")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Development and Evaluation of in-situ Nasal Gel Formulations of Nanosized Transferosomal Sumatriptan: Design, Optimization, in vitro and in vivo Evaluation
Authors Omar MM , Eleraky NE, El Sisi AM, Ali Hasan O
Received 17 October 2019
Accepted for publication 5 December 2019
Published 27 December 2019 Volume 2019:13 Pages 4413—4430
DOI https://doi.org/10.2147/DDDT.S235004
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Yan Zhu
This paper has been retracted.
Mahmoud M Omar,1,2 Nermin E Eleraky,3 Amani M El Sisi,4 Omiya Ali Hasan1,2
1Department of Pharmaceutics and Industrial Pharmacy, Deraya University, El-Minia, Egypt; 2Department of Pharmaceutics, Sohag University, Sohag, Egypt; 3Faculty of Pharmacy, Assiut University, Assiut, Arab Republic of Egypt; 4Department of Pharmaceutics and Industrial Pharmacy, Beni-Suef University, Beni-Suef, Egypt
Correspondence: Mahmoud M Omar
Pharmaceutics and Industrial Department, Deraya University, Deraya Square Street, Minia, New-Minia 61768, Egypt
Tel +20 10 0933 2419
Email [email protected]
Background: Sumatriptan succinate (SUT) is a potent drug used for relieving or ending migraine and cluster headaches. SUT bioavailability is low (15%) when it is taken orally owing to its gastric breakdown and bloodstream before reaching the target arteries.
Aim: The aim of the study was to enhance SUT bioavailability through developing an intranasal transferosomal mucoadhesive gel.
Methods: SUT-loaded nanotransferosomes were prepared by thin film hydration method and characterized for various parameters such as vesicle diameter, percent entrapment efficiency (%EE), in vitro release and ex vivo permeation studies. The in-situ gels were prepared using various ratios of poloxamer 407, poloxamer 188, and carrageenan and characterized for gelation temperature, mucoadhesive strength, and rheological properties.
Results: The prepared transferosomes exhibited percent entrapment efficiencies (%EE) of 40.41±3.02 to 77.47±2.85%, mean diameters of 97.25 to 245.01 nm, sustained drug release over 6 hours, and acceptable ex vivo permeation findings. The optimum formulae were incorporated into poloxamer 407 and poloxamer 188-based thermosensitive in-situ gel using carrageenan as a mucoadhesive polymer. Pharmacokinetic evaluation showed that the prepared in-situ gel of SUT-loaded nano-transferosomes gave enhanced bioavailability, 4.09-fold, as compared to oral drug solution.
Conclusion: Based on enhancing the bioavailability and sustaining the drug release, it can be concluded that the in-situ gel of SUT-loaded nano-transferosomes were developed as a promising non-invasive drug delivery system for treating migraine.
Keywords: nanotransferosomes, sumatriptan succinate, SUT, thermosensitive in-situ gel and intranasal drug delivery system
Introduction
Migraine is a neurological disorder, which is often characterized with recurrent attacks accompanied with primary symptoms, gastrointestinal, headache, neurologic, and sometime aural symptoms.1,2 Migraine is one of the most common disorders in the world.3 It is the second most common cause of short-period absence of non-manual workers.4 Migraine has been treated by many drug formulations; however, accompanied limitations with drug delivery systems have proved a major obstacle. The nasal drug delivery system may be affected by many factors; the capacity of the nasal cavity for the drug volume (<0.2 mL), anterior leakage, and mucociliary clearance.5 Two types of drugs can be used for treating migraine; one of them is preventive and the other is abortive. Abortive drugs, including triptans (e.g. sumatriptan citrate), target serotonin receptor (5-HT receptor). Moreover, sumatriptan succinate (SUT) inhibits calcitonin gene-related peptide.6 SUT is administered in different routes such as oral, intranasal and subcutaneous (s.c) routes. However, its oral administration or intranasal application is limited because of low absolute bioavailability, pre-systemic breakdown, and incomplete absorption. Despite the absolute bioavailability of parenteral formulation of SUT being high (96%), its parenteral formulation is inconvenient. In this sense, intranasal formulations can be developed to overcome reasons of low bioavailability of SUT. High vascular mucous membranes of the nose facilitate rapid absorption of un-metabolized drug to the central nervous system.7,8 Recently, novel studies have been carried out to enhance the bioavailability, such as solid dispersion,9 liposomes,10 chitosan microparticles,11 polymeric lipid-core nanocapsules,12 and lipid vesicles.13,15
Lipid vesicles, as a tool for drug delivery of SUT, have been studied.16 Transferosomes are ultra-flexible and very deformable vesicles. They are composed of phospholipids and permeation enhancers. The presence of permeation enhancers softens the lipid bilayers of the prepared transferosomes, making them very deformable vesicles. The ability of these vesicles to change their shapes and intracellularly squeeze improves the permeation.17
In the present study, transferosomal vesicles were incorporated into in-situ gel as an alternative tool for intranasal administering of SUT. Another goal of the study was to improve the bioavailability of the drug and target the brain using optimum in-situ gel containing transferosomal formulations. The prepared transferosomes were evaluated for different parameters such as particle-size, encapsulation efficiency, in vitro SUT release and kinetics analysis of the release data, elasticity. In-situ gels were prepared and characterized. The optimum prepared transferosomes were incorporated into the selected gel and evaluated for different properties such as in vitro release, stability study, in vitro tolerability of sheep nasal mucosa; histopathological evaluation and in vivo pharmacokinetics study.
Materials and Methods
Materials
SUT was purchased from Natco Fine Pharmacis Pvt Ltd (Hyderabad, India). Soybean phospholipids, cholesterol, tween 80, sodium caprate, and sodium cholate were purchased from Aladdin (Shanghai, China). Poloxamer 407 (PLX 407), poloxamer 188 (PLX 188), and carrageenan were purchased from (BASF, Ludwigshafen am Rhein, Germany). Clostridium perfringens enterotoxin (CPE) was purchased from MyBiosource, Inc. (Southern California, San Diego, CA). Acetonitrile, methanol, and chloroform were provided by Aladdin. All reagents were of high-performance liquid chromatography (HPLC) or analytical grade. Rabbits and rats were purchased from the animal house of the Faculty of Medicine, Assiut University, Egypt.
Methods
Formulation of SUT-Loaded Transferosomes
SUT-loaded transferosomes were prepared using a thin-film hydration method, which was reported as a good method for preparing lipid vesicles.14 Briefly, SUT, soybean phospholipids, and surfactant (docosahexaenoic acid (DHA) or sodium cholate) were dissolved in 10 mL of 2:3 (v/v) chloroform/methanol mixture, as presented in Table 1. Dried thin lipid films were established under vacuum at 70ºC, using Rotavapor® (type Hei-VAP manufactured by Heidolph Instruments GmbH & Co. KG, Schwabach, Germany). To eliminate any organic solvent, thin lipid films were stored in a desiccator under reduced pressure (100 mbar) for 24 hours. Simulated nasal fluid (SNF; pH 5.5, 10 mL) containing permeation enhancer; Clostridium perfringens enterotoxin (CPE) or sodium caprate (Sod C) was used to hydrate the prepared thin dried film. The resultant vesicles were allowed to swell for 24 hours, then they were sonicated using a sonicator (powerson I C405, Hwashin Co., Shanghai, China) for 30 minutes, forming smaller vesicles. SUT-loaded transferosomes (free from untrapped drug) were extruded through a 200-nm Sartorius polycarbonate membrane filter (Sartorius Instrument, Nieuwegein, Netherlands) four times to decrease their diameter. SUT-loaded transferosomes were separated from the free SUT by high-speed centrifugation at 18,000 rpm for 0.5 hours at −5°C using a cooling high speed centrifuge (High-Speed Refrigerated Centrifuge, CR22N; Hitachi Ltd., Tokyo, Japan). Precipitates were re-suspended with SNF (pH 5.5, 10 mL). Supernatants were collected to calculate free SUT. SUT-loaded transferosomes (free from untrapped drug) were kept at 4°C in clean glass containers. All processes were carried out under aseptic conditions using laminar flow (horizontal laminar flow hood, BZ Series, model BZ-3SS RX; Germfree, Ormond Beach, FL).
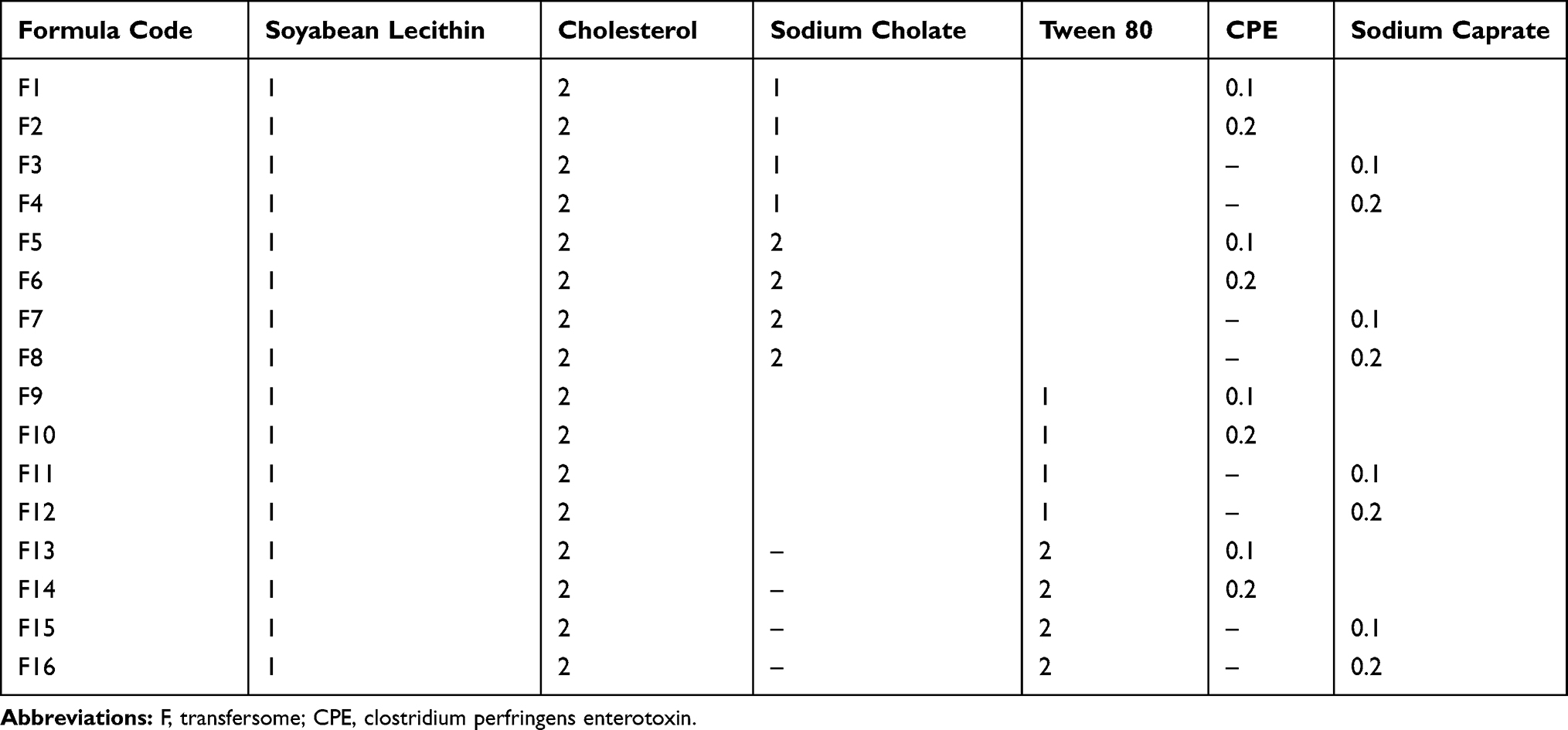 |
Table 1 Composition of the Prepared Transferosomes in Molar Ratio |
Differential Scanning Calorimetry (DSC)
DSC examination was carried out using the DSC 204 (Netzsch, Hanau, Germany). The heating rate of the samples was 10°C/min over a temperature range of 40–300°C.
The sample was taken for analysis; with an aluminum empty pan used as a reference. DSC profiles of SUT, tween 80, cholesterol, soybean phospholipids, physical mixtures of SUT and tween 80, physical mixture of SUT and cholesterol, physical mixture of SUT and sodium cholate, and physical mixture of SUT and soybean phospholipids were performed.
Characterization of SUT-Loaded Transferosomes
Vesicle Size, Polydispersity Index (PDI), and Electric Potential Analysis
To evaluate the zeta potential, mean diameter, and size distribution curve of the prepared vesicles; samples of transferosomal dispersion (100 μL) were diluted with purified water (900 μL) and measured through dynamic light scattering method using a Malvern Zetasizer (Malvern Instruments Corp; Nano ZS ZEN 3600, Worcestershire, UK). The measurements were repeated in triplicate (n=3).
Determination of Transferosomal SUT Entrapment Efficiency
Indirect Method
The collected supernatants from the preparation step were assayed using a UV-spectrophotometer (Shimadzu, Kyoto, Japan). As the amounts of free SUT were determined, amounts of the encapsulated SUT were calculated as follows,
Eq (1) Eq (2)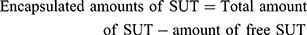
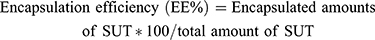
Evaluation of Elasticity of the Prepared Transferosomes
Elasticity of the prepared transferosomes is an important parameter of elastic vesicles, which is characterized by the capability of remaining intact as they permeate through intranasal mucous membranes. Measuring of the vesicles elasticity (E) was carried out by measuring the amount of vesicles permeation through semi-permeable membrane (J), mean vesicles size (rv), and membrane pore size (rp). Elasticity parameter was calculated as reported in literature using the following equation.18
Eq (3)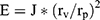
Amount of transferosomal suspension (J), which permeated through pores of the membrane (50 nm in diameter) under high pressure (5 bars) (Sartorius Instrumenten, Nieuwegein, The Netherlands) during 5 minutes, was weighed.
Morphology of SUT-Loaded Transferosomes
To investigate the morphology of SUT-loaded transferosomal formula (F5), transmission electron microscopy (TEM) (JEOLJEM-1400, Tokyo, Japan) was used. A drop of freshly prepared transferosomal formula was used to cover carbon–copper grid and left to dry, allowing the vesicles to stick to the surface of the carbon–copper grid. Phospho-tungstic acid dye was used to stain the dried vesicles. Photographs of the stained vesicles were captured using TEM through an accelerating voltage of 80 kV.13
In-vitro Release Study of SUT-Loaded Transferosomes
SUT release from the prepared transferosomes was studied using a dialysis method, which was previously prescribed in the literature.19 Transferosomal suspension (10 mg drug equivalent) and SUT solution (10 mg drug equivalent, control) were inserted into dialysis bags (donor compartment) with a molecular weight cut-off of 12,000 kDa. Release study was carried out using USP dissolution apparatus type II; paddle rotation was kept to 100 rpm. The dialysis bags were fully immersed under the surface of 100 mL of SNF as a receptor medium (pH 5.5 at 37ºC) to keep a sink condition. Samples of release medium (2 mL) were taken at definite time intervals (0, 0.25, 0.5, 1, 2, 4, and 6 hours) and replaced with 2 mL of fresh prepared SNF. Taken samples were analyzed UV-spectrophotometrically at 282 nm (Shimadzu UV-1800). The experiments were repeated in triplicate. Percent release of SUT at a time (t) was calculated as follows,
Eq (4)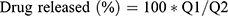
where Q1 and Q2 are the initial quantity of drug entrapped in the prepared transferosomes and the quantity of drug released at time t, respectively.
Stability Study of the Optimized Transferosomes
The stability of the prepared transferosomes was evaluated based on aggregation of the lipid vesicles and leakage of SUT from them. A protocol developed by Du Plessis et al20 was applied with minor changes. The examined transferosomes (10 mg of SUT equivalent) were kept in amber vials and stored in a refrigerator at 4ºC and at a room temperature of 4±2°C, 25±2°C, and 40±2°C for 3 months. A physical stability study was carried out based on examining the clarity, pH, and percent encapsulation efficiency (EE%). Clarity of the examined dispersions was measured using Abbe’s refractometer in terms of refractive index. Adjustment of refractometer was carried out in such a way that the cross wire of the telescope was exactly on the boundary between the dark and bright spaces. Calibration of the refractometer was performed using water as a reference standard. Any visible changes, such as sedimentation, creaming or color changes were recorded.
Stability Study of the Optimized in-situ Gels
The stability study was carried out to investigate drug content of the prepared in-situ gels (G5/F9, G5/F10, G5/F11, and G5/F12). The examined gels were kept in amber-colored bottles covered with an aluminum cap at 4±2°C, 25±2°C, and 40±2°C at relative humidity 75±5% for 3 months. Gel samples (1 mL) were taken every month to be analyzed for drug content. One milliliter of the examined gels was digested using triton X-100 (3 mL, 1% v/v), which was diluted in double distilled water up to 10 mL to release the drug content and filtered using polycarbonate membrane filter (0.2 μm) (Whatman International Ltd, Springfield Mill, UK). One milliliter of the filtrate was transferred to 10 mL volumetric flask and diluted in distilled water to the final volume 10 mL. The taken samples were analyzed to measure their UV-absorbance using a UV-spectrophotometrer at 282 nm.
Preparation of Mucoadhesive Nasal Gel
Mucoadhesive intranasal in-situ gels containing SUT were prepared using PLX 407, PLX 188, and mucoadhesive agent (carrageenan) (Table 2). Mucoadhesive gels were prepared in accordance to the modified cold method.21 Carrageenan was dissolved in hot water and then cooled. PLX 407 and PLX 188 were stirred in cold distilled water (4°C) until a clear solution was formed. PLX 407/188 solution was mixed with carrageenan gel. The prepared mixtures were kept in a refrigerator for 48 hours. Then, they were evaluated based on gelation temperature, gelation time, viscosity, and syringeability of the formulations.
 |
Table 2 Composition, pH, and Mucoadhesive Strength of PLX 407, PLX 188, and Carrageenan in-situ Gel |
Characterization of the Prepared Gels
Gelation Temperature
Gelation temperature and gelation time are critical factors to optimize thermo-sensitive gel. Gelation temperature is the lowest temperature degree at which the prepared formulation transits to gel state. Rheological evaluations were conducted using a thermostatically programmable Brookfield rheometer (MCR 302; Anton Paar, Graz, Austria). To determine precisely the gelling temperature, therheometer was fitted with a CP-52 spindle and cone/plate geometry. The diameter of the cone was 2.4 cm and an angle of 3º. Moreover, the shear stress was adjusted to keep a shearing rate at 10 s−1. The temperature was increased at a rate of 0.5ºC/min within the range of 20–40ºC. The obtained viscosity (mPa.s) values were plotted against the temperature. The gelation temperature was estimated graphically using the plotted graph and was defined as the temperature point on the viscosity-temperature plot at which the sudden increase of the apparent viscosity was recorded. The gels were evaluated three times to calculate standard deviation (±SD) and confirm the repeatability.
Evaluation of Rheological Properties of SUT Loaded Transferosomal in-situ Gels
Rheological evaluation of the prepared gels was carried out using a rheometer equipped with a cone (0.8◦) and parallel stainless plate geometry (40 mm/diameter) (Bohlin Gemini HR nano, Malvern instruments, UK). The formulations were examined in triplicate.
Determination of pH
The prepared samples pH was measured using a pH meter (SP-701, Suntex Company, New Taipei City, Taiwan), to ensure compatibility of the gels with nasal mucosa at room temperature. Ten milliliters of each formulation was withdrawn into a suitable container, dipping the glass electrode of a pH meter into the formulations samples to measure the pH.
Preparation of Nasal Mucosa
Nasal mucosa membrane was isolated from the fresh nasal tissue of sheep, obtained from a local slaughterhouse, within 1 hour of slaughtering the sheep. Fatty tissues and different tissues were removed gently from the isolated nasal mucosa membrane. The clean nasal mucosa was kept into isotonic saline solution at −20°C. The procedures of the experiment were approved by the Animal Ethical Committee of the Faculty of Medicine, Assiut University.
Mucoadhesive Strength
Mucoadhesive strength is equivalent to the detachment force needed to separate the gel from the nasal mucosa tissue. To measure the mucoadhesive strength of the prepared gels, two parts of the nasal mucosa (20*25 mm) were attached to two different glass slides using glue. One of the two glass slides was attached to the base of the used balance beneath its pan. The other one was tied to the under surface of the balance pan in an inverted position in such a way that the tissue has been just facing and beneath upper nasal tissue. About 0.5 g of the examined gel was positioned between two nasal membranes and kept in contact for 5 minutes. Using a water filled burette, a definite water volume was added to the other pan, allowing the two parts of the nasal membrane to be detached from each other.22 Finally, the mucoadhesive strength was defined as the minimum force in dyne/cm2 able to detach the mucosal membrane from the examined gel and calculated based on the following equation:22
Eq (5)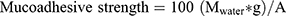
where M is the amount of water in grams that equals the released water in milliliters multiplied in water density, g is gravitational acceleration (980 cm/s2), and A is surface area of the examined nasal mucosa in cm2.
Drug Content
The formulations (G5/F9, G5/F10, G5/F11, and G5/F12) were analyzed to measure drug contents in triplicate by using a double beam UV-visible spectrophotometer (Shimadzu UV-1800).23 One milliliter of the formulation was treated as mentioned in the stability study, then 1 mL of the filtrate was transferred to a 10 mL volumetric flask and diluted in distilled water to a final volume of 10 mL. The taken samples were analyzed to measure their UV-absorbance using UV-spectrophotometry at 282 nm.
In vitro Release of SUT from the Prepared Gel
To investigate the release rate of SUT from transferosomal SUT containing gel, G5/F9, G5/F10, G5/F11, and G5/F12 formulations were utilized for conducting the in vitro release study as reported in the literature.24 Briefly, a certain amount of the examined formulation (10 mg SUT equivalent) was placed into cellophane dialysis tubing (a molecular weight cut-off 12,000–14,000 kDa, Heidelberg, Germany). The tube was submerged under the surface of SNF (300 mL, pH 5.5, rotation speed 75 rpm, and 37±2°C) in a dissolution flask. Aliquots (5 mL) were withdrawn and replaced with equal volume of freshly prepared SNF (pH 5.5, 5 mL, 37ºC) and analyzed spectrophotometrically at λmax282 nm. The experiment was performed independently three times. The percent cumulative drug permeated was calculated and represented against the time.
Ex vivo Permeation Studies
Transferosomes (F9, F10, F11, and F12) were selected to be incorporated into the optimum in-situ gel (G5) based on the previous characterization. The experiment was conducted according to the previously described method with some modification.25 Briefly, loaded transferosomes in-situ gel (G5/F9, G5/F10, G5/F11, and G5/F12), SUT solution (control I), and SUT-loaded transferosomes, F9 (control II) equivalent to 20 mg were placed into an open-sided tube that was sealed with the treated rat abdomen skin and the other side reinforced into 100 mL of SNF (pH 5.5 at 37ºC) as a receptor compartment; USP dissolution apparatus type II. Its paddle was stirred at 100 rpm. Samples (2 mL) were taken at a definite time (0, 0.5, 1, 2, 3, 4, 5, and 6 hours) and replaced with equal volume of a freshly prepared NSF. Taken samples were analyzed spectrophotometrically at 282 nm for determining the cumulative permeated SUT in triplicate manner. Lag time was determined from the plot of cumulative permeated drug against the time as the X-axis intercept of the linear portion. Permeation parameters were calculated as follows:26
Eq (6) Eq (7) Eq (8)
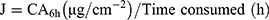
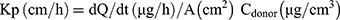
where Q6h is the cumulative amount of the permeated drug over 6 hours, J is the permeation flux, Kp is the permeation coefficient, and dQ/dt equals the amount of drug/time obtained from the slope of the straight portion of the plot.
Histopathology for Nasal Mucosal Toxicity and Tolerability
The histopathological investigation was carried out to examine the effect of the optimized transferosomal SUT-loaded in-situ gels (G5/F9) and (G5/F11) on the integrity of nasal mucosa. The study was achieved, in accordance with Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, revised 2011) and were approved by the local animal ethical committee of the Faculty of Medicine, Assiut University. Fifteen adult New Zealand White rabbits (2−2.2 Kg) were distributed into three groups (five rabbits were included into each group; n=5). The first group, A, was kept without application of any formula (the normal one, n=5), the second group, B (n=5), received intranasal transferosomal SUT in-situ gel (G5/F9), while the third group, C (n=5), received intranasal transferosomal SUT in-situ gel (G5/F11) for 10 consecutive days twice. After that, the examined rabbits were humanely scarified to isolate intact nasal mucosa; whereas the membranes were cleaned off from adipose tissues and bones and were fixed and dehydrated using formaldehyde (10% v/v) and ethanol (95% v/v), respectively. Hard block of the tissue, prepared with a hard paraffin/beeswax mixture 2:3 (w/w), was sliced at 5 µm using a microtome. Sliced membranes were deparaffinized, then they were stained using eosin and hematoxylin stains. Light microscopy was used to observe and record any changes in the examined tissues.
In vivo Pharmacokinetic Study
Sixty-three New Zealand White rabbits (2.8–3 kg) were used within the pharmacokinetics study. Procedures of the study were also in compliance with the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, revised 2011) and were approved by the local animal ethical committee of the Faculty of Medicine, Assiut University. The rabbits were provided by the animal house of the Faculty of Medicine of Assiut University and were randomly distributed into three groups (n=21). The first group members received 20 mg equivalent of SUT orally, while the second and the third group received 20 mg equivalent of transferosomal SUT containing gels intranasally, (G5/F9) and (G5/F11), respectively. Blood samples (2 mL) were taken from the marginal ear vein at determined times (0, 0.25, 0.5, 1, 2, 4, 6, and 12 hours) and kept in heparinized tubes. Three animals from each group were sacrificed humanely at each time interval, then brain tissue was collected. Plasma samples were isolated by centrifuging the blood samples at 14,000 rpm, 5°C for 20 minutes.
The collected brain tissues were flushed with normal saline solution (0.9 w/v) and maintained in filter paper to remove the blood excess. The brain tissues were mixed with normal saline solution and homogenized using a tissue homogenizer. The supernatant was separated to be analyzed. The tissue homogenate samples and plasma were stored at −20ºC, until the analysis was carried out.
The calibration curve and measurement of SUT plasma concentration, based on UV-HPLC (Schimadzu Instruments, Japan), was carried out as reported in the literature.27 Briefly in a clean tube, plasma sample (200 µL) or 500 μL volume of brain tissue homogenate, 20 µL of the paracetamol as an internal standard (1 µg/mL) and 1.5 mL of ethyl acetate were vortexed for 5 minutes. The mixture was centrifuged at 13,000 rpm, for 10 minutes at 5°C, to separate the precipitated protein. In another clean tube, the supernatant was dried under a vacuum in a SpeedVac vacuum evaporator (Savant Instruments, Holbrook, NY) at 45°C for 60 minutes. The dried residue was reconstituted with 300 µL of the mobile phase (acetonitrile: 0.05 M KH2PO4, 16:84 (v/v), pH 3) and vortexed for 10 minutes, then it was kept to conduct the UV-HPLC analysis. Determination of drug concentration was conducted, using the Phenomenex column (a Hypersil BDS C18, 5 µm, 4.6×250 mm) and eluted isocratically with the mobile phase at 1 mL/min and 30°C, with UV detection at 282 nm. Different pharmacokinetic parameters (PK) such as peak plasma concentration (Cmax) and area under the curve (AUC) were calculated by using WinNonlin software 5.0 (M/s Pharsight, CA). To evaluate the brain-targeting efficiency of the prepared in-situ nasal gels formulations of nanosized transferosomal SUT, the drug-targeting index (DTI) was calculated based of the following equation:
Eq (9)
Statistical Analysis
All experiments were conducted in triplicate. The standard differences were examined for statistical significance at P<0.05 using Student’s t-test.
Results and Discussion
In the present examination, many endeavors were exerted to prepare the sustained release transferosomal SUT in-situ gel forming intranasal solution using polymers such as PLX 407, PLX 188, and carrageenan. PLX 407/188 and carrageenan mixture novel intranasal gel-forming mucoadhesive polymer, which gets converted to gel at body temperature, was used as the gelling agent.
Differential Scanning Calorimetry (DSC)
The DSC curve of SUT displayed a sharp endothermic peak at 168.89°C due to melting of crystalline SUT (Figure 1A), which is consistent with the literature report. The thermograms of tween 80 (Figure 1B) and physical mixture of SUT and tween 80 (Figure 1C) revealed a broad endothermic peak at 168.89°C, which indicates a dilution effect as a result of mixing. The thermograms of soybean phospholipids (Figure 1D) and physical mixture of SUT and phospholipids (Figure 1E) revealed a broad endothermic peak at 121.41°C (ΔH0253.62 J/g), which indicates melting of soybean phospholipids and another shallow peak at 167.43°C (ΔH060.40 J/g) correlate to melting of amorphous SUT and also due to the dilution effect. The thermogram of sodium cholate displayed an endothermic peak at 198.89°C due to melting of sodium cholate (Figure 1F) and physical mixture of SUT, and sodium cholate (Figure 1G) revealed a broad endothermic peak at 166.15°C which indicates a mild shift of SUT melting point. The thermogram of chloesterol displayed an endothermic peak at 147.89°C due to melting of chloesterol (Figure 1H). The physical mixture of SUT and cholesterol was an additive thermogram (Figure 1J) of broad endothermic peak at 168.70°C for SUT along with a broad peak of chloesterol at 146.9°C (ΔH0 108.56 J/g). The shift towards lower temperatures was the result of mixing of two components.
 |
Figure 1 DSC curves for SUT (A), tween 80 (B), physical mixture of tween 80 with SUT (C), soybean phospholipids (D), physical mixture of soybean phospholipids and SUT (E), sodium cholate (F), physical mixture of sodium cholate and SUT (G), cholesterol (H), and physical mixture of cholesterol and SUT (J). Abbreviations: DSC, differential scanning calorimetry; SUT, sumatriptan. |
Characterization of the Prepared Transferosomes
Particle size, the polydispersity index (PDI), zeta potential, and encapsulation efficiency of the prepared SUT-loaded transferosomes were measured and represented in Table 3. The size of the prepared transferosomes ranged from 97.25±3.14 (F9) to 245.01±3.4 (F5). When the surfactant concentrations were increased, the vesicles sizes were decreased (Table 3). Those findings were explained on the basis of interfacial tension, whereas lower interfacial tension was created at higher surfactant concentration, leading to form small-sized vesicles.28 Many parameters may affect the vesicle size such as hydrophilic-lipophilic balance (HLB), molecular structure, and ionic nature of the used surfactant as reported in the literature.29 HLB values are 18 and 15 for sodium cholate and tween 80, respectively. As the HLB of the used surfactant decreased, the particle size of the vesicle increased as a result of interaction of the surfactant with lipid chains of the membrane, leading to increase packaging density and the surface free energy. However, increasing HLB value of the used surfactant led to a decrease in the vesicle size as a result of interaction of the surfactant with the inner aqueous phase, leading to a decrease in the vesicle size. The results obtained with sodium cholate-based vesicles seem to deviate from the aforementioned explanation, because anionic nature of sodium cholate generates a strong repulsive force between the lamellae as a result of negative charge formation on the vesicles, leading to an increase in the internal aqueous core. The present study was concurrent with the previous finding that explained the effect of HLB and the vesicle size of water soluble loaded-transferosomes.30 However, the present findings were in contrast to the findings of the other studies formulating water-insoluble drugs.31 Polydispersity index is an important indicator regarding homogeneity of the prepared transferosomes (F1–F16), whereas PDI of all the prepared transferosomes had values in the range between 0.178–0.312. All of the prepared transferosomes that had PDI less than 0.25 were mono-dispersed suspensions.32 They are preferable because of their high stability and less probability of aggregation of the suspended vesicles. Being partition coefficient (Log k 1.2), SUT was incorporated in both internal aqueous core and lipid bilayer. The EE% of SUT in the prepared transferosomes was in the range of 40.41±3.02 to 77.47±2.85. EE% changed related to variation of surfactant type and its concentration. Regarding the surfactant concentration, the ratio 1:2:1 (w/w/w) gave higher EE% than the ratio 1:2:2 (w/w/w). The aforementioned results could be attributed to an increase of the vesicle size as outcomes of incorporation of surfactant into lipid bilayer. Initially, the increase of vesicle size accompanied with high drug loading. Lastly, pores in the lipid bilayer were created facilitating the drug escape and the decrease of EE%. Regarding the surfactant type, tween 80 based transferosomes gave higher EE% than sodium cholate based transferosomes. These results could be attributed to HLB value, the lower HLB, and the higher EE%. These findings were concurrent with those reported in the literature.19 However, these results disagreed with those reported with González-Rodríguez et al.26
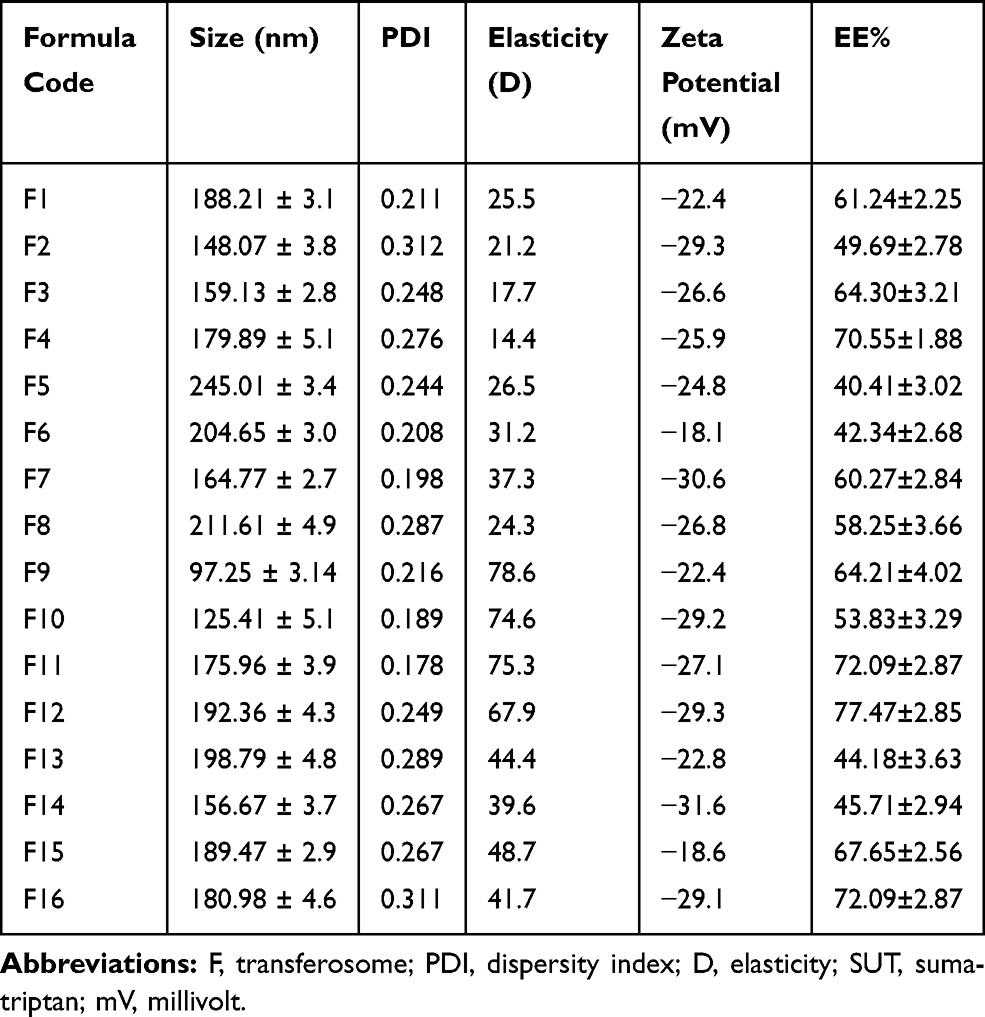 |
Table 3 Vesicle Size, Poly-Dispersity Index (PDI), and Elasticity (D), Zeta Potential and Percent Entrapment Efficiency of SUT-Loaded Transferosomes |
Regarding elasticity results, tween 80 based transferosomes were more elastic than sodium cholate based transferosomes. However, an increase of tween 80 ratios in formulae (F13–F16) led to decrease the elasticity of transferosomes as compared to the elasticity of transferosomes (F9–F12). The elasticity parameter is very important for permeating the vesicles across the epithelium of the nasal membrane barriers; because intradermal permeation of vesicles is a function of elasticity of the vesicle membrane.33
Morphology of SUT-Loaded Transferosomes
Transmission electron micrograph (TEM) analysis has been used for evaluating the morphology of colloidal systems and confirming the obtained results regarding vesicle size, as reported in the literature. TEM of optimized SUT-loaded transferosomes has outlined non-aggregated small spherical vesicles with a well-defined bilayer, Figure 2.
 |
Figure 2 Transmission electron micrograph of the prepared transferosomes F5 when stained with uranyl acetate (10%) with scale 500 nm. |
In vitro Release Study of Transferosomes
As compared to SUT solution as a control, the percentages of SUT released from the prepared transferosomes are represented in Figures 3A–D. The drug release from control was the highest one at 6 hours post beginning the experiment (99.21%), as compared to the drug release from different transferosomes. Those results could be attributed to the reservoir effect as a result of the vesicular encapsulation of the drug. Also, the sustained release of the drug from different transferosomes (F1–F16) was not as a result of using cellophane membrane. The release of SUT from the transferosomes (Figures 3A–D) were biphasic processes. The first process over the first hour was fast followed by the slower one. This result could be explained on the basis of a property of the transferosome structure or fast loss of surface associated drug, and then slow release of the drug from the core was carried out. Moreover, a certain drug amount may disable to be accommodated within the lipid bilayer of the transferosome, occurring at burst effect. Drug release from transferosomes (F5–F8, F13–F16) with high surfactant ratio 1:2:2 (w/w/w) was slower than from that oftransferosomes (F1–F4, F9–F12) with low surfactant ratio (1:2:1 w/w/w). This finding could be attributed to the ability of the surfactant molecules to render the bilayer of transferosomes more ordered and less leaky forms, hindering the release of the drug. Moreover, using a high surfactant ratio may lead to the formation of mixed micelles which were less sensitive to the concentration gradient.34 However, the drug release from sodium cholate based transferosomes (F1–F4) was slower than from that of tween 80 based transferosomes (F9–F12). This result was elucidated based on the alkyl chain length, whereas the higher the surfactant chain length, the slower the drug release. Moreover, the surfactant chain length and shape might affect the ordering of the lipid bilayer, making the variations of the drug release. Permeation enhancer was an insignificant effective factor on the drug release (P>0.05). Linear regression analysis of the release data proved that the diffusion controlled mechanism (R2>0.981) was the predominant driving force, except F1, F4, F5, F13, and F16, which followed first-order kinetics (R2>0.969).
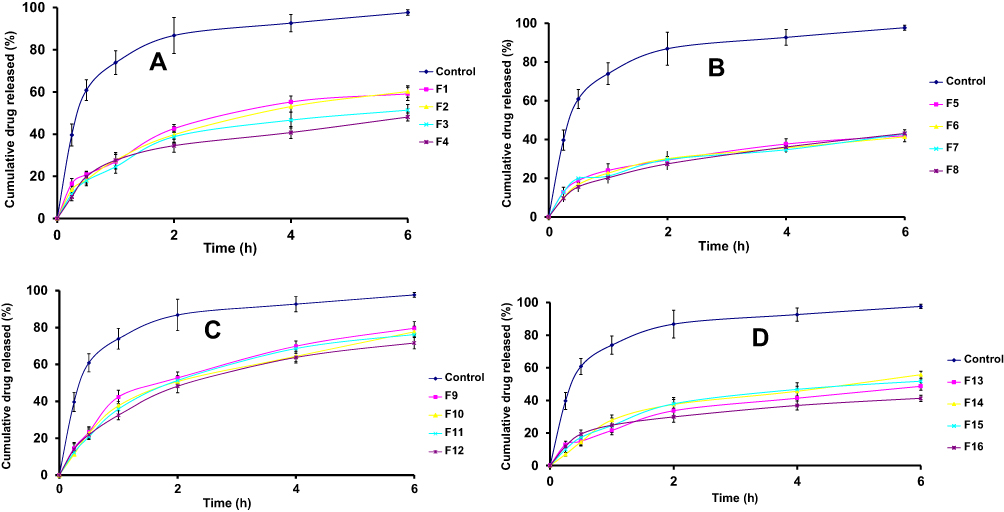 |
Figure 3 SUT release profiles (A) (F1–F4), (B) (F5–F8), (C) (F9–F12), and (D) (F13–F16) from SUT-loaded transferosomes versus SUT solution. Abbreviations: F, transferosome; SUT, sumatriptan. |
Stability Study of the Optimized Transferosomes
The optimized transferosomal formulations (F9, F10, F11, and F12) showed insignificant difference (P<0.05) in pH, clarity, color, and the percent encapsulation efficiency within 3 months at refrigerator temperature 4±2ºC and 25±2ºC, which proves the stability of the examined transferosomes (Table 4). However, these formulations kept at 40±2ºC were unstable with respect to drug content because of improving fluidity of the vesicular membrane. Moreover, the elevated temperature may affect liquid transition of the lipid bilayers of the gel and cause the phospholipids chemical degradation, facilitating the drug escape.35 The study suggests that storage of these formulations at 40±2ºC is prohibited to avoid dramatically changing the desired properties of the prepared transferosomes.
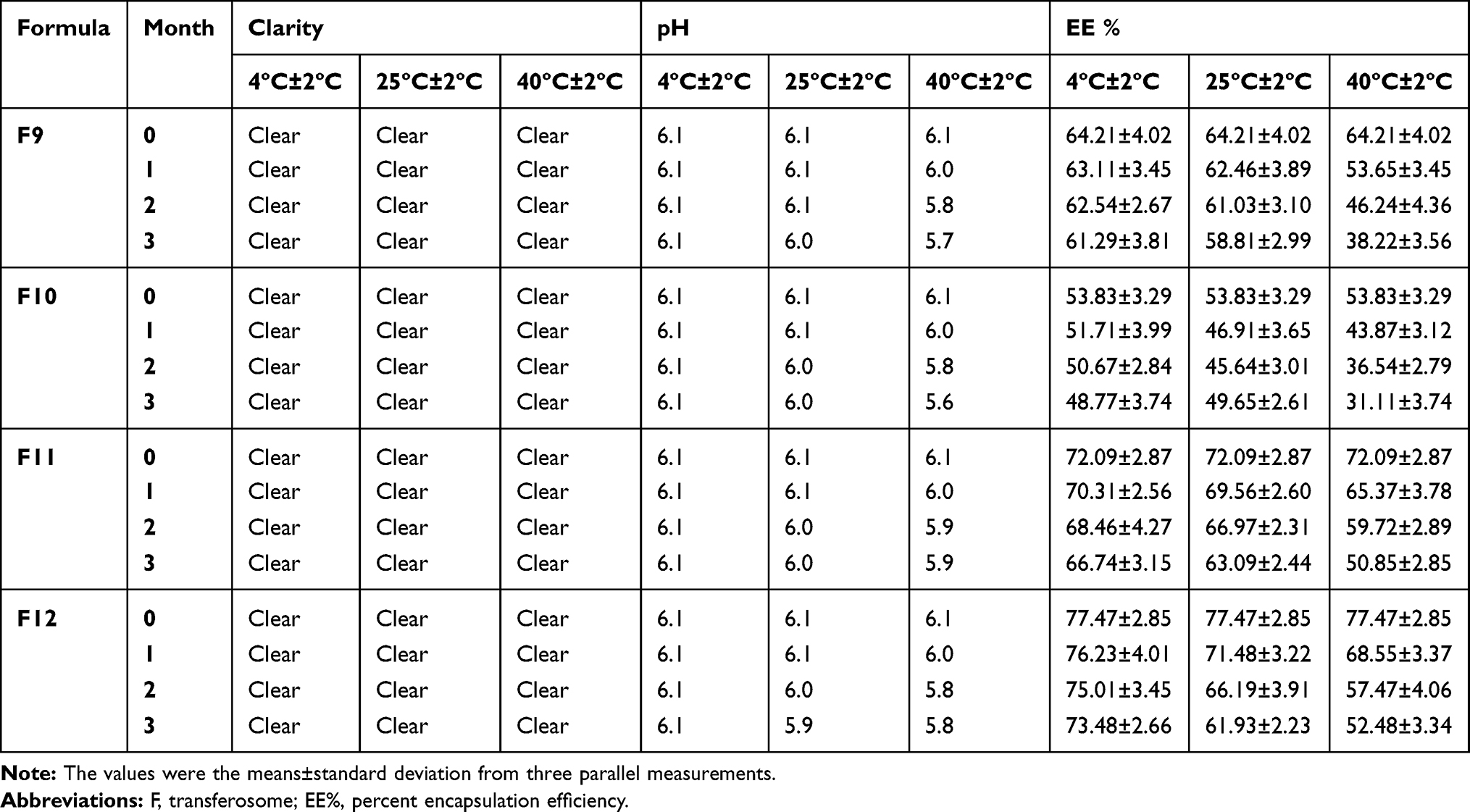 |
Table 4 Clarity, pH, and Percent Encapsulation Efficiency (EE%) of the Prepared Transferosomal Vesicles During Storage at 4±2ºC, 25±2ºC, and 40±2ºC Over a Period of 3 Months |
Stability Study of the Optimized in-situ Gels
The prepared in-situ gels (G5/F9, G5/F10, G5/F11, and G5/F12) were examined based on susceptible variation in the drug content. The results of examination showed an insignificant decrease (P<0.05) in the drug content over a period of 3 months for the in-situ gels stored at 4±2ºC, 25±2ºC, and 40±2ºC (Table 5). Decrease of drug content was detected as a function of an increase in temperature. These findings suggest that storing of transferosomal SUT in-situ gel at low temperature is a must, to overcome the drug decomposition.
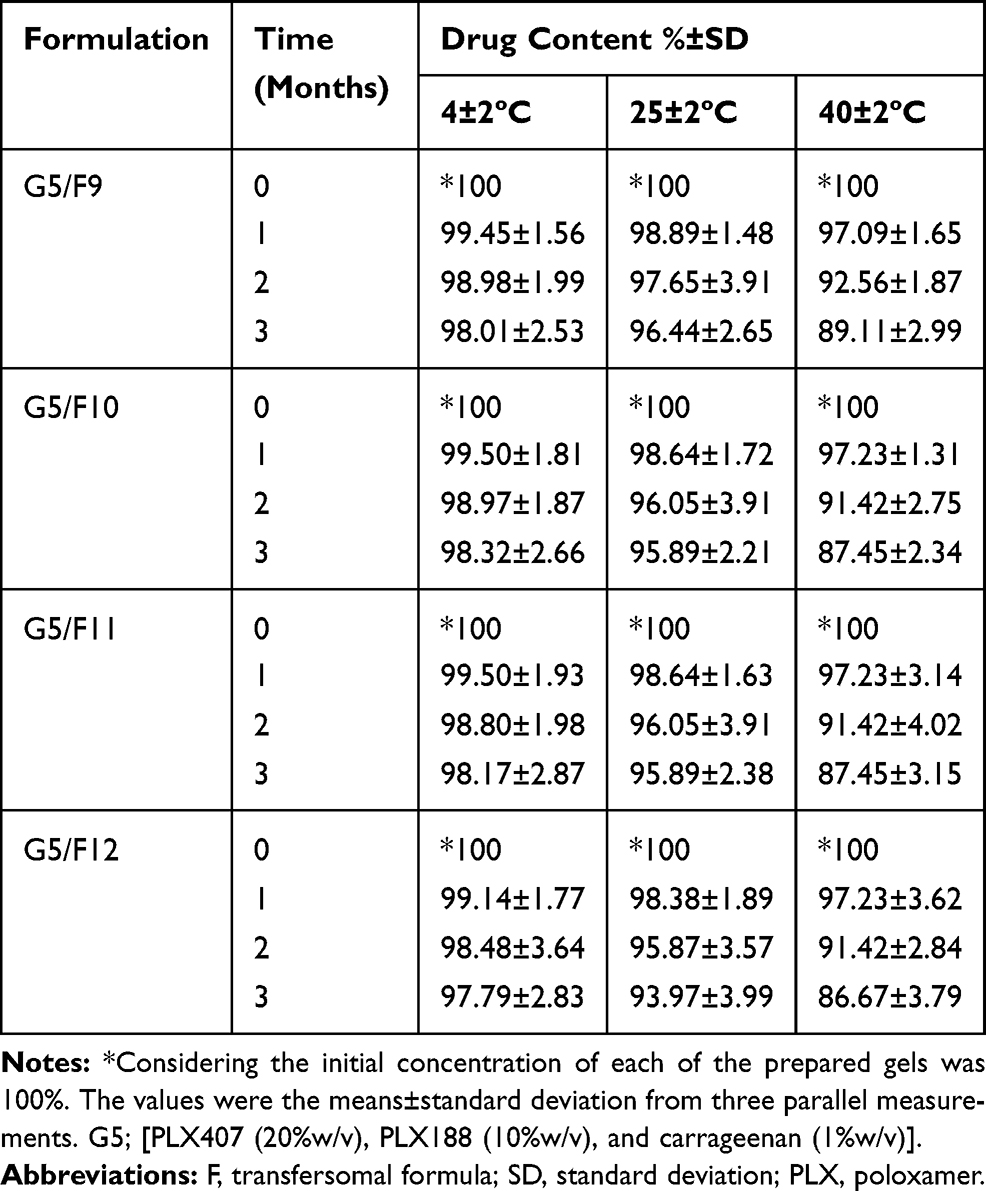 |
Table 5 The Percent Drug Content (%) of the Prepared in-situ Gels During Storage at 4±2°C, 25±2°C, and 40±2°C/75±5% Relative Humidity, RH Over a Period of 3 Months |
Gelling Temperature
Thermosensitive in-situ gel formulation characterized with a reversible change of its viscosity as a function of temperature change. As a result of increased temperature, polymer-based liquid formulations convert to be gels, delaying the nasal clearance of the administered formulations. To avoid the conversion of the formulation from liquid to gel state before its application, the gelling temperature should be more than room temperature. Because the temperature of the nasal cavity is 34ºC,36 the acceptable gelling temperature should be in the range of 26–34ºC. If the gelling temperature is lower than 26ºC, difficulty in manufacturing and application is predicted. But if the gelling temperature is higher than 34ºC, the applied formulation will be cleared early from the nasal cavity.
In the gel preformulation study, the formulations containing PLX 407 higher than 20% were highly viscous, accordingly they were difficult to be administered intranasally.
All gelling temperatures of the formulations containing 18% or 20% PLX 407 were in the range of 23.7ºC to 33.9ºC (Figure 4) confirming their capability to be administered intranasally. Elucidation of the PLX formulation gelation depends on the change of micellar number as a function of temperature. As a result of the negative solubility coefficient of the block copolymer, the formed micelles number increases when the temperature increases, leading to formation of the tightly packed micelles. So, the solution becomes gel.37 Packing and entanglements of the polymer may be another explanation for forming the gel.38 Moreover, as a result of the increase of temperature, the side chains methyl groups of the polymer chains, forming the inner core of the micelles, reoriented conformationally, expelling the water from the micelles, and the gelation phenomenon occurred.39
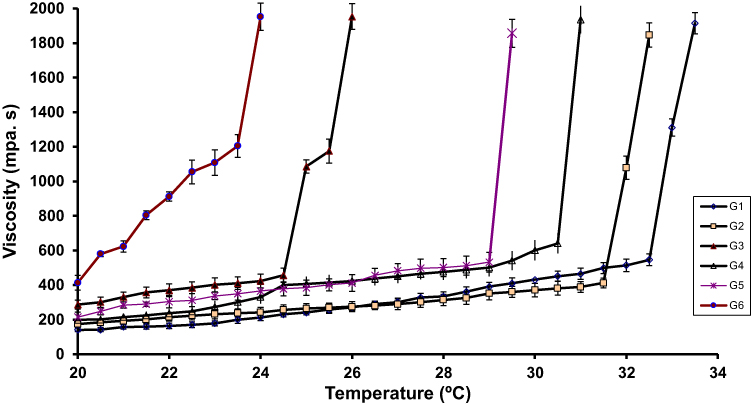 |
Figure 4 Effect of temperature on viscosity of various PLX 407 gels (18% and 20%) with varying concentrations of PLX 188 (5%, 10%, and 15%) and carrageenan (1.5%, 1%, and 0.5%) determined at 10 s−1 shear rate. Values are represented as mean±SD (n=3). Notes: G1; gel [PLX407 (18%w/v), PLX188 (5%w/v), and carrageenan (1.5%w/v)], G2; gel [PLX407 (18%w/v), PLX188 (10%w/v), and carrageenan (1%w/v)], G3; gel [PLX407 (18%w/v), PLX188 (15%w/v), and carrageenan (0.5%w/v)], G4; [PLX407 (20%w/v), PLX188 (5%w/v), and carrageenan (1.5%w/v)], G5 [PLX407 (20%w/v), PLX188 (10%w/v), and carrageenan (1%w/v)], and G6; [PLX407 (20%w/v), PLX188 (15%w/v), and carrageenan (0.5%w/v)].Abbreviation: PLX, poloxamer. |
Evaluation of pH of the Prepared Gel
The prepared in-situ gels were examined to measure pH and kept (Table 2). The pH of prepared in-situ gels ranged from 5.9–6.2. These findings prove that those gels were acceptable and physiologically compatible to be used in the nose cavity.
Mucoadhesive Strength
Evaluation of mucoadhesive strength is very important because of its great impact on elongation of residence time and decrease of formulation leakage. Mucoadhesive strength is defined as a quantity of formulation binding to the mucous membrane at nose temperature; 34ºC. To overcome nasal clearance, the mucoadhesive strength of the intranasal formulation should be high enough. Otherwise, the mucous membrane can be damaged when the mucoadhesive strength is too high.40
In the experiment process, the optimum contact time for giving the optimum mucoadhesive strength was 2 minutes. Any decrease in the contact time led to a sharp decrease in the mucoadhesive strength due to incomplete polymers chains entanglement with mucin. However, an increase of contact time has an insignificant effect on the mucoadhesive strength. A tensile test was conducted to measure the maximum force required to detach a piece of mucosal membrane from the prepared PLX gels. Our experimental findings showed sufficient mucoadhesive strength for all prepared gels, as represented in Figure 5. The highest mucoadhesive strength was recorded by the gel formula G5. So, this was selected to be the gel base for the optimized transferosomes F9, F10, F11, and F12 producing in-situ gels G5/F9, G5/F10, G5/F11, and G5/F12.
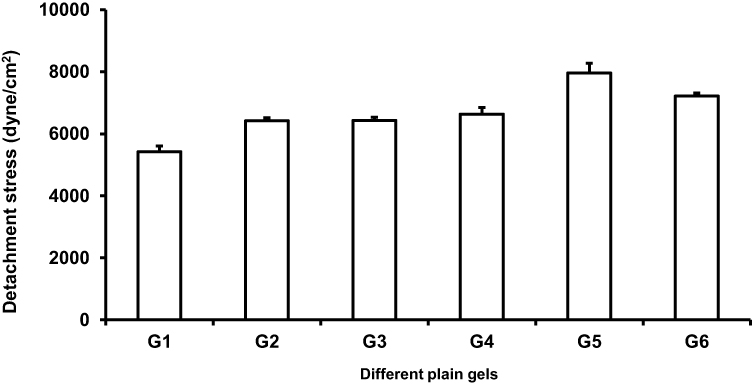 |
Figure 5 Influence of various PLX 407 gels (18% and 20%) with varying concentrations of PLX 188 (5%, 10%, and 15%) and Carrageenan (0.5%, 10%, and 15%) on the detachment stress measured in vitro. Measured values are represented as mean±SD (n=3). Notes: G1 composed of PLX407 (18%w/v), PLX188 (5%w/v), and carrageenan (1.5%w/v), G2 composed of PLX407 (18%w/v), PLX188 (10%w/v), and carrageenan (1%w/v), G3 composed of PLX407 (18%w/v), PLX188 (15%w/v), and carrageenan (0.5%w/v), G4 composed of PLX407 (20%w/v), PLX188 (5%w/v), and carrageenan (1.5%w/v), G5 composed of PLX407 (20%w/v), PLX188 (10%w/v), and carrageenan (1%w/v), and G6 composed of PLX407 (20%w/v), PLX188 (15%w/v), and carrageenan (0.5%w/v).Abbreviation: PLX, poloxamer. |
Drug Content in the Prepared in-situ Gels
The drug content of the examined formulations (G5/F9, G5/F10, G5/F11, and G5/F12) were 7,140, 6,132, 7,990, and 8,500 µg/cm3. The drug content variation in the in-situ gel was due to variation of encapsulation efficiency of the used transferosomes (F9, F10, F11, and F12).
In vitro Release of SUT from the Prepared Gel
Release of SUT from different transferosomes (F9, F10, F11, and F12) embedded within the prepared hydrogel (G5) was measured and is represented in Figure 6. The release rate from free drug embedded within G5 was higher thanfrom that of various transferosomes incorporated within G5. The release order of formulations can be arranged in descending order as follows: G5/free drug>G5/F9>G5/F10>G5/F11>G5/F12. Statistical analysis of release values proved that there were significant differences (P<0.05, paired two-tailed t-test) among the examined formulations.
The release of SUT was changed as the transferosomes were imbedded into the in situ gel. The slow SUT release from the prepared gel can be explained on the basis of difficult diffusion of the drug from the gel, where a number of barriers impeded the release of SUT from gel. First, the drugs penetrate across vesicular membranes, then diffuse within the gel space and finally to dissolution medium across dialysis cellophane membrane, consuming a longer time as compared to the free drug.24
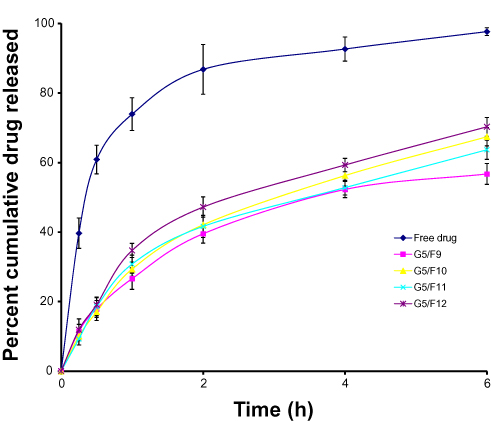 |
Figure 6 In vitro release profiles of SUT from both free SUT solution and various transferosomes (F9, F10, F11, and F12) incorporated into in situ gel G5 (PLX 407 (20%), PLX 188 (%w/v), carrageenan (%w/v)) at 37ºC. Notes: G5, gel formula composed of PLX407 (20%w/v), PLX188 (10%w/v), and carrageenan (1%w/v).Abbreviations: PLX, poloxamer; SUT, sumatriptan. |
Ex vivo Permeation Studies
The present experiments showed the effect of clostridium perfringens enterotoxin and sodium caprate as penetration enhancers formulated into transferosomes (F9, F10, F11, and F12). Permeation of a molecule across the biological membrane is a challenge multistep process. Many factors such as chemical structure, physical properties, and biological interactions can affect the permeation of the molecule. The permeation efficiency of the examined transferosomes against SUT solution (control I) and SUT-loaded transferosomes, F9 (control II), were evaluated using the calculated permeation parameters (Table 6). Based on the cumulative permeated SUT over 6 hours (Q6h), the order of the examined formula was arranged as follows; G5/F9>G5/F11>G5/F10>G5/F12>control II>control I. Permeation efficiency of any one of the prepared transferosomes containing in-situ gels was higher than permeation efficiency of control SUT solution. This finding could be attributed to the constituents of transferosomes, whereas the phospholipids and the used surfactant reduced the interfacial tension at the surface of the skin. Moreover, phospholipids have high affinity to the biological membrane.41 Also, deformability of the transferosomes bilayer was enhanced as a result of including the surfactant, enhancing the permeation across the nasal membrane. The prepared gels may play a role for enhancing the permeation because of their composition of anionic polymers Plx407, Plx188, and carrageenan. These polymers have great Ca2+ binding ability. Unexpectedly, Q6h of transferosomes with low ratios of permeation enhancers (F9 and F11) are higher than Q6h of transferosomes with high ratios of permeation enhancers (F10 and F12), respectively. Moreover, Q6h of transferosomes with permeation enhancer C-CPE (F9) is higher than Q6h of transferosomes with permeation enhancer Sodium Caprate (F10). These results can be attributed to action mode of the used enhancers. Whereas the C-terminal part of CPE can affect the second extracellular loop of claudin-3 and claudin-4 which are the most important components of the tight junction among cells, making pore formation in the plasma membrane of the cell.42 Modulation of the tight junction was reported as an effective technique for paracellular permeation of drugs. Moreover, the action mode of sodium caprate depends on opening the paracellular passage of SUT, as a result of retrieving of claudin-5 from bicellular tight junctions, causing a decrease in paracellular resistance and transepithelial resistance. The selective and rapid effect of sodium caprate on the paracellular barriers may also enhance the permeability of macromolecules.43 The strength of the present approach was mainly due to the use of surfactant, phospholipids, the effect of the gel forming polymers as chelators and high tissue specificity tight junction modulators. Moreover, the size and the vesicular shape of transferosomes may have a great impact on the permeability of the loaded drug.
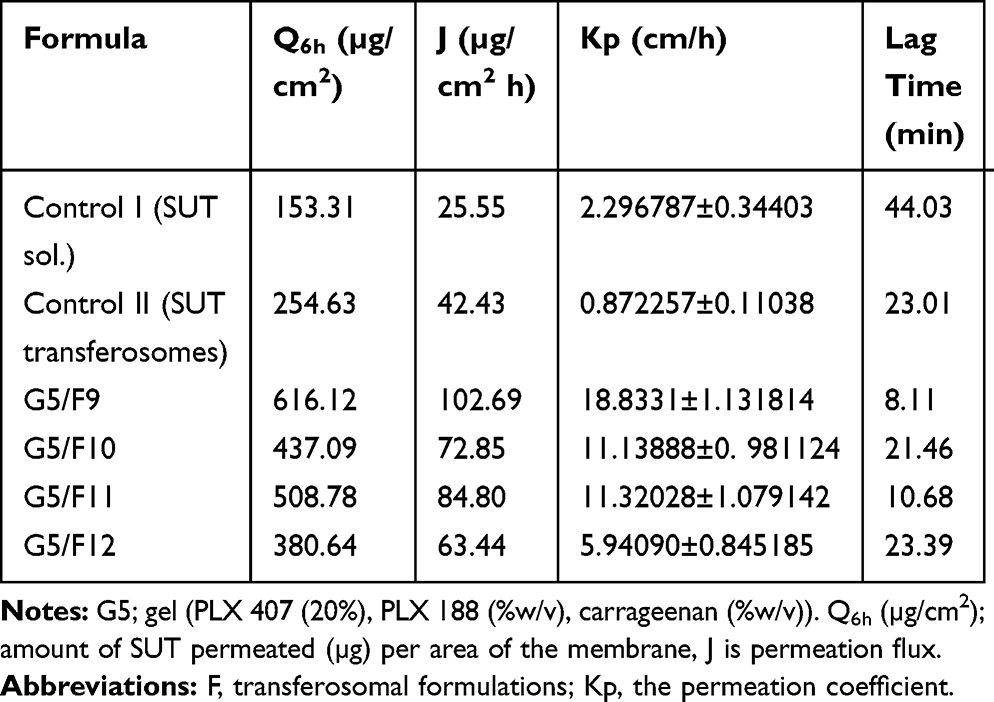 |
Table 6 Ex vivo Permeation Parameters of SUT-Loaded Transferosomes versus SUT Solution |
Histopathological Study
The histopathological study was conducted to examine safety of the prepared transferosomal SUT in-situ gel G5/F9 and G5/F11 as compared to the control (Figure 7). The safety was evaluated through determining any abnormalities, such as damage, irritation, or bleeding in nasal epithelial membrane barriers (Figure 7). In the first group, no abnormalities were detected (Figure 7A). In the second group, nasal epithelial membrane barriers showed intact and no abnormalities or damage (Figure 7B). However, in the third group, mild epithelial disruption and partial loss of cellular and ciliary identity as well as prevalence of extracellular debris were detected (Figure 7C). These results suggest that transferosomal SUT-loaded in-situ gel might be regarded as a safe with respect to nasal administration.
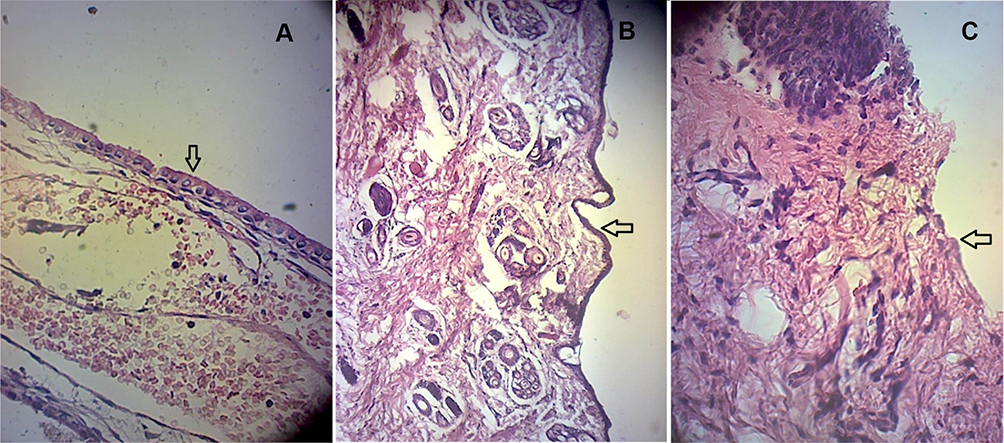 |
Figure 7 Light photomicrographs of (A) untreated rabbit nasal mucosal membrane, (B) rabbit nasal mucosal membrane treated with SUT-transferosome gel (G5/F9), and (C) rabbit nasal mucosal membrane treated with SUT-transferosome gel (G5/F11), stained with eosin and hematoxylin at magnification power 10×40, arrows denote mucosal membrane change. Notes: G5; gel formula composed of poloxamer 407 PLX407 (20%w/v), poloxamer 188; PLX188 (10%w/v) and carrageenan (1%w/v).Abbreviations: SUT; sumatriptan, F, transfersomal1 formula. |
In vivo Pharmacokinetic Study
The pharmacokinetic parameters of SUT in rabbit plasma were determined to examine the in vivo behavior of the G5/F9 and G5/F11 formulae as compared to oral SUT solution. The ultraviolet liquid chromatography was confirmed with good linearity within the used range of 1–300 ng/mL. The mean plasma concentrations of SUT have been plotted against the time (Figure 8), and the corresponding calculated pharmacokinetic parameters are represented in Table 7. Oral SUT was absorbed rapidly and reached the maximum value at 1 hour. However, SUT plasma concentration decreased obviously within the following few hours.
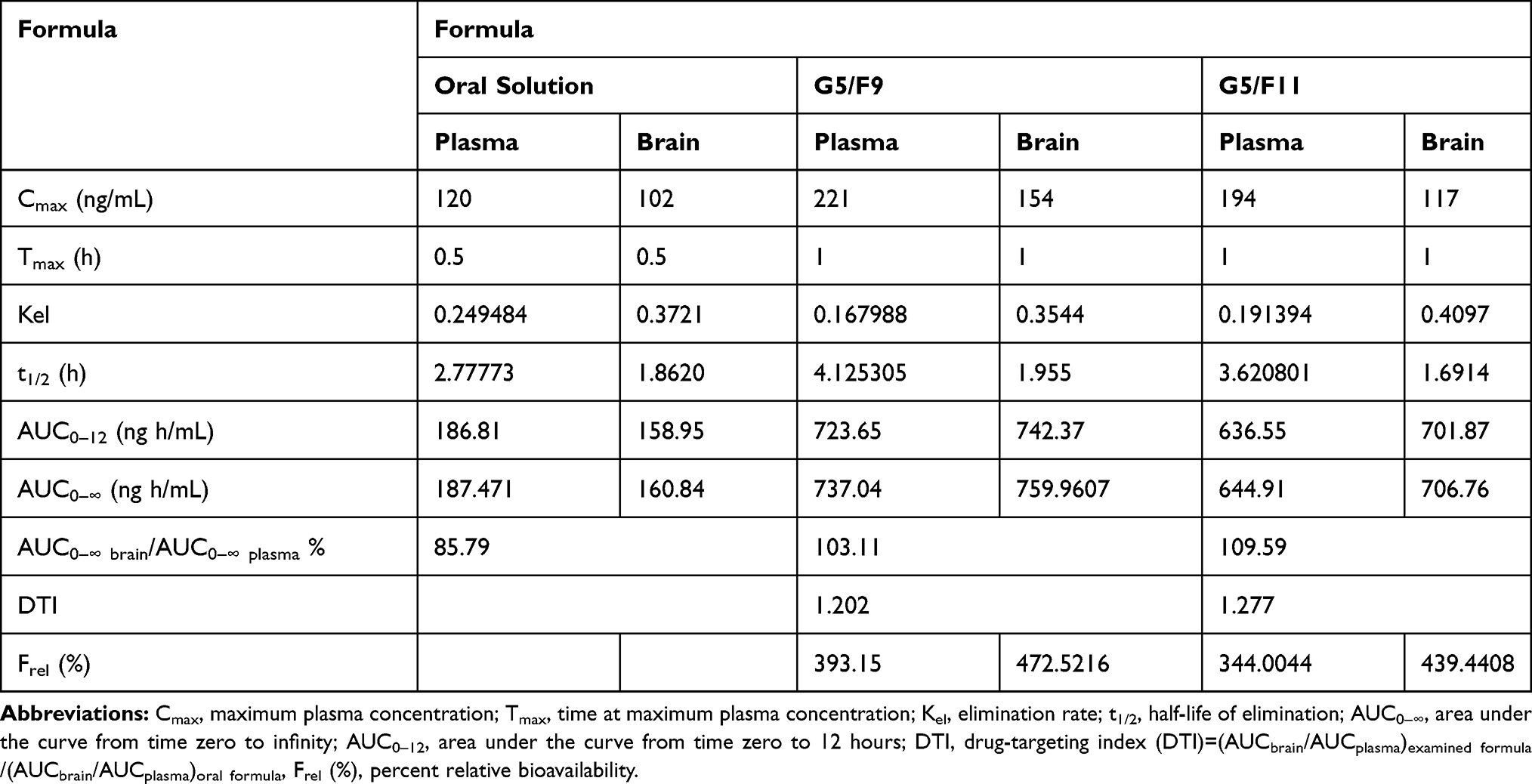 |
Table 7 Pharmacokinetic Parameters of SUT in Rabbit Plasma Following Administration of Oral Solution, and Transferosomal SUT in-situ Gels G5/F9 and G5/F11 |
The maximum plasma concentrations of SUT of 221 ng/mL and 194 ng/mL were determined at 1 hour for G5/F9 and G5/F11, respectively. The relative plasma bioavailability of G5/F9 and G5/F11 based on AUC0–∞ of oral formula were 393.15% and 344.00%, respectively. The enhanced bioavailability of those formulas was attributed to the great permeation power of the formulated transferosomes. Moreover, the role of perfringens enterotoxin as permeation enhancer was more efficient than sodium caprate. Among the transferosomal SUT in-situ gels, the perfringens enterotoxin based transferosomes showed the best efficacy in intranasal permeation of the drug due to a great ability of perfringens enterotoxin to open the tight junctions of mucosal tissues. The size of the prepared transferosomes within the nano-range may play a role for enhancing the permeability. The significant increase of half-life of the prepared formulae G5/F9 and G5/F11 may be explained on the basis of capability of the nano-transferosomes to escape from the metabolism. Among the transferosomal SUT in-situ gels, the perfringens enterotoxin based transferosomes showed the best efficacy in intranasal permeation of the drug due to the great ability of perfringens enterotoxin to open the tight junctions of mucosal tissues. Regarding SUT concentration in brain tissues, the AUC0–12 (ng.h/mL), Cmax of the groups received nasal gels (G5/F9 and G5/F11) were found to be greater than the groups receiving orally (Table 7). The ratio of (AUC brain tissue/AUC plasma) % for nasal gels (G5/F9 and G5/F11) and oral groups were found to be 85.79, 103.11, and 109.59%, respectively. It is concluded that intranasal administration of SUT loaded transferosomes as in-situ gel led to improve AUC values for brain tissues compared to oral administration (Table 7). Drug targeting index of SUT in brain tissues post intranasal administration of in-situ gel of SUT loaded transferosomes (G5/F9) and (G5/F11) was 1.202 and 1.277, respectively. The findings of pharmacokinetics study have correlated to ex vivo findings based on the Wagner-Nelson model.44 Finally, high relative bioavailability values (Frel) and increased half-lives of the prepared transferosomes of SUT incorporated into in-situ gel confirmed that a great bioavailability and sustained release of the drug were done.
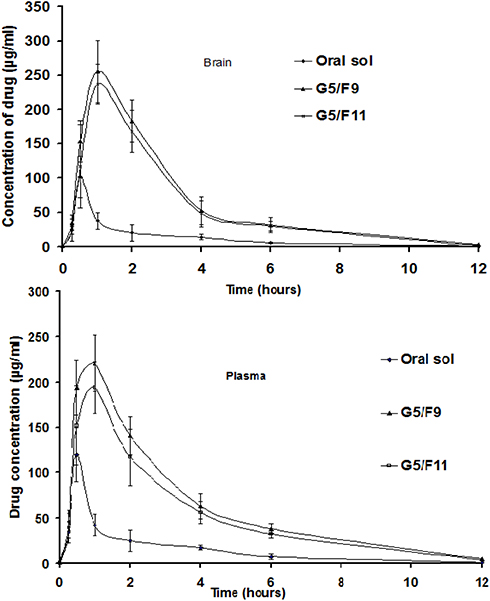 |
Figure 8 Mean SUT concentrations in rabbit plasma and brain tissue post administration of oral solution (20 mg) and application of nasal transferosomal SUT (20 mg) in-situ gel (G5/F9) and (G5/F11). Notes: G5; gel (PLX 407 (20%), PLX 188 (%w/v), carrageenan (%w/v)).Abbreviations: F, transferosomal formulations; Oral sol, oral solution. |
Conclusion
The best formula (F9) with clostridium perfringens enterotoxin showed a considerably high EE%, small vesicle size and sustained release of SUT over 6 hours. The pharmacokinetic results confirmed that the optimum gel G5/F9 enhanced the relative bioavailability of SUT 3.93- and 4.09-fold as compared to oral SUT solution in plasma and brain tissues, respectively. It also showed a sustained drug release with elimination half-life (t0.5) of 4.12 and 1.96 hours in plasma and brain tissues, respectively. Moreover, the relative bioavailability of the gel G5/F11 of SUT was 3.44- and 4.39-fold as compared to oral SUT solution in plasma and brain tissues, respectively. From the aforementioned comparison, the prepared transferosomal SUT in-situ gels succeeded as a non-invasive drug delivery system through the nasal route. Finally, it concluded that a promising non-invasive drug delivery with an improved patient compliance was developed. It is evident from the study that there is feasibility of delivering SUT through transferosomal transdermal gel. Thus, the developed transdermal transferosomal formulation may prove to be a promising carrier for SUT, especially due to their simple production and simplistic scale-up.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Silberstein SD, Lipton RB. Overview of diagnosis and treatment of migraine. Neurology. 1994;44(10):6–16.
2. Lipton R, Stewart W, Diamond S, Diamond M, Reed M. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. J Headache Pain. 2001;41(7):646–657. doi:10.1046/j.1526-4610.2001.041007646.x
3. Timothy J, Lars JS, Gretchen L. Migraine: the seventh disabler. J Headache Pain. 2013;14(1):1–2. doi:10.1186/1129-2377-14-1
4. Steiner TJ, Scher AI, Stewart WF, Kolodner K, Liberman J, Lipton RB. The prevalence and disability burden of adult migraine in England and their relationships to age, gender and ethnicity. Cephalalgia. 2003;23(7):519–527. doi:10.1046/j.1468-2982.2003.00568.x
5. Capkova Z, Vitkova Z, Subova M. Formulation of loratadine into hydrogels. Acta Facult Pharm Univ Comenianae. 2005;52:73–78.
6. Tso A, Goadsby P. Anti-CGRP monoclonal antibodies: the next era of migraine prevention? Curr Treat Options Neurol. 2017;19(8):27–38. doi:10.1007/s11940-017-0463-4
7. Ullah I, Chung K, Oh J, et al. Intranasal delivery of a fas-blocking peptide attenuates fas-mediated apoptosis in brain ischemia. Sci Rep. 2018;8(1):15041. doi:10.1038/s41598-018-33296-z
8. William H, Frey I. Bypassing the blood–brain barrier to delivery therapeutic agents to the brain and spinal cord. Drug Delivery Technol. 2002;2(5):46–49.
9. Omar M, Hasan O. Improving dissolution and oral bioavailability of fenofibrate by microwave induced fusion technique using HPMC E5 LV. Lat Am J Pharm. 2019;38(3):587–598.
10. Salem H, Ahmed S, Omar M. Liposomal flucytosine capped with gold nanoparticle formulations for improved ocular delivery. Drug Des Dev Ther. 2016;10:277. doi:10.2147/DDDT.S91730
11. Scalia S, Trotta V, Iannuccelli V, Bianchi A. Enhancement of in vivo human skin penetration of resveratrol by chitosan-coated lipid microparticles. Colloids Surf B Biointerfaces. 2015;135:42–49. doi:10.1016/j.colsurfb.2015.07.043
12. Friedrich RB, Kann B, Coradini K, Offerhaus HL, Beck R, Windbergs M. Skin penetration behavior of lipid-core nanocapsules for simultaneous delivery of resveratrol and curcumin. Eur J Pharm Sci. 2015;78:204–213. doi:10.1016/j.ejps.2015.07.018
13. Salem H, Ahmed S, Hassaballah A, Omar M. Targeting brain cells with glutathione-modulated nanoliposomes: in vitro and in vivo study. Drug Des Dev Ther. 2015;9:3705–3727. doi:10.2147/DDDT.S85302
14. Omar M, Hasan O, El Sisi A. Preparation and optimization of lidocaine transferosomal gel containing permeation enhancers: a promising approach for enhancement of skin permeation. Int J Nanomed. 2019;14:1551–1562. doi:10.2147/IJN.S201356
15. Abdel-Rashid RS, Helal DA, Omar MM, El Sisi AM. Nanogel loaded with surfactant based nanovesicles for enhanced ocular delivery of acetazolamide. Int J Nanomed. 2019;14:2973–2983. doi:10.2147/IJN.S201891
16. Villasmil-Sánchez S, Drhimeur W, Ospino SC, Rabasco Alvarez AM, González-Rodríguez ML. Positively and negatively charged liposomes as carriers for transdermal delivery of sumatriptan: in vitro characterization. Drug Dev Ind Pharm. 2010;36(6):666–675. doi:10.3109/03639040903419640
17. Ghanbarzadeh S, Arami S. Formulation and evaluation of piroxicam transferosomal gel: an approach for penetration enhancement. J Drug Deliv Sci Technol. 2013;23(6):587–590. doi:10.1016/S1773-2247(13)50089-X
18. van den Bergh B, Wertz P, Junginger H, Bouwstra J. Elasticity of vesicles assessed by electron spin resonance, electron microscopy and extrusion measurements. Int J Pharm. 2001;217(1–2):13–24. doi:10.1016/S0378-5173(01)00576-2
19. Hao Y, Zhao F, Li N, Yang Y, Li K. Studies on a high encapsulation of colchicine by a niosome system. Pharm J Int. 2002;244(1–2):73–80. doi:10.1016/S0378-5173(02)00301-0
20. Du Plessis J, Ramachandran C, Weiner N, Müller D. The influence of lipid composition and lamellarity of liposomes on the physical stability of liposomes upon storage. Pharm J Int. 1996;127(2):273–278. doi:10.1016/0378-5173(95)04281-4
21. Choi H-G, Jung J-H, Ryu J-M, Yoon S-J, Oh Y-K, Kim C-K. Development of in situ-gelling and mucoadhesive acetaminophen liquid suppository. Int J Pharm. 1998;165(1):33–44. doi:10.1016/S0378-5173(97)00386-4
22. Shelke S, Shahi S, Jadhav K, Dhamecha D, Tiwari R, Patil H. Thermoreversible nanoethosomal gel for the intranasal delivery of eletriptan hydrobromide. J Mater Sci Mater Med. 2016;27(6):103. doi:10.1007/s10856-016-5713-6
23. Pourmand M, Azar M, Aghavalijamaat M. Development of validated UV spectrophotometric method for in vitro analysis of sumatriptan in pharmaceutical preparations in comparison with HPLC. Pharm Chem J. 2011;44(10):585–589. doi:10.1007/s11094-011-0522-1
24. Majumder P, Baxa U, Walsh SR, Schneider JP. Design of a multicompartment hydrogel that facilitates time‐resolved delivery of combination therapy and synergized killing of glioblastoma. Angew Chem. 2018;130(46):15260–15264. doi:10.1002/ange.201806483
25. Kamel R, Basha M, Abd El-Alim S. Development of a novel vesicular system using a binary mixture of sorbitan monostearate and polyethylene glycol fatty acid esters for rectal delivery of rutin. J Liposome Res. 2013;23:28–36. doi:10.3109/08982104.2012.727422
26. Gonza´lez-Rodrı´guez M, Arroyo C, Co´zar-Bernal M, et al. Deformability properties of timolol-loaded transferosomes based on the extrusion mechanism. Statistical optimization of the process. Drug Dev Ind Pharm. 2016;42:1683–1694. doi:10.3109/03639045.2016.1165691
27. Lappin G, Shishikura Y, Jochemsen R, et al. Comparative pharmacokinetics between a microdose and therapeutic dose for clarithromycin, sumatriptan, propafenone, paracetamol (acetaminophen), and phenobarbital in human volunteers. Eur J Pharm Sci. 2011;43(3):141–150. doi:10.1016/j.ejps.2011.04.009
28. Dora CP, Singh SK, Kumar S, Datusalia AK, Deep A. Development and characterization of nanoparticles of glibenclamide by solvent displacement method. Acta Pol Pharm. 2010;283–90:283–290.
29. El Zaafarany G, Awad G, Holayel S, Mortada N. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int J Pharm. 2010;397:164–172. doi:10.1016/j.ijpharm.2010.06.034
30. Abdelbary G, El-Gendy N. Niosome-encapsulated gentamicin for ophthalmic controlled delivery. AAPS PharmSciTech. 2008;9:740–747. doi:10.1208/s12249-008-9105-1
31. Yusuf M, Sharma V, Pathak K. Nanovesicles for transdermal delivery of felodipine: development, characterization, and pharmacokinetics. Int J Pharm Investig. 2014;4:119–130. doi:10.4103/2230-973X.138342
32. Ourique AF, Pohlmann AR, Guterres SS, Beck RCR. Tretinoin-loaded nanocapsules: preparation, physicochemical characterization, and photostability study. Int J Pharm. 2008;352(1–2):1–4. doi:10.1016/j.ijpharm.2007.12.035
33. Cristina B, Andres F, Virginia M, et al. Elastic vesicles of sumatriptan succinate for transdermal administration: characterization and in vitro permeation studies. J Liposome Res. 2011;21(1):55–59. doi:10.3109/08982101003736002
34. Jain S, Jain P, Uma Maheshwari R, Jain N. Transferosomes—a novel vesicular carrier for enhanced transdermal delivery: development, characterization, and performance evaluation. Drug Dev Ind Pharm. 2003;29:1013–1026. doi:10.1081/DDC-120025458
35. Wasankar S, Faizi S, Deshmuk A. Formulation and development of liposomal gel for topical drug delivery system. Int J Pharm Sci Res. 2012;3(11):4461.
36. Keck T, Leiacker R, Riechelmann H, Reittinger G. Temperature profile in the nasal cavity. Laryngoscope. 2000;110:651–654. doi:10.1097/00005537-200004000-00021
37. Kabanov A, Batrakova E, Alakhov V. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release. 2002;82:189–212. doi:10.1016/S0168-3659(02)00009-3
38. Cabana A, AitKadi A, Juhasz J. Study of the gelation process of polyethylene oxide a-polypropylene oxide b-polyethylene oxide a copolymer (poloxamer 407) aqueous solutions. J Colloid Interface Sci. 1997;190:307–312. doi:10.1006/jcis.1997.4880
39. Rassing J, Attwood D. Ultrasonic velocity and light scattering studies on polyoxyethylene-polyoxypropylene copolymer PF127 in aqueous solution. Int J Pharm. 1982;13:47–55. doi:10.1016/0378-5173(82)90141-7
40. Yong C, Choi J, Quan Q, et al. Effect of sodium chloride on the release, absorption and safety of diclofenac sodium delivered by poloxamer gel. Int J Pharm. 2001;263:195–205. doi:10.1016/S0378-5173(01)00809-2
41. Law SL, Huang KJ, Chou HY. Preparation of desmopressin-containing liposomes for intranasal delivery. J Control Release. 2001;70(3):375–382. doi:10.1016/S0168-3659(00)00369-2
42. Anna V, Jonas P, Jan R, Ingolf E, Gerd K, Joerg P. On the interaction of clostridium perfringens enterotoxin with claudins. Toxins. 2010;2:1336–1356. doi:10.3390/toxins2061336
43. Susanne M, Maren A, Isabel D, Ilya C, Michael F, Salah A. Sodium caprate as an enhancer of macromolecule permeation across tricellular tight junctions of intestinal cells. Biomaterials. 2013;34:275–282. doi:10.1016/j.biomaterials.2012.09.051
44. Wagner JG. Application of the Wagner-Nelson absorption method to the two-compartment open model. J Pharmacokinet Pharmacodyn. 1974;2(6):469–486.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.