")
Back to Journals » International Journal of Nanomedicine » Volume 15
Development and in vitro Evaluation of Gastro-protective Aceclofenac-loaded Self-emulsifying Drug Delivery System
Authors Jianxian C, Saleem K, Ijaz M , Ur-Rehman M, Murtaza G, Asim MH
Received 17 February 2020
Accepted for publication 15 June 2020
Published 23 July 2020 Volume 2020:15 Pages 5217—5226
DOI https://doi.org/10.2147/IJN.S250242
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Chen Jianxian,1,2 Kalsoom Saleem,3 Muhammad Ijaz,4 Masood Ur-Rehman,3 Ghulam Murtaza,4 Mulazim Hussain Asim5
1School of Economics, Capital University of Economics and Business, Beijing, People’s Republic of China; 2Chapter of traditional Chinese Medicine, China Information Industry Association, Beijing, China; 3Riphah Institute of Pharmaceutical Sciences, Islamabad, Pakistan; 4COMSATS University Islamabad, Lahore Campus, Lahore 54000, Pakistan; 5Institute of Pharmaceutical Technology, University of Innsbruck, Innsbruck 6020, Austria
Correspondence: Muhammad Ijaz
Lahore Campus, COMSATS University Islamabad, Lahore 54000, Pakistan
Tel +0092-42-99204787
Email [email protected]
Masood Ur-Rehman
Riphah Institute of Pharmaceutical Sciences, Islamabad, Pakistan
Email [email protected]
Aim: Chronic use of oral nonsteroidal anti-inflammatory drugs (NSAIDs) is commonly associated with gastric irritation and gastric ulceration. Therefore, the aim of study was to develop a novel oral drug delivery system with minimum gastric effects and improved dissolution rate for aceclofenac (ACF), a model BCS class-II drug.
Methods: Self-emulsifying drug delivery systems (SEDDS) were formulated to increase the solubility and ultimately the oral bioavailability of ACF. Oleic acid was used as an oil phase, Tween 80 (T80) and Kolliphor EL (KEL) were used as surfactants, whereas, polyethylene glycol 400 (PEG 400) and propylene glycol (PG) were employed as co-surfactants. Optimized formulations (F1, F2, F3 and F4) were analyzed for droplet size, poly dispersity index (PDI), cell viability studies, in vitro dissolution in both simulated gastric fluid and simulated intestinal fluid, ex vivo permeation studies and thermodynamic stability.
Results: The optimized formulations showed mean droplet sizes in the range of 111.3 ± 3.2 nm and 470.9 ± 12.52 nm, PDI from 244.6 nm to 389.4 ± 6.51 and zeta-potential from − 33 ± 4.86 mV to − 38.5 ± 5.15 mV. Cell viability studies support the safety profile of all formulations for oral administration. The in vitro dissolution studies and ex vivo permeation analysis revealed significantly improved drug release ranging from 95.68 ± 0.02% to 98.15 ± 0.71% when compared with control. The thermodynamic stability studies confirmed that all formulations remain active and stable for a longer period.
Conclusion: In conclusion, development of oral SEDDS might be a promising tool to improve the dissolution of BCS class-II drugs along with significantly reduced exposure to gastric mucosa.
Keywords: self-emulsification, BCS class-II, solubility, gastric irritation, gastro-protective, nano-emulsion
Introduction
Aceclofenac (ACF) is a nonsteroidal anti-inflammatory drug (NSAID) which belongs to BCS Class II with marked anti-inflammatory and analgesic properties, finding its use in the management of pain-related disorders including rheumatoid arthritis, osteoarthritis and ankylosing spondylitis.1,2 Like many other NSAIDs when administered orally for a prolonged time, ACF can lead to gastric ulcers and gastric mucosal irritation.3 Prolonged exposure of NSAIDs at the gastric mucosal membrane can cause local irritation and inhibition of prostaglandin synthesis, ultimately leading to the gastric ulceration. Gastric intolerance can be minimized significantly, when the direct contact of drug with gastric mucosal membrane is minimized.4 Numerous studies have been reported to tackle the problem of gastric irritation and intolerance, these studies have investigated the use of combinations of extended release ACF with immediate release prostaglandins, use of soft gelatin capsules loaded with drug and solubilizer, use of solid dispersion systems, use of surfactants, and complexation of drug with cyclodextrin.5–8
All the practices reported by the above mentioned studies look very promising in cases of improving the drug dissolution, but not a single study had addressed the concern of gastric side effects. With the increased drug dissolution, it is likely that more and more free drug becomes available for direct contact with gastric mucosa, which could aggravate the irritation and lead to increased drug intolerance. Therefore, there is a dire need of a formulation that not only improves the dissolution of ACF, but also reduces the direct contact time of the active drug contents with the gastric mucosa. Furthermore, such formulation should have a simple method of preparation so that it can be easily adopted at commercial level.
Lipid-based drug delivery systems have gained attention in recent years for improving the solubility of poorly water-soluble drugs as they have been found advantageous in enhancing the oral bioavailability of lipophilic drugs. The commonly used lipid drug delivery systems include lipid-based nanoparticles (nanoemulsions, solid lipid nanoparticles, lipid drug conjugates, nanosuspensions, liposomes, lipid nanocapsules and liquid crystalline nanoparticles).9 Among the lipid-based drug delivery systems, SEDDS are promising and innovative delivery systems. Self-emulsifying drug delivery systems (SEDDS) have emerged as potential drug delivery systems for solubility of poorly soluble drugs by increasing the surface area of drug particles and increasing the permeability through the membranes.10 During the last two decades SEDDS-based registered drugs gained a significantly high share in the markets in the US, UK, and Japan.11 SEDDS have been characterized as isotropic mixtures of natural or synthetic oil, surface active agents or, alternatively, a single or several hydrophilic solvents and co-solvents with a drug that spontaneously forms oil-in-water nanoemulsions in aqueous media following gentle stirring.12,13 The lipophilic drugs are easily encapsulated within small diameter droplets, which provide a large surface area for absorption resulting in increased bioavailability.14 In addition, lipid-based formulations provide protection against the harsh environment of the gastrointestinal tract. SEDDS have also demonstrated significantly enhanced oral bioavailability of easily metabolized and poorly soluble drugs.15
The present study is aimed at designing ACF-loaded SEDDS formulations to enhance solubility of ACF with significantly reduced exposure to the gastric mucosa. Analytical and qualitative characterization of optimized ACF-loaded SEDDS were carried out. The in vitro dissolution studies and ex vivo permeation studies were carried out to ascertain the release and permeation/diffusion of the drug for developed ACF-SEDDS.
Materials and Methods
Material
ACF 97.99% was donated from Global Pharmaceutical Co., Ltd (Islamabad, Pakistan). Capryol 90 (Gattefosse SAS, France), Tween 80, Span 80 (Sigma-Aldrich Co., St Louis, MO, USA), kolliphor EL, transcutol P (Glentham Life Sciences, Wiltshire, UK), propylene glycol, PEG 400 (VMR Chemicals, UK), miglyol 812, clove oil, oleic acid and ethyl oleate (Aladdin Industrial Corporation, Shanghai, China). Caco-2 cell line was purchased from Sigma-Aldrich Co., Germany. All chemicals were of analytical grades.
Solubility Studies
To find appropriate compositions of the SEDDS formulations, solubility profile of ACF in various oils, surfactants and co-surfactants was measured. In brief, excessive quantity of ACF was added into 5 mL of each vehicle in different glass vials. After sealing the glass vials, the systems were stirred at 37 ± 0.5°C for 72 h, followed by ultrasonic treatment for three minutes and then centrifuged at 3000 rpm for 10 min. The supernatants were filtered using 0.45 μm polytetrafluoroethylene (PTFE)) membrane filters. The supernatant samples were diluted with methanol and drug concentrations were determined by UV spectroscopy at λmax of 276 nm. Each sample was analyzed in triplicate.16
Selection of Excipients Based on Solubility Studies
All the excipients of the optimized SEDDS formulations were selected based on solubility studies of ACF. Oleic acid displayed improved solubility and was selected as the oil phase, Tween 80 and kolliphor EL were selected as surfactants and PEG 400 and propylene glycol (PG) as co-surfactants. All the selected excipients were gently mixed into two combinations for the development of self-emulsifying systems. The combinations of selected excipients were, Group I (oleic acid, Tween 80, and PEG 400), and Group II (oleic acid, kolliphor EL and PG). The selected surfactants and co-surfactants were blended in three weight ratios of 1:1, 2:1, and 3:1 at room temperature. The mixture of surfactant and co-surfactant is called as Smix, also known as Km.17 In the second step of formulation development, oils and Smix were mixed thoroughly in different weight ratios ranging from 1:1 to 1:9 separately, in different 20 mL stoppered glass vials. The systems (oils + Smix) were slowly stirred avoiding any bubble formation while addition of aqueous phase was done using micropipette. The clarity of the developed self-emulsifying systems was visually assessed using the following grading to identify the uniform and clear nanosystems.
- Nanoemulsion (oil in water) (NE): clear, transparent, isotropic mixtures, easily flowable
- Nanoemulsion gel (NG): clear and highly viscous gel-like mixture
- Macroemulsion (ME): milky or cloudy mixture
- Emulgel (EG): milky and highly viscous gel18
Systems that appeared nanoemulsions were assigned for further studies.
Preparation of ACF-loaded SEDDS
The SEDDS formulations displaying clear nanoemulsions (Type A) were then loaded with ACF. Each 1 mL of system was loaded with 100 mg of ACF. The systems were mixed thoroughly under constant stirring of 1200 rpm at 37 ± 0.5°C for 15 min or until the appearance of clear systems and filtered through 0.45 μm membrane filter. ACF-SEDDS formulations were then diluted with methanol, centrifuged and analyzed on UV-visible spectrophotometer at a λ max of 276 nm and the entrapment efficiency was calculated. Each sample was analyzed in triplicate. These formulations were then stored at ambient temperature for further studies. The composition of different optimized formulations is shown in Table 1. The percentage entrapment efficiency and loading capacity of all the formulations were determined by the following formula.
 |
Table 1 Composition of the Optimized Formulations Displaying the Ratio of Components used for the Development of Self-emulsifying Systems Loaded with ACF (ACF-SEDDS) |
Entrapment Efficiency
% EE = [(total drug – free drug)/total drug] *100
Loading Capacity
% LC = [entrapped drug/drug loaded nanoemulsion weight] * 100
Particle Size, Zeta-potential and Polydispersity Index (PDI) Analysis
The estimation of zeta potential, particle size and PDI for selected ACF-SEDDS formulations was performed using ZS 90 Zetasizer (Malvern Instruments, Worcestershire, UK). Each sample was analyzed in triplicate. For the determination of mean droplet size and size distribution, photon correlation spectroscopy was carried out at 25°C. Each formulation was diluted to the appropriate concentration with distilled water to clear nanoemulsions. The PDI values indicate the size distribution. Thereby, lower PDI values point out a close size distribution providing good stability of nanoemulsions due to the reduced Ostwald ripening.19 All the measurements were done in triplicate and mean standard deviation was calculated.
Self-emulsification Time Determination
The efficiency of the developed SEDDS formulations for the emulsification time was tested using a standard USP dissolution apparatus type II (Galvano Scientific, Lahore, 53700, Pakistan). Briefly, 250 µL of selected SEDDS formulations were added to 500 mL of distilled water maintained at 37 ± 0.5°C and 50 rpm. Systems were then evaluated visually according to rate of emulsification or emulsification time and appearance of final nanoemulsion. All formulations were assessed using following grading system.
- Grade A: clear nanoemulsion (<1 min)
- Grade B: translucent/less clear nanoemulsion (>1 min)
- Grade C: slightly translucent nanoemulsion with bluish white appearance (within 2 min)
- Grade D: bright milky/white emulsion (longer than 2 min)
- Grade E: emulsions with either poor emulsification possessing large oil globules on the surface or no emulsion formation.20
Measurement of Cloud Point
The optimized ACF-SEDDS formulations were diluted 100 times with distilled water and kept in a water bath at 37 ± 0.5°C in different beakers. The temperature of water bath was gradually raised at increments of 1°C. The formulations were cooled to an ambient temperature and the test was repeated to check reproducibility. Similarly, all the formulations were diluted with phosphate buffer pH 6.8 at 37 ± 0.5°C and temperature was gradually raised to evaluate the cloud points.
Thermodynamic Stability Studies
Heating Cooling Cycle
For the thermodynamic stability of the formulations, six cycles between refrigerator temperature (4°C) and 45°C were carried out for each formulation for 48 h. Stable formulations were further taken for centrifugation test.
Centrifugation Test
Centrifugation test was carried out to put the nanoemulsion under a more stressful environment. Formulations having passed heating and cooling cycles were then subjected to centrifugation cycles at 6000 rpm for 30 min. Formulations showing no phase separation were subjected to freeze–thaw cycle.
Freeze–Thaw Cycle
Freeze–thaw cycle is the last stress factor that is employed to assess the very harsh conditions of freezing temperature. Three freeze–thaw cycles between −21°C and 25°C with storage at each temperature for not less than 48 h were carried out for the formulations that passed the centrifugation test.
Cell Viability Studies
For the toxicity evaluation of the excipients and the developed formulations of SEDDS, the resazurin assay was employed. The reducing environment of metabolically active living Caco-2 cells converts the blue colored nonfluorescent resazurin to red color fluorescent resorufin. In brief, the test was completed on Caco-2 cells, which were cultivated in 24-well plates for 14 days in the MEM medium and environment. Test solutions (0.15% m/v capryol 90, kolliphor EL, transcutol P, propylene glycol, PEG 400, oleic acid, ethyl oleate and 0.5 % m/v SEDDS), MEM without phenol red used as negative control and 4% m/v Triton™ X-100 as a positive control were added into the Transwell plates in 500 µL quantities. Afterward, the treated cells were kept under standard incubation conditions for three hours, the supernatant was removed and the cells were washed twice with PBS.To each Transwell, 250 µL of 2.2 µM resazurin solution was added and cells were incubated for further three hours. The aliquots of supernatants were observed at 540 nm with background subtraction at 590 nm with Tecan Group Ltd. Männedorf, Switzerland Infinite M200.
In vitro Drug Release Study
The in vitro dissolution studies were performed using USP type II dissolution apparatus by following the previously established methods. In short, the regenerated cellulose dialysis membranes with molecular weight cut-off 3500 Da (Spectrum Laboratories, Inc., Rancho Dominguez CA, USA) were soaked in buffer medium pH 6.8 for two hours prior to use. For the release studies, 2 mL of ACF-SEDDS (40 mg ACF) formulations and 2 mL ACF suspension in buffer pH 6.8 (40 mg ACF) in PEG 400 as control were sealed in dialysis membrane tubes. Afterward the dialysis tubes were suspended in 500 mL of simulated gastric fluid (SGF ie 0.05M HCl pH 1.2) or simulated intestinal fluid (SIF ie, PBS, pH 6.8) or distilled water as release medium maintained at 37 ± 0.5°C at 100 rpm. Aliquots (3 mL) were withdrawn periodically at predetermined time intervals and replenished with an equal volume of fresh medium. Aliquots were filtered with 0.45 µm PTFE membrane filters and analyzed with UV spectroscopy at a wavelength of 276 nm.21 Each sample was analyzed in triplicate.
Ex vivo Drug Permeation Studies
Sprague–Dawley male rats (250–300 g) were used for ex-vivo permeability studies. Institutional ethical approval was obtained from the Animal and Scientific ethical review board of the Ripha International University Islamabad, Pakistan. Ex-vivo studies were performed according to guidelines provided by NIH. Briefly, the rats were sacrificed, duodenal intestine was isolated, cleansed with normal saline and cleaned intestine was cut into pieces of 3.5 cm length. To remove the mucous and lumen content, intestinal pieces were washed with Ringer’s solution pre-warmed at 37 °C. Reconstituted ACF suspension (control) containing 5 mg/mL was prepared by mixing 50 mg of drug with 10 mL PBS (pH 6.8) on vortex mixer. ACF-SEDDS formulations were also reconstituted with PBS pH 6.8 to produce formulations equivalent to 5 mg/mL and injected into the lumen of the duodenum using a syringe while another side of the lumen was tightly closed with the thread. The prepared tissues were placed in a chamber of organ bath with continuous aeration and a constant temperature of 37 ± 0.5 °C. The compartment was filled with 30 mL of PBS pH 6.8. Aliquots of 3 mL were withdrawn at predetermined time intervals and replenished with fresh PBS medium. The samples were analyzed via UV spectroscopy at wavelength 276 nm for the drug contents dialyzed across the membrane. Each sample was analyzed in triplicates and results were presented as mean ± SD.22
Statistical Analysis
All the studies were done in triplicates and the results are presented as mean ± SD. Statistical analysis of evaluated data was done using DD-Solver 1.0. Difference between evaluated data was done using one-way analysis of variance (ANOVA) and Bonferroni correction post hoc t-test. Significance of data was determined at 5% level of significance (α) and p value <0.05.
Results
Solubility Studies
Solubility of ACF is very low which results in significantly low bioavailability. The log P-value of ACF is 2.170.23 Increased solubility increases the absorption of active drug molecules at the site of action. For efficient loading of lipophilic drug molecules into SEDDS formulations, solubility of (ACF) a lipophilic drug was evaluated in lipophilic solvents. Of all the five solvents (oils) evaluated for solubility, Oleic acid showed the highest solubility of ACF (185.136 ± 0.73 mg/mL) and ethyl oleate displayed the least solubility (0.7995 ± 0.68 mg/mL). From the various surfactants investigated, higher solubility of ACF was seen in Tween 80 (310 ± 0.39 mg/mL) as compared to Kolliphor EL (250.69 ± 1.05 mg/mL). Transcutol P used as a co-surfactant exhibited maximum solubility for ACF (207.57 ± 1.002 mg/mL) as compared to PEG-400 (108.68 ± 0.99 mg/mL) and PG (63.55 ± 0.24 mg/mL) as displayed in Figure 1. Excipients with better solubility properties were used for further study. Synergistic effect of all excipients resulted in significantly improved solubility of ACF.
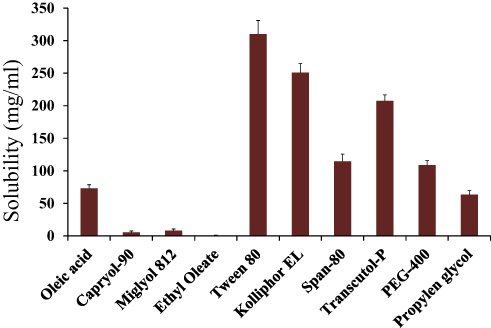 |
Figure 1 Comparative analysis of the solubility of lipophilic drug (ACF) in different type of oils, surfactants and co-surfactants performed at room temperature. |
Selection of Excipients Based on Solubility Studies
All the excipients were selected on bases of the solubility studies. The compatibility between selected surfactants, co-surfactants and oils was investigated to select the best possible combination of excipients for the formulations. All selected surfactants, co-surfactants and oils in combinations groups described previously, resulted in clear and homogenous mixtures.24 These combinations were then further mixed in different weight ratios ranging from 1:1 to 1:9. The Smix prepared in combination ratios of 1:7, 1:8 and 1:9 when mixed with oils in 2:1 ratio resulted in larger emulsification region. The nano-emulsion region narrows down as the combination ratios of Smix decrease. These systems were observed visually for emulsification region and characterized according to grading system described in “Selection of Excipients Based on Solubility Studies”.
Preparation of ACF-Loaded SEDDS
The entrapment efficiency (EE) of the optimized formulations was determined and F3 showed the highest entrapment efficiency of 76% while formulation F2 showed the highest drug loading (DL) capacity of 7.33% as presented in Table 2.
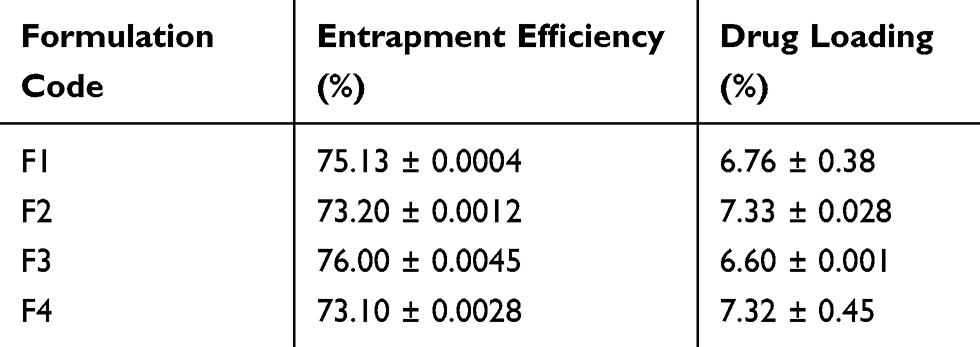 |
Table 2 Drug Loading (DL) and Percentage Entrapment Efficiency (EE) of Different Formulations of ACF-SNEDDS Determined at Room Temperature |
Particle Size and Zeta-Potential Measurements
Droplet size, zeta potential and PDI of optimized SEDDS are presented in Table 3. The zeta potential of ACF-SEDDS was found to range between −31.1 mV and −38.5 mV. The average globule size ranged between 111.3 ± 3.2 nm and 470.9 ± 12.52 nm.
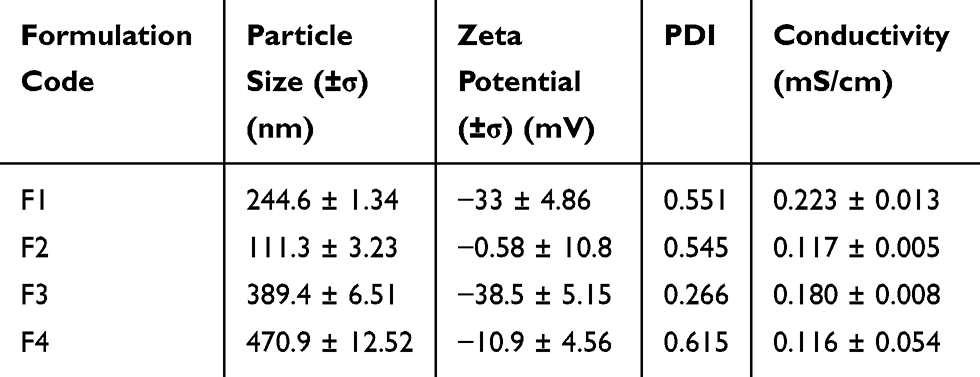 |
Table 3 Particle Size, Zeta Potential and PDI of 100 µg Optimized ACF-SNEDDS Determined in Phosphate Buffer pH 6.8 at Room Temperature |
Self-Emulsification Time Determination
The in vitro performance of ACF-SEDDS systems, were visually assessed using the grading system mentioned in Table 4. Systems were evaluated for any phase separation upon dilution. Visual observations showed that all ACF-SEDDS systems were found to be of grade A. The results are correlated with previously reported studies.20,25 Shorter emulsification time leads to stable and clear emulsions when the formulations are administered orally. SEDDS formulations loaded with ACF are immediately converted to the nano-emulsions as soon as the formulations contact with gastric fluids.26
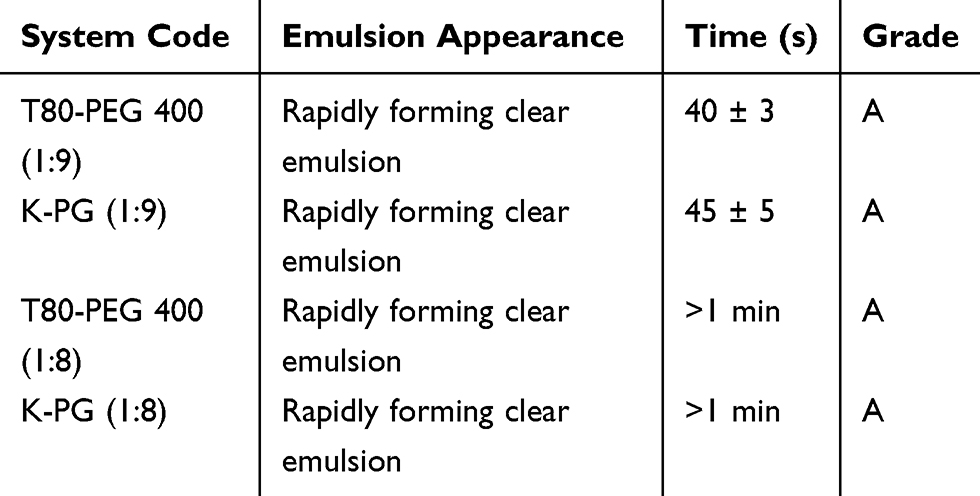 |
Table 4 Visual Observations for Disperse-Ability Test for Various SEDDS Systems |
Thermodynamic Stability Studies
All ACF-SEDDS formulations showed no sign of precipitation, cloudiness or separation after three heat-cool cycles, centrifugation and three freeze-thaw cycles, which ensured the stability of all SEDDS formulations on different temperatures and environmental fluctuations.27 Visual observation indicated that there was no phase separation or flocculation in any formulation. The results are presented in Table 5. Thus, the overall stability of all SEDDS formulations under these stress condition was found to be acceptable.
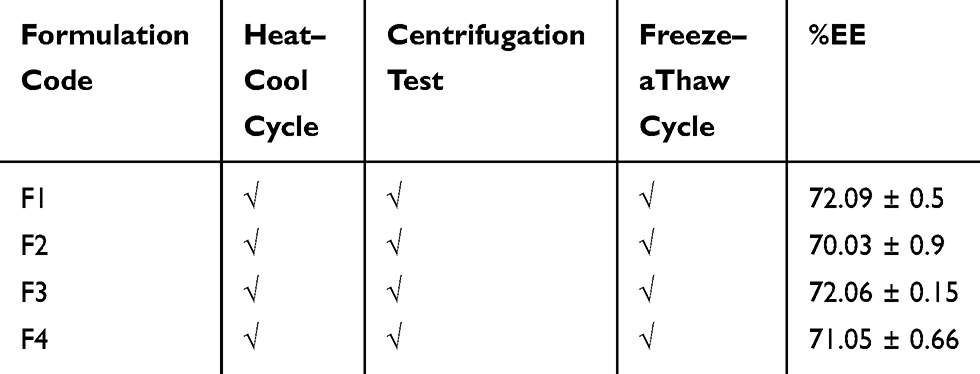 |
Table 5 Thermodynamic Stability Studies of All ACF-SEDDS Formulations Evaluated Under Various Stress Conditions |
Cell Viability Studies
Cell viability studies were carried out on Caco-2 cells by utilizing resazurin assay. All the excipients and the developed SEDDS formulations showed non-significant cytotoxic effects. Moreover, during the 24 h of the assay under the experimental conditions, cells viability of more than 80% was considered as non-toxic.10 All the excipients used for SEDDS development and all SEDDS formulations demonstrated less than 20% cell cytotoxicity as illustrated in Figure 2.
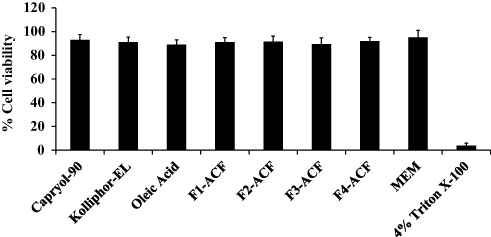 |
Figure 2 In vitro cell viability studies performed on Caco-2 cells for 24 h at pH 6.8 and 37°C, (mean ±SD, n=3). |
In vitro Drug Release
The in vitro release profile of different ACF-SEDDS formulations were carried out in SGF and SIF media at 37 °C along with pure ACF suspension as control. Normally, the reported residence time for lipid-based diet in stomach is up to 2 h and in intestine lipase enzymes degrade the lipids and digestion takes place.28 In the current study, we assessed the drug release in SGF and SIF for 6 h period to evaluate the influence of lipids on drug release. The in vitro release profile of ACF in both SGF and SIF are presented in Figures 3 and 4. As illustrated in Figure 4, in vitro release profile of ACF from SEDDS formulations produced a constantly superior drug release in PBS at pH 6.8 but on the other hand, drug release was halted in 0.05M HCL buffer at pH 1.2, as compared to pure ACF suspension as shown in Figure 3. Within the initial 2h of in vitro release study, only 24.81 ± 0.97% ACF was released in SGF and 2.43 ± 0.73% in PBS from ACF suspension whereas, in SIF medium, 74 ± 0.49% and 88.15 ± 0.71% of ACF was released from F1 and F3 formulations, respectively, and 95.68 ± 0.02% and 95.83 ± 0.04% from F2 and F4 formulations, respectively. ACF-SEDDS formulation F1 showed the improved drug release of 98.13 ± 0.2 % within 4h, F3 showed 98.83 ± 0.11% release in PBS within 6h while F2 and F4 showed 97.75 ± 0.22% and 95.73 ± 0.38% within 6h, respectively. While on the contrary, drug release in gastric media SGF pH 1.2 was significantly low. The dissolution studies were continued for 6h to detect any precipitation or variation that may occur over a period. Statistical comparison of formulations revealed that ANOVA was significant and Bonferroni corrected post hoc t-test indicating F1 was significantly higher than F2, F3 and F4 both in SGF and SIF, respectively. All samples were analyzed in triplicate and results were presented as mean ± SD.
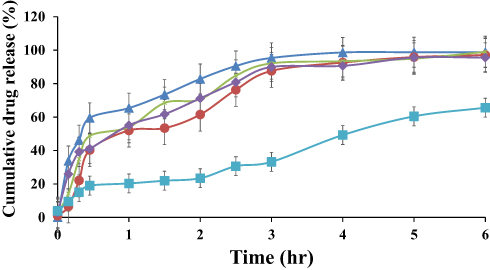 |
Figure 3 Dissolution Profile of ACF-SEDDS formulations (F1 ▲, F2 ●, F3 ×, F4 ♦) and pure ACF suspension (C, ■) carried out for six hours at 37 ± 0.5°C in SGF (0.01 MHCl, pH 1.2) (mean ±SD, n=3). |
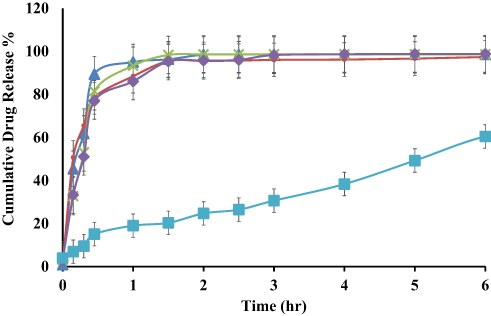 |
Figure 4 Dissolution profile of ACF-SEDDS formulations (F1 ▲, F2 ●, F3 ×, F4 ♦) and pure ACF suspension (C, ■) carried out in SIF (PBS, pH 6.8) at 37 ± 0.5°C for seven hours (mean ± SD, n=3). |
Ex vivo Drug Permeation Studies
The cumulative drug perfused through a rat’s intestine is presented in Figure 5 for ACF-SEDDS formulations and pure ACF suspension as control. It was found that cumulative drug diffused through rat intestine from all ACF-SEDDS formulations was higher than cumulative drug diffused through pure ACF suspension. The amount of drug diffused through rat intestine from all formulations can be arranged in following descending order F1>F3>F2>F4>C. The cumulative amount of drug diffused after 90 min of study from F1 was 98.70 ± 0.01%, F3 was 98.43 ± 0.33% after 150 min, F2 and F4 were 97.20 ± 0.25% and 97.25 ± 0.11% after 180 min, respectively, while alternatively the amount of drug diffused from control was 68.52 ± 0.67% after 360 min study. All formulations showed a significantly improved drug release profile compared to control (p<0.05). Statistical analysis revealed significant difference in drug release among the ACF-SEDDS formulations and the control formulation, yet Students t-test indicating F1 was significantly higher than all other formulations.
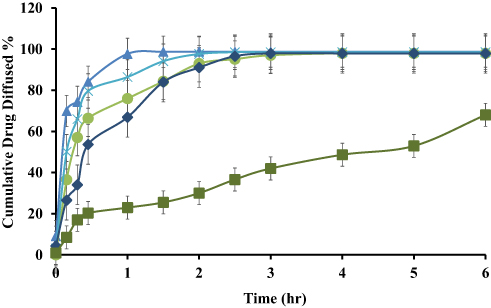 |
Figure 5 Ex vivo drug diffusion study of reconstituted ACF-SEDDS formulations (F1 ▲, F2 ●, F3 ×, F4 ♦) and pure ACF suspension (C, ■) performed in PBS, pH 6.8 at 37 ± 0.5°C using rat intestine (mean ±SD, n=3). |
Discussion
ACF is a highly potent NSAID being used for the relief of severe pain caused by rheumatoid arthritis, osteoarthritis and ankylosing spondylitis. ACF is a selective COX-2 inhibitor and it inhibits the production of inflammatory cytokines like interleukin 1, TNF and prostaglandin E2. Like other NSAIDS, ACF has very serious effects on the gastric mucus membrane due to the inhibitory effect on prostaglandin E2 and direct contact with the mucus membrane. Gastric irritation and intolerance are not only related with the prostaglandin inhibitory effects of NSAIDS, but can be related with the direct contact of these drugs with gastric mucosal membranes. SEDDS are lipid-based drug delivery systems, which not only improve the dissolution profile of lipophilic drugs, but also have additional protective properties for drug molecules against the harsh environment of the gut. SEDDS could also be used to prevent the local mucosal irritation side effects of drug molecules by hindering the immediate release of drug (ACF) in the stomach.29,30 To design the SEDDS formulations with desired physicochemical characteristics, the selection of components is the most crucial part. The drug loading capacity of SEDDS formulation depends upon the solubility of ACF in different components, which was based upon solubility studies. Solubility studies aimed at identifying the suitable oil, surfactant and co-surfactant having maximum solubilizing capacity to achieve optimum drug loading.30 Oils remain the main component/excipient of SEDDS formulation as they can increase the solubility of lipophilic drugs because they increase the fraction of drug transported via the lymphatic system and, hence, increase the absorption of lipophilic drugs. Surfactants have a vital role in stabilizing SEDDS formulation, its nature and amount determining globule size and stability.10 Nonionic surfactants are commonly preferred compared to ionic and amphiphilic surfactants because nonionic surfactants have lower toxicity and higher stability to pH and ions.31
By comparing the results for a selection of excipients, it appeared that the self-emulsifying properties of mixtures with Tween 80 are better than with kolliphor EL. Although these surfactants possess high HLB values, the difference in self-emulsifying properties can be attributed to the difference in structure and chain lengths.32
The efficiency of ACF-SEDDS could be estimated by rate of emulsification as emulsification is considered a rate limiting step in drug absorption. The results presented in Table 3 showed that short self-emulsification time indicated their ability for easy and rapid emulsification. The results also revealed that the self-emulsification is dependent on composition of system and proportion of oil to surfactant and co-surfactant. From the self-emulsification time results, it was concluded that the self-emulsification process was spontaneous and this time was decreased with the increase in surfactant concentration. It means that the developed ACF-SEDDS formulations could disperse quickly and easily under gentle agitation. Droplet size is the fundamental factor in determining the rate and extent of drug release and drug absorption. It has been reported that smaller droplet size allows faster dissolution and provides a larger surface area for drug absorption.33 The particle size of ACF-loaded SEDDS were in the range of 111.3 ± 3.23 to 470.9 ± 12.52 showing that the developed ACF-SEDDS formulations are in the range of nanoemulsions. Another crucial factor, usually considered in measuring the particle homogeneity is PDI, which varies from 0.0 to 1.0. The small PDI value indicates more uniform nanoemulsion with higher physical stability. Zeta-potential gives indication of stability of nanoemulsion. Large negative or positive zeta-potential value indicates greater repulsion between droplets creating emulsion stability.34 The zeta-potentials of selected ACF-SEDDS were −33 ± 4.86 mV and −38.5 ± 5.15 mV showing that these formulations are stable. Various ions are present in the physiological environment of the GI tract. These ions minimize the surface charge of nanoemulsions generated from self-emulsifying formulations. This is the reason that SGF shielded the negative charge of SEDDS. The zeta potential of near zero results in insufficient repulsion between droplets and subsequent aggregation, confirming the larger droplets in SGF as compared with water. A previous study also confirmed that the stomach favors nanoparticulate fusion because of its acidity and high ionic strength.35 Likewise, the negative charge of the gastric mucus layer would repel the negatively charged SEDDS formulations, shortening the gastric emptying time. Reduced gastric emptying time leads to rapid passage of formulations from the stomach which results in reduced drug release in the stomach. Reduced drug released in the stomach will ultimately reduced the exposure to gastric mucosa with reduced gastric side effects. The results from thermodynamic stability for all SEDDS formulations showed good stability under stress conditions. The results indicated that there was no sign of precipitation, crystallization or flocculation.
The cloud point is important for predicting stability and phase separation of developed formulations. It also predicts whether surfactants precipitate at higher temperatures or not because at higher temperatures, there is a possibility those surfactants may lose water from molecules causing gelling of formulation and loss of emulsification property. It was determined at temperatures higher than physiological temperature. The results conclude that all the formulations would be stable in in vivo conditions. F1 SEDDS formulation showed highest cloud point.
The enhancement in ACF in vitro release rate and extent can be attributed to spontaneous formation of emulsions during the dissolution process. Thus, this dissolution of ACF from ACF-SEDDS could lead to higher absorption and higher oral bioavailability. A good correlation between droplet size and in vitro release of ACF has been observed. Thus, this rapid drug release in the intestinal fluid was promoted by the large interfacial areas present in nanoemulsion.36 The poor performance of pure ACF suspension may be attributed to poor aqueous solubility and poor wettability of drug. Among all formulations, F1 showed significantly improved drug release with highest level of significance. The difference in drug permeation from all ACF-SEDDS formulations and pure ACF suspension may be contributed due to difference in particle size and due to higher concentration of surfactant used.
Conclusion
Various lipid excipients in different combinations and amounts were evaluated for the self-emulsifying properties based on emulsification times, droplets size, in vitro dissolution and ex vivo permeation. In the view of above results, it can be seen that SEDDS formulations exhibited highest self-emulsification in a short period of time and were thermodynamically stable which further confirmed that formulations could withstand various storage conditions with excellent stability. ACF-SEDDS formulations prepared with Tween 80, kolliphor EL showed reduced drug release in SGF media, which could be assumed for reduced gastric irritation effects when administered orally, on the other hand, significantly improved drug release was observed in SIF.Likewise, ACF-SEDDS formulations displayed improved diffusion across the ex vivo intestinal membranes compared to control and therefore, it could lead to an increase in solubility and oral bioavailability of ACF. Based on the above results, the present ACF-SEDDS would be a promising novel system for improving the solubility of lipophilic drugs with additional benefits of reduced gastric irritation.
Acknowledgments
Authors are thankful to the Dean and Chairman Riphah Institute of Pharmaceutical Sciences for providing necessary facilities to complete this work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Samuel AR, Nazar N. Efficacy of Bromelain versus Aceclofenac on post-operative analgesia following dental extractions-a randomised controlled clinical trail. J Pharm Sci Res. 2019;11(8):3006–3010.
2. Garg NK, Tyagi RK, Sharma G, et al. Functionalized lipid–polymer hybrid nanoparticles mediated codelivery of methotrexate and aceclofenac: a synergistic effect in breast cancer with improved pharmacokinetics attributes. Mol Pharm. 2017;14(6):1883–1897. doi:10.1021/acs.molpharmaceut.6b01148
3. Magliocca S, De Caro C, Lazzarato L, et al. Aceclofenac–galactose conjugate: design, synthesis, characterization, and pharmacological and toxicological evaluations. Mol Pharm. 2018;15(8):3101–3110. doi:10.1021/acs.molpharmaceut.8b00195
4. Bhatia N, Katkar K, Ashtekar S. Formulation and evaluation of co-prodrug of flurbiprofen and methocarbamol. Asian J Pharm Sci. 2016;11(3):449–458. doi:10.1016/j.ajps.2015.10.031
5. Jana S, Sen KK. Chitosan—Locust bean gum interpenetrating polymeric network nanocomposites for delivery of aceclofenac. Int J Biol Macromol. 2017;102:878–884. doi:10.1016/j.ijbiomac.2017.04.097
6. Shi L-B, Tang P-F, Zhang W, Zhao Y-P, Zhang L-C, Zhang H. Aceclofenac-hydroxypropyl-β-cyclodextrin complex for prolonged and improved drug delivery for orthopedic applications. J Biomater Tissue Eng. 2017;7(4):327–332. doi:10.1166/jbt.2017.1571
7. Kim T, Thapa S, Lee D, et al. Pharmacokinetics and anti-gastric ulceration activity of oral administration of aceclofenac and esomeprazole in rats. Pharmaceutics. 2018;10(3):152. doi:10.3390/pharmaceutics10030152
8. Sarangi M, Singh N. A comparative study of solubility enhancement of aceclofenac by solid dispersion technique using several polymers. J Appl Pharm. 2018;10(1).
9. Kalepu S, Manthina M, Padavala V. Oral lipid-based drug delivery systems–an overview. Acta Pharm Sin B. 2013;3(6):361–372. doi:10.1016/j.apsb.2013.10.001
10. Ijaz M, Bonengel S, Zupančič O, et al. Development of oral self nano-emulsifying delivery system (s) of lanreotide with improved stability against presystemic thiol-disulfide exchange reactions. Expert Opin Drug Deliv. 2016;13(7):923–929. doi:10.1517/17425247.2016.1167034
11. McClements DJ. Advances in nanoparticle and microparticle delivery systems for increasing the dispersibility, stability, and bioactivity of phytochemicals. Biotechnol Adv. 2018;38:107287.
12. Chen X-Q, Ziemba T, Huang C, et al. Oral delivery of highly lipophilic, poorly water-soluble drugs: self-emulsifying drug delivery systems to improve oral absorption and enable high-dose toxicology studies of a cholesteryl ester transfer protein inhibitor in preclinical species. J Pharm Sci. 2018;107(5):1352–1360. doi:10.1016/j.xphs.2018.01.003
13. Vithani K, Jannin V, Pouton CW, Boyd BJ. Colloidal aspects of dispersion and digestion of self-dispersing lipid-based formulations for poorly water-soluble drugs. Adv Drug Deliv Rev. 2019;142:16–34. doi:10.1016/j.addr.2019.01.008
14. Kazi M, Al-Qarni H, Alanazi FK. Development of oral solid self-emulsifying lipid formulations of risperidone with improved in vitro dissolution and digestion. Eur J Pharm Biopharm. 2017;114:239–249. doi:10.1016/j.ejpb.2017.01.015
15. Suchaoin W, Bernkop-Schnürch A. Nanocarriers protecting toward an intestinal pre-uptake metabolism. Nanomedicine. 2017;12(3):255–269. doi:10.2217/nnm-2016-0331
16. Eleftheriadis GK, Mantelou P, Karavasili C, et al. Development and characterization of a self-nanoemulsifying drug delivery system comprised of rice bran oil for poorly soluble drugs. AAPS PharmSciTech. 2019;20(2):78. doi:10.1208/s12249-018-1274-y
17. Nesamony J, Kalra A, Majrad MS, et al. Development and characterization of nanostructured mists with potential for actively targeting poorly water-soluble compounds into the lungs. Pharm Res. 2013;30(10):2625–2639. doi:10.1007/s11095-013-1088-2
18. Shafiq-un-Nabi S, Shakeel F, Talegaonkar S, et al. Formulation development and optimization using nanoemulsion technique: a technical note. AAPS PharmSciTech. 2007;8(2):E12–E7. doi:10.1208/pt0802028
19. Yilmaz E, Borchert -H-H. Design of a phytosphingosine-containing, positively-charged nanoemulsion as a colloidal carrier system for dermal application of ceramides. Eur J Pharm Biopharm. 2005;60(1):91–98. doi:10.1016/j.ejpb.2004.11.009
20. Nasr A, Gardouh A, Ghorab M. Novel solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of olmesartan medoxomil: design, formulation, pharmacokinetic and bioavailability evaluation. Pharmaceutics. 2016;8(3):20. doi:10.3390/pharmaceutics8030020
21. Dohare N, Khan AB, Maurya N, et al. An insight into the binding of aceclofenac with bovine serum albumin at physiological condition: a spectroscopic and computational approach. J Biomol Struct Dyn. 2018;36(2):398–406. doi:10.1080/07391102.2017.1278722
22. Bendas ER, Tadros MI. Enhanced transdermal delivery of salbutamol sulfate via ethosomes. AAPS PharmSciTech. 2007;8(4):213. doi:10.1208/pt0804107
23. Narayan R, Pednekar A, Bhuyan D, Gowda C, Koteshwara K, Nayak UY. A top-down technique to improve the solubility and bioavailability of aceclofenac: in vitro and in vivo studies. Int J Nanomedicine. 2017;12:4921. doi:10.2147/IJN.S141504
24. Sigward E, Mignet N, Rat P, et al. Formulation and cytotoxicity evaluation of new self-emulsifying multiple W/O/W nanoemulsions. Int J Nanomedicine. 2013;8:611. doi:10.2147/IJN.S37465
25. Mahmoud EA, Bendas ER, Mohamed MI. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS PharmSciTech. 2009;10(1):183–192. doi:10.1208/s12249-009-9192-7
26. Ibrahim TM, Abdallah MH, El-Megrab NA, El-Nahas HM. Upgrading of dissolution and anti-hypertensive effect of Carvedilol via two combined approaches: self-emulsification and liquisolid techniques. Drug Dev Ind Pharm. 2018;44(6):873–885. doi:10.1080/03639045.2017.1417421
27. Dou Y-X, Zhou J-T, Wang -T-T, et al. Self-nanoemulsifying drug delivery system of bruceine D: a new approach for anti-ulcerative colitis. Int J Nanomedicine. 2018;13:5887. doi:10.2147/IJN.S174146
28. Khatri P, Shao J. Impact of digestion on the transport of dextran-loaded self-emulsified nanoemulsion through MDCK epithelial cell monolayer and rat intestines. Int J Pharm. 2018;536(1):353–359. doi:10.1016/j.ijpharm.2017.12.009
29. Zidan AS, Aljaeid BM, Mokhtar M, Shehata TM. Taste-masked orodispersible tablets of cyclosporine self-nanoemulsion lyophilized with dry silica. Pharm Dev Technol. 2015;20(6):652–661. doi:10.3109/10837450.2014.908307
30. Akula S, Gurram AK, Devireddy SR. Self-microemulsifying drug delivery systems: an attractive strategy for enhanced therapeutic profile. Int Sch Res Notices. 2014;2014:1–11. doi:10.1155/2014/964051
31. Hanke U, May K, Rozehnal V, Nagel S, Siegmund W, Weitschies W. Commonly used nonionic surfactants interact differently with the human efflux transporters ABCB1 (p-glycoprotein) and ABCC2 (MRP2). Eur J Pharm Biopharm. 2010;76(2):260–268. doi:10.1016/j.ejpb.2010.06.008
32. Date AA, Nagarsenker M. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329(1–2):166–172. doi:10.1016/j.ijpharm.2006.08.038
33. Weerapol Y, Limmatvapirat S, Jansakul C, Takeuchi H, Sriamornsak P. Enhanced dissolution and oral bioavailability of nifedipine by spontaneous emulsifying powders: effect of solid carriers and dietary state. Eur J Pharm Biopharm. 2015;91:25–34. doi:10.1016/j.ejpb.2015.01.011
34. Clogston JD, Patri AK. Zeta potential measurement. In: McNeil Scott E. editors. Characterization of Nanoparticles Intended for Drug Delivery. Springer; 2011:63–70.
35. Chen C-H, Chang -C-C, Shih T-H, Aljuffali IA, Yeh T-S, Fang J-Y. Self-nanoemulsifying drug delivery systems ameliorate the oral delivery of silymarin in rats with Roux-en-Y gastric bypass surgery. Int J Nanomedicine. 2015;10:2403.
36. Araújo F, Kelmann R, Araújo B, Finatto R, Teixeira H, Koester L. Development and characterization of parenteral nanoemulsions containing thalidomide. Eur J Pharm Sci. 2011;42(3):238–245. doi:10.1016/j.ejps.2010.11.014
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.