng. 2018;226:22–30. doi:10.1016/j.jfoodeng.2018.01.017
45. Lau E. 5 - preformulation studies. In: Ahuja S, Scypinski S, editors. Separation Science and Technology. Vol. 3. Academic Press; 2001:173–233.
46. Sherrington LA, Sherrington A. Guaifenesin. In: Brittain HG, editor. Analytical Profiles of Drug Substances and Excipients. Vol. 25. Academic Press; 1998:121–164.
47. Du S, Wang Y, Wu S, et al. Two novel cocrystals of lamotrigine with isomeric bipyridines and in situ monitoring of the cocrystallization. Eur J Pharm Sci. 2017;110:19–25. doi:10.1016/j.ejps.2017.06.001
48. Cheney ML, Shan N, Healey ER, et al. Effects of crystal form on solubility and pharmacokinetics: a crystal engineering case study of lamotrigine. Cryst Growth Des. 2010;10(1):394–405. doi:10.1021/cg901010v
49. Singh H, Nathani S, Singh N, et al. Development and characterization of solid-SNEDDS formulation of DHA using hydrophilic carrier with improved shelf life, oxidative stability and therapeutic activity. J Drug Deliv Sci Technol. 2019;54:101326. doi:10.1016/j.jddst.2019.101326
50. Paarakh MP, Jose PA, Setty C, Peter G. Release kinetics–concepts and applications. Int J Pharm Res Technol. 2018;8:12–20.
51. Jaiswal P, Aggarwal G, Harikumar SL, Singh K. Development of self-microemulsifying drug delivery system and solid-self-microemulsifying drug delivery system of telmisartan. Int J Pharm Investig. 2014;4(4):195. doi:10.4103/2230-973X.143123
52. Salem HF, Kharshoum RM, Halawa AKA, Naguib DM. Preparation and optimization of tablets containing a self-nano-emulsifying drug delivery system loaded with rosuvastatin. J Liposome Res. 2018;28(2):149–160. doi:10.1080/08982104.2017.1295990
53. Atef E, Belmonte AA. Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system (SEDDS). Eur J Pharm Sci. 2008;35(4):257–263. doi:10.1016/j.ejps.2008.07.004
54. Sagar K, Kendre P, Pande V, Chaundhari V. Design, development, and characterization of Self Nanoemulsifying Drug Delivery System (SNEDDS) of nateglinide. World J Pharm Pharm Sci. 2014;3(8):794–811.
55. Mohan A, Madhavi M, Swetha G, Jyosthna P. Preparation, in vitro and in vivo characterization of solid dispersions of lamotrigine using solvent evaporation technique. IOSR J Pharm. 2015;5(1):54–59.
56. Bary AA, El-Gazayerly ON, Alburyhi MM. Formulation of immediate release lamotrigine tablets and bioequivalence study. J Chem Pharm Res. 2013;5(10):266–271.
57. Hotha KK, Kumar SS, Bharathi DV, Venkateswarulu V. Rapid and sensitive LC-MS/MS method for quantification of lamotrigine in human plasma: application to a human pharmacokinetic study. Biomed Chromatogr. 2012;26(4):491–496. doi:10.1002/bmc.1692
58. Homayoun M, Seghatoleslam M, Pourzaki M, Shafieian R, Hosseini M, Bideskan AE. Anticonvulsant and neuroprotective effects of rosa damascena hydro-alcoholic extract on rat hippocampus. Avicenna J Phytomed. 2015;5(3):260.
59. Bahr TA, Rodriguez D, Beaumont C, Allred K. The effects of various essential oils on epilepsy and acute seizure: a systematic review. Evid Based Complement Alternat Med. 2019;2019:1–14. doi:10.1155/2019/6216745
60. Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60(6):625–637. doi:10.1016/j.addr.2007.10.010
61. Constantinides PP, Lancaster CM, Marcello J, et al. Enhanced intestinal absorption of an RGD peptide from water-in-oil microemulsions of different composition and particle size. J Control Release. 1995;34(2):109–116. doi:10.1016/0168-3659(94)00129-I
62. Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. doi:10.1016/j.addr.2007.10.014
63. Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–1572. doi:10.1023/A:1016268311867
64. Kang JH, Oh DH, Oh Y-K, Yong CS, Choi H-G. Effects of solid carriers on the crystalline properties, dissolution and bioavailability of flurbiprofen in solid self-nanoemulsifying drug delivery system (solid SNEDDS). Eur J Pharm Biopharm. 2012;80(2):289–297. doi:10.1016/j.ejpb.2011.11.005
65. Singh S, Shyale S, Karade P. Formulation and evaluation of orally disintegrating tablets of lamotrigine. Int J Pharm Sci Nanotech. 2015;8(2):2881–2888.
66. Kalamkar R, Wadher S. Formulation and pharmacokinetic evaluation of phosal based zaltoprofen solid self-nanoemulsifying drug delivery system. Pharm Nanotechnol. 2019;7(4):328–338. doi:10.2174/2211738507666190802141754
67. Naseef MA, Ibrahim HK, Nour SAE-K. Solid form of lipid-based self-nanoemulsifying drug delivery systems for minimization of diacerein adverse effects: development and bioequivalence evaluation in albino rabbits. AAPS PharmSciTech. 2018;19(7):3097–3109. doi:10.1208/s12249-018-1138-5
68. Inugala S, Eedara BB, Sunkavalli S, et al. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: in vitro and in vivo evaluation. Eur J Pharm Sci. 2015;74:1–10. doi:10.1016/j.ejps.2015.03.024
69. Rahman Z, Zidan AS, Samy R, Sayeed VA, Khan MA. Improvement of physicochemical properties of an antiepileptic drug by salt engineering. AAPS PharmSciTech. 2012;13(3):793–801. doi:10.1208/s12249-012-9800-9
70. Wei Q, Keck CM, Müller RH. Preparation and tableting of long-term stable amorphous rutin using porous silica. Eur J Pharm Biopharm. 2017;113:97–107. doi:10.1016/j.ejpb.2016.11.009
71. Weerapol Y, Limmatvapirat S, Nunthanid J, Sriamornsak P. Self-nanoemulsifying drug delivery system of nifedipine: impact of hydrophilic-lipophilic balance and molecular structure of mixed surfactants. AAPS PharmSciTech. 2014;15(2):456–464. doi:10.1208/s12249-014-0078-y
72. Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. doi:10.1016/S0928-0987(01)00095-1
73. Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol Pharm. 2010;67(3):217–223.
74. Li F, Hu R, Wang B, et al. Self-microemulsifying drug delivery system for improving the bioavailability of huperzine A by lymphatic uptake. Acta Pharm Sin B. 2017;7(3):353–360. doi:10.1016/j.apsb.2017.02.002
75. Basalious EB, Shawky N, Badr-Eldin SM. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: development and optimization. Int J Pharm. 2010;391(1–2):203–211. doi:10.1016/j.ijpharm.2010.03.008
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.