")
Back to Journals » OncoTargets and Therapy » Volume 10
Developing a novel dual PI3K–mTOR inhibitor from the prodrug of a metabolite
Authors Zhou Y, Zhang GY, Wang F, Wang J, Ding YW, Li XY, Shi CT, Li JK, Shih CK, You S
Received 24 May 2017
Accepted for publication 23 August 2017
Published 20 October 2017 Volume 2017:10 Pages 5077—5087
DOI https://doi.org/10.2147/OTT.S142492
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Yan Zhou,1,2 Genyan Zhang,2 Feng Wang,2 Jin Wang,2 Yanwei Ding,2 Xinyu Li,2 Chongtie Shi,2 Jiakui Li,2 Chengkon Shih,2 Song You1
1The School of Life Science and Biopharmaceutical, Shenyang Pharmaceutical University, Shenyang, 2Department of Project Management, Medicinal Chemistry, Pharmacology, Drug Metabolism, and Pharmacokinetics, Toxicology, Xuanzhu Pharma, Jinan, China
Abstract: This study presents a process of developing a novel PI3K–mTOR inhibitor through the prodrug of a metabolite. The lead compound (compound 1) was identified with similar efficacy as that of NVP-BEZ235 in a tumor xenograft model, but the exposure of compound 1 was much lower than that of NVP-BEZ235. After reanalysis of the blood sample, a major metabolite (compound 2) was identified. Compound 2 exerted similar in vitro activity as compound 1, which indicated that compound 2 was an active metabolite and that the in vivo efficacy in the animal model came from compound 2 instead of compound 1. However, compound 1 was metabolized into compound 2 predominantly in the liver microsomes of mouse, but not in the liver microsomes of rat, dog, or human. In order to translate the efficacy in the animal model into clinical development or predict the pharmacokinetic/pharmacodynamic parameters in the clinical study using a preclinical model, we developed the metabolite (compound 2) instead of compound 1. Due to the low bioavailability of compound 2, its prodrug (compound 3) was designed and synthesized to improve the solubility. The prodrug was quickly converted to compound 2 through both intravenous and oral administrations. Because the prodrug (compound 3) did not improve the oral exposure of compound 2, developing compound 3 as an intravenous drug was considered by our team, and the latest results will be reported in the future.
Keywords: PI3K, mTOR, NVP-BEZ235, prodrug, metabolite, antitumor
Introduction
With rapid progress in molecular biology and translational medicine, the focus of malignant-tumor treatment has moved from conventional chemotherapy to targeted therapy, especially in cases with multiple alterations, overexpression, and mutations of genes, such as EGFR, ALK, MEK, BRAF, VEGFR, and MET.1 A great amount of effort has also been put into exploring the PI3K–Akt–mTOR signaling pathway for cancer therapy. This pathway not only plays a major role in normal physiological function but also impacts the progress of growth, proliferation, and metastasis of tumor cells.2 The PI3K family are lipid kinases. Their function is the phosphorylation of phosphoinositides on 3-OH when it is activated by the upstream receptor tyrosine kinases.3 mTOR is a serine/threonine kinase that belongs to the series of PI3K-related protein kinases. First identified in yeast in 1994, mTOR is made up of multiple proteins that drive the process of the cell life cycle. Since PI3K–Akt–mTOR is the downstream element of growth-factor receptor tyrosine kinases, when the mutation or amplification of the key encoding genes in this pathway occurs, it will cause abnormal signal transcription or translation and lead ultimately to cancer.4,5 It has been reported that in breast cancer, the most frequent mutation occurs in the PI3K gene, and overall abnormal activation on the PI3K–Akt–mTOR signaling pathway reaches approximately 70% for these patients.6,7
Extensive research has been conducted on targeting the PI3K–Akt–mTOR pathway in oncology therapy, especially in breast and ovarian cancers.8 Many new chemical entities are being assessed in preclinical or clinical stages. Rapamycin and everolimus, successful mTOR-inhibitor drugs, have been approved by the US Food and Drug Administration (FDA).9 The pioneer dual inhibitor of PI3K–mTOR, NVP-BEZ235, was developed by Novartis and is currently in clinical Phase II trials. Other dual inhibitors, such as apitolisib (Genentech), PQR-309 (Universität Basel), and voxtalisib (Exelixis), are also being developed and are in different clinical phases.10–12 Most dual inhibitors are designed by modifying the structure of NVP-BEZ235. NVP-BEZ235 structure is shown in Figure 1.
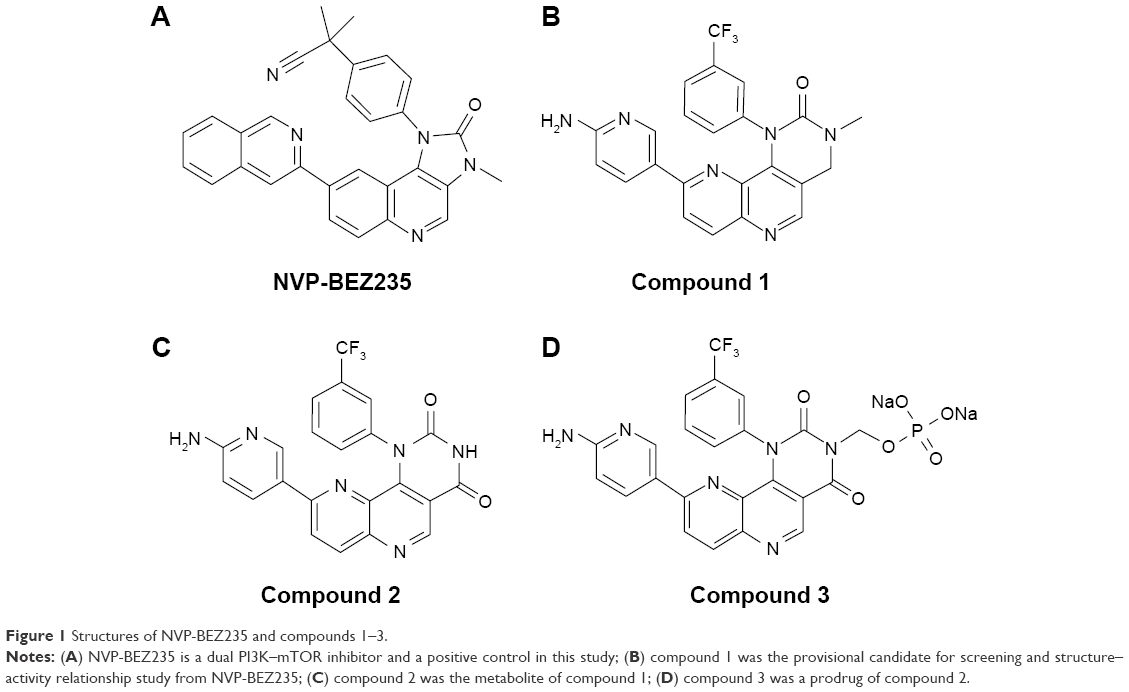 | Figure 1 Structures of NVP-BEZ235 and compounds 1–3. |
It is well established that drug tolerance is one of the key challenges for dual PI3K–mTOR inhibitors, which explains the slow progress of research and development of these inhibitors. Currently, only eleven new dual PI3K–mTOR inhibitors are being evaluated actively in the clinical stage, but none has been approved by the FDA. Although significant therapeutic effects against cancer have been demonstrated, drugs targeting the PI3K–Akt–mTOR pathway, (eg, everolimus) have shown significant toxicity, including stomatitis, noninfectious pneumonitis, rash, hyperglycemia, and immunosuppression.13 Therefore, developing new drug candidates with reduced side effects is necessary. In addition, dual PI3K–mTOR inhibitors combined with other pathway inhibitors could have a synergistic effect in treating malignant tumors.14,15 Indeed, as an example, the combination of PD98059 (MEK inhibitor) with NVP-BEZ235 induces cell-cycle arrest and apoptosis more effectively than single inhibitors.
Furthermore, there are large unmet medical needs for the development of new drugs with activity on the PI3K–Akt–mTOR pathway. In our earlier work, a series of compounds were designed and synthesized with a similar scaffold as NVP-BEZ235. After evaluation of the structure–activity relationship, absorption, distribution, metabolism, and excretion properties, and in vivo efficacy, a lead compound (compound 1) was identified and selected for further development. Figure 1 illustrates the chemical structure of compound 1. In this paper, we disclose the process of discovering a metabolite of compound 1 and developing it into a novel dual PI3K–mTOR inhibitor through the prodrug strategy (Figures 2–4 and Tables 1–7).
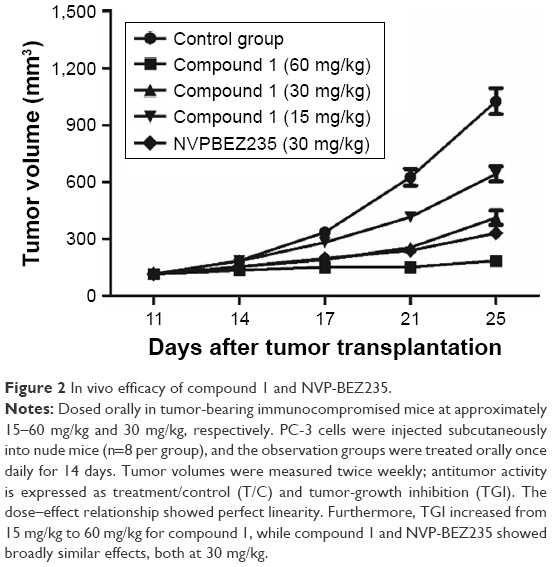 | Figure 2 In vivo efficacy of compound 1 and NVP-BEZ235. |
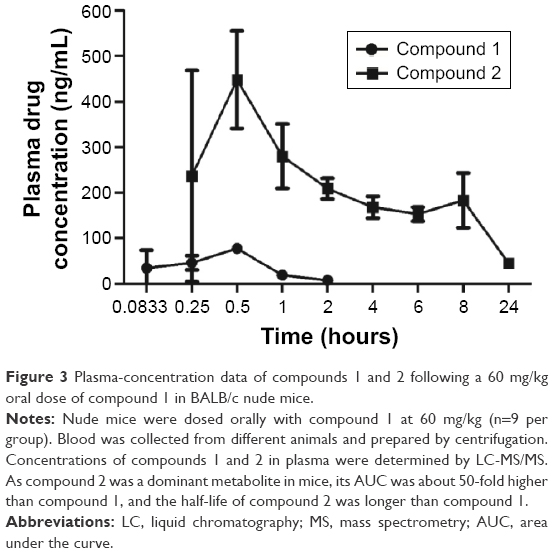 | Figure 3 Plasma-concentration data of compounds 1 and 2 following a 60 mg/kg oral dose of compound 1 in BALB/c nude mice. |
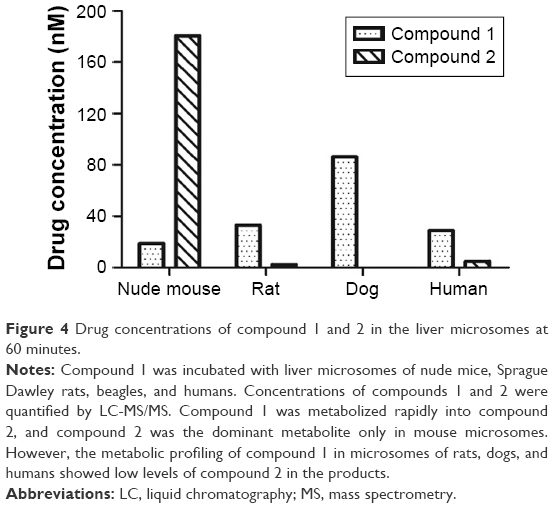 | Figure 4 Drug concentrations of compound 1 and 2 in the liver microsomes at 60 minutes. |
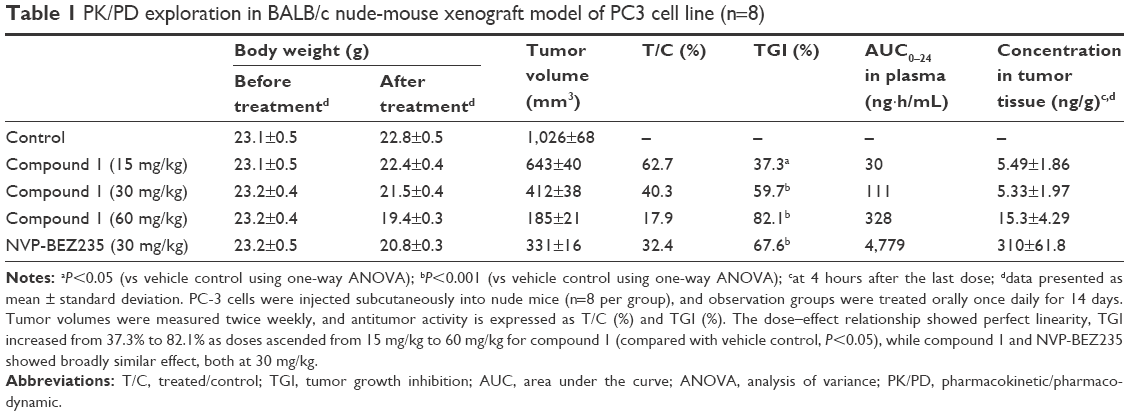 | Table 1 PK/PD exploration in BALB/c nude-mouse xenograft model of PC3 cell line (n=8) |
 | Table 2 Pharmacokinetic parameters of compound 1 and its metabolite compound 2 after oral dosing of compound 1 in nude mice (60 mg/kg) |
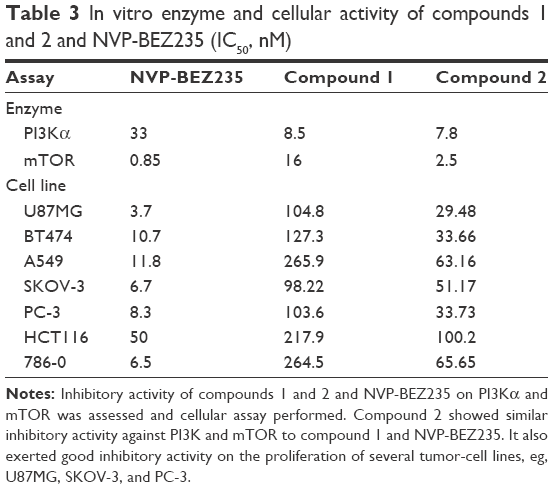 | Table 3 In vitro enzyme and cellular activity of compounds 1 and 2 and NVP-BEZ235 (IC50, nM) |
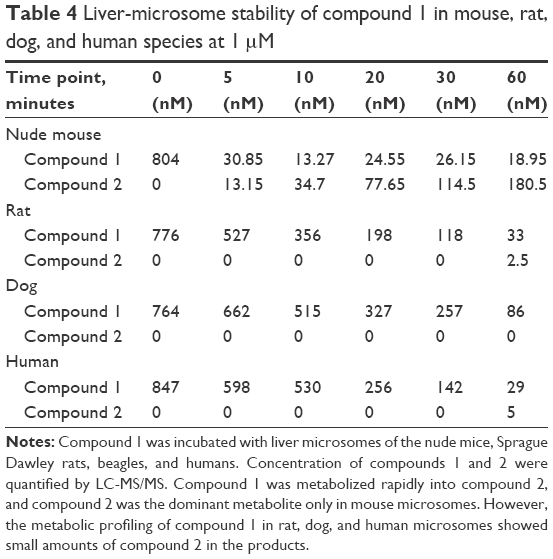 | Table 4 Liver-microsome stability of compound 1 in mouse, rat, dog, and human species at 1 μM |
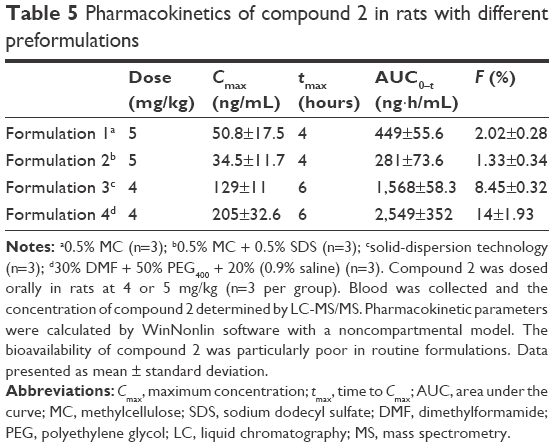 | Table 5 Pharmacokinetics of compound 2 in rats with different preformulations |
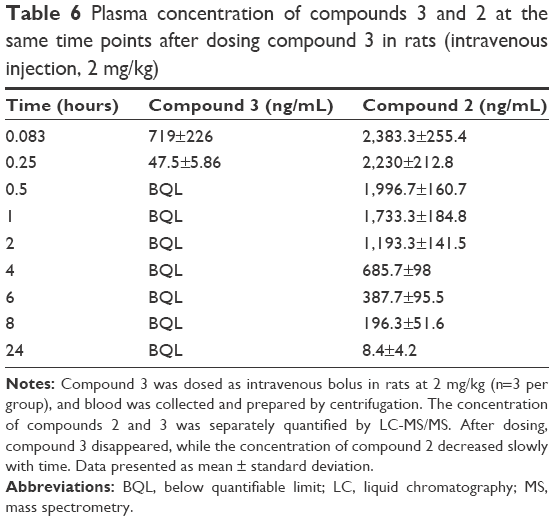 | Table 6 Plasma concentration of compounds 3 and 2 at the same time points after dosing compound 3 in rats (intravenous injection, 2 mg/kg) |
 | Table 7 Pharmacokinetics in rats of compound 2 after IV and PO dosing of compound 3 |
Materials and methods
Chemicals and materials
NVP-BEZ235 was purchased from Selleckchem (S1009). Compounds 1–3 were synthesized in the laboratory at Xuanzhu Pharma, and the synthetic routes of the three compounds are shown in Figures 5–7, respectively. In addition, detailed synthetic routes have been described in the patents.16–18 All three compounds were characterized by nuclear magnetic resonance (NMR) and mass spectrometry (MS) and their structures further qualified. Other materials – β-cyclodextrin, HEPES, Tween 20, penicillin, streptomycin, MgCl2, and β-NADPH – were all of analytical grade.
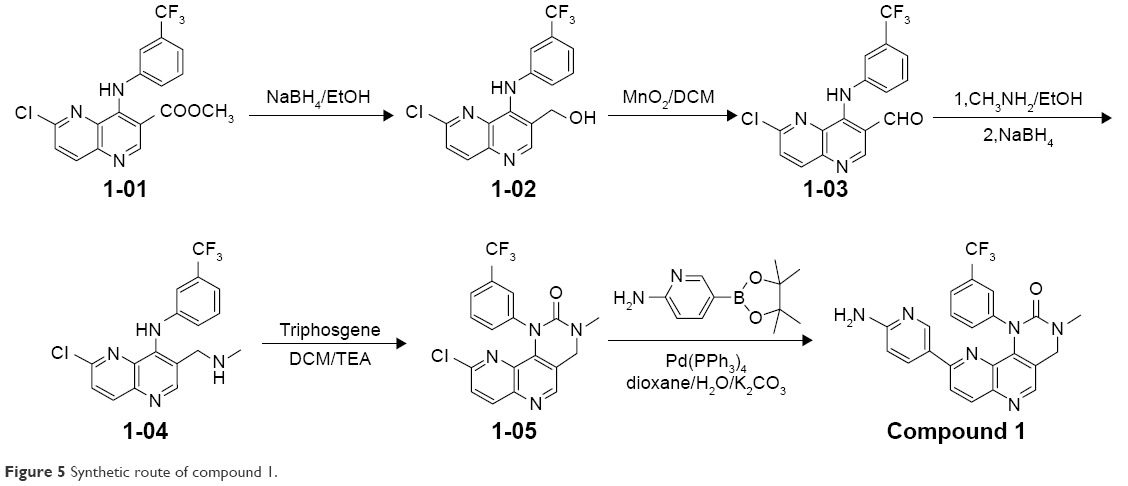 | Figure 5 Synthetic route of compound 1. |
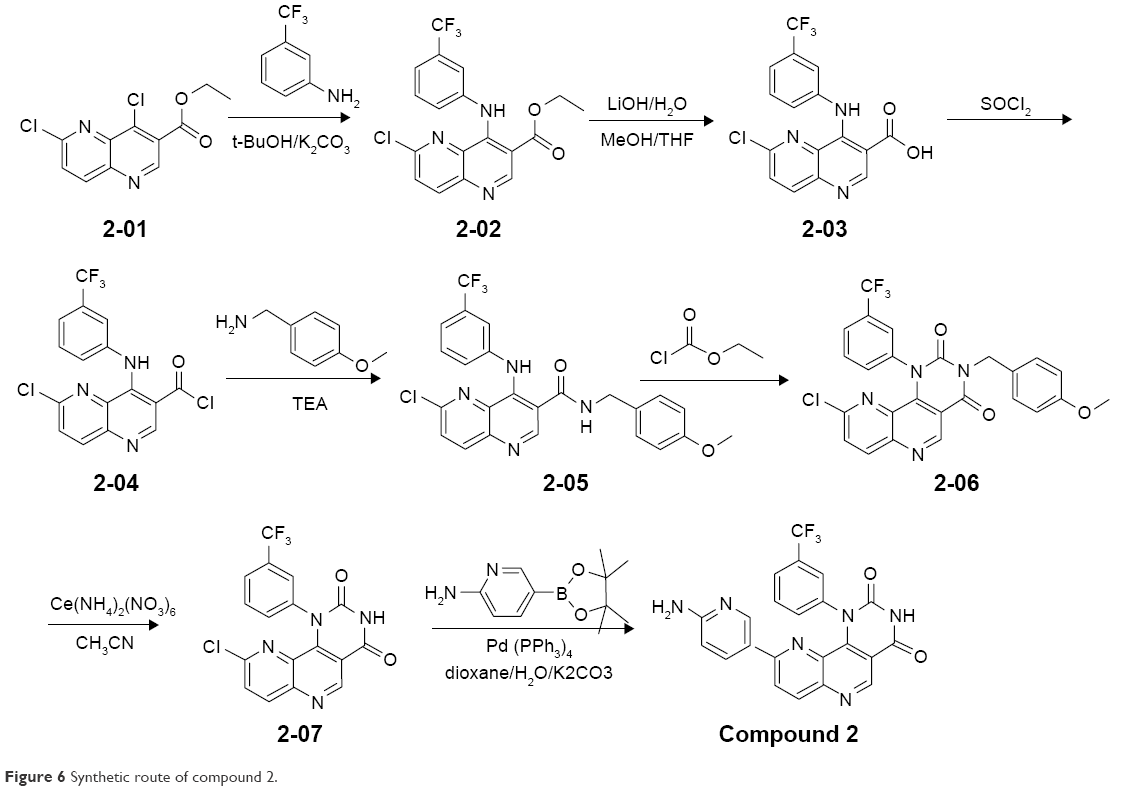 | Figure 6 Synthetic route of compound 2. |
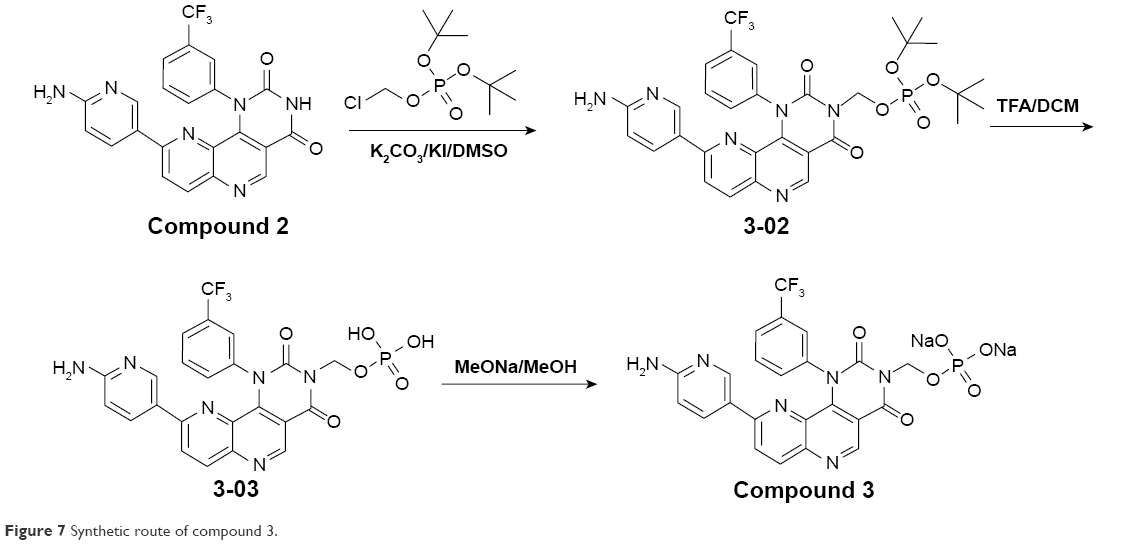 | Figure 7 Synthetic route of compound 3. |
PI3K and mTOR enzymes were purchased from Thermo Fisher Scientific (A11815) and EMD Millipore (14-770), respectively. A Kinase-Glo Plus luminescent kinase-assay kit was purchased from Promega (V3771). U87MG, BT474, A549, SKOV-3, PC-3, HCT116, and 786-0 cell lines were purchased from ATCC. RPMI- 1640 medium for 786-0/BT474, McCoy’s 5A medium for HCT116/SKOV-3, and DMEM for U87MG were purchased from GE Healthcare. Ham’s F-12K medium for PC-3/A549 was purchased from Thermo Fisher Scientific, and FBS was purchased from GE Healthcare. All the liver microsomes were purchased from the Research Institute for Liver Diseases. BALB/c nude mice were obtained from Huafukang Biological Technology, and Sprague Dawley rats were purchased from Vital River Laboratories.
Ethics statement
All animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals. The protocol was approved by the institutional animal care and use committee of Xuanzhu Pharma. All experiments were carried out in strict compliance with the regulations. Animals were monitored daily for physical appearance and mobility and were killed when they met specific criteria or showed signs of distress. Animals were acclimatized for more than 3 days before experiments and were housed under controlled temperature and humidity with a 12/12-hour light/dark cycle with ad libitum feeding in an Association for Assessment and Accreditation of Laboratory Animal Care-certified animal facility. All procedures in the pharmacokinetic (PK) experiments in rats and mice were performed under isoflurane anesthesia, and all efforts were made to minimize suffering.
In vitro PI3K- and mTOR-inhibition assays
To evaluate the inhibitory activities of the compounds on PI3Ks and mTOR, in vitro kinase assays were performed at ChemPartner. Compounds were serially diluted (ten concentrations) and prepared as source plate and assay plate. Kinase (4×) and (2×) substrate solutions were also prepared, and the assay plate was subsequently incubated at room temperature for 1 hour. After kinase detection, data were collected on an EnVision (Caliper reading). Lance signal (665 nm) values were copied and subsequently converted to percentage-inhibition values:
|
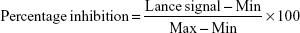
where min indicates the Lance signal of no enzyme control and max indicates the Lance signal of the dimethyl sulfoxide control. Compounds 1 and 2 were tested for their potency against class I PI3K, mTOR, and a panel of representative kinases using Kinase-Glo assays for PI3Kα, Lance Ultra kinase assay for mTOR, and Caliper for other kinases, with ATP concentrations in Km (Michaelis–Menten constant).
Tumor-cell growth-inhibition assays
In vitro compound potency was determined using different tumor-cell lines and methods that have been described in the patents.16,17 Cells were cultured in medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C under a 5% CO2 atmosphere. Cells that were 80% confluent were used in the assay. The suspension of freshly trypsinized cultured cells was spun at 1,000 rpm for 4 minutes and subsequently resuspended in fresh medium supplemented with 10% FBS. After adjustment of cell density, cells were seeded in 96-well plates. After 24 hours, test-compound solutions of different concentrations were prepared with culture medium, and 100 μL of each compound solution was transferred to each well according to the plate maps. Cells were treated for 72 hours at 37°C under a 5% CO2 atmosphere. After the medium had been removed, 150 μL of XTT reagent was added to each well. The plate was then incubated at 37°C for 120 minutes and the testing wells read at a wavelength of 450 nm. All tests were performed in duplicate.
In vivo efficacy studies in tumor xenograft model
Tumor-growth-inhibition studies were performed in BALB/c nude mice. Initial body weight of mice on arrival was 18–22 g. Briefly, PC3 cells were grown in RPMI 1650 medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C under 5% CO2 atmosphere. Resuspended PC3 cells (5×106 cells per animal) were injected subcutaneously into the right flank in 90 mice. When the average tumor volume reached 100 mm3, animals were randomly assigned to four groups (n=8 each). Animals were treated with compound 1 (15, 30, and 60 mg/kg) and vehicle (β-cyclodextrin) orally once daily for 14 days. Tumor volume and body weight were measured twice a week. The treatment:control (T/C) ratio (tumor-volume ratio [%]) and tumor-growth inhibition (TGI; %) were calculated, and data are presented as mean ± SEM. The experiment was terminated when the mean tumor volume of the vehicle groups reached 1,500 mm3.
Plasma and tumor-tissue samples were collected for PK analysis. Fresh tumor tissue obtained from nude mice was collected and placed in cold 0.9% NaCl solution. Samples were homogenized under standardized conditions (tumor:buffer [v:v] 1:4). After homogenization, samples were centrifuged at 22,000 g for 5 minutes at 4°C. The supernatant was transferred into a clean microcentrifuge tube and kept in ice-cold conditions until assayed.
In vivo pharmacokinetics in animals
Compound 1 was dosed orally (60 mg/kg) with a formulation of 10% β-cyclodextrin in BALB/c nude (n=9 male animals/group). Subsequently, 200 μL whole blood was collected at 0.083, 1, and 6 hours for animals 1–3, 0.25, 2, and 8 hours for animals 4–6, and 0.5, 4, 24 hours for animals 7–9.
Compound 2 in different formulations was administered orally to male Sprague Dawley rats (four groups, three male animals/group). Formulations were: formulation 1, 5 mg/kg of compound 2 in 0.5% methylcellulose; formulation 2, 5 mg/kg of compound 2 in 0.5% methylcellulose +0.5% sodium dodecyl sulfate; formulation 3, 4 mg/kg of compound 2 (solid-dispersion technology); and formulation 4, 30% dimethylformamide +50% polyethylene glycol 400 +20% saline. A total of 200 μL whole blood was collected at 0.167, 0.5, 1, 2, 4, 6, 8, and 24 hours.
The PK parameters of compound 2 via dosing with compound 3 were studied in male Sprague Dawley rats (two groups, three male animals/group). Compound 3 was intravenously dosed at 2 mg/kg with a formulation of 30% dimethylformamide and 70% sterile water. Subsequently, 200 μL whole blood was collected at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours. Compound 3 was also orally dosed with the same formulation at 4 mg/kg, and blood-collection time points were 0.167, 0.5, 1, 2, 4, 6, 8, and 24 hours.
All the aforementioned blood samples were collected into K2-EDTA-coated (2.5%) tubes at the designated time points and prepared by centrifugation. Blood was centrifuged at 4,500 rpm for 5 minutes at 4°C, and plasma was subsequently harvested and stored at −20°C until further analysis. Finally, animals were killed. Compound concentrations in the plasma samples were determined by liquid chromatography (LC)–MS/MS. PK parameters were calculated by WinNonlin software with a noncompartmental model.
In vitro metabolic stability assay with multiple-species liver microsomes
In vitro metabolic stabilities of compound 1 in different species were tested by incubating the compound with liver microsomes of nude mice, Sprague Dawley rats, beagles, and humans. The incubated system was composed of PBS (100 mM), MgCl2 (20 mM), liver microsomes (20 mg protein/mL), compound 1 (1 μM), and β-NADPH (10 mM). Incubation of compound 1 and liver microsomes in the absence of NADPH served as a negative control. Incubation was terminated by acetonitrile at different incubation times (0, 5, 10, 20, 30, and 60 minutes). Subsequently, samples were centrifuged for 5 minutes at 12,000 rpm to precipitate the proteins. Supernatants were analyzed using optimized LC-MS/MS methods, and concentrations of compound 1 and compound 2 were monitored at the same time.
Procedure of LC-MS/MS
All biological samples were analyzed by LC-MS/MS using a Shimadzu high-performance liquid-chromatography system and MS (API 4000 equipped with an electrospray ionization probe in positive-ion mode). The column used was a Waters XBridge C18 (2.1×50 mm, 5 μm). The binary-gradient method consists of mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile) with a flow rate of 0.50 mL/min. The gradient used was B held at 95% for 1 minute, then decreased linearly to 10% in 4.5 minutes. After B had been held at 10% for another 0.5 minutes, it was brought back to 95% in 0.5 minutes, followed by a 1-minute re-equilibration. The total cycle duration was 7.5 minutes. The injection volume was 3 μL. MS with electrospray ionization was used to quantify compounds by multiple-reaction monitoring. Curtain, nebulizing, auxiliary, and collision gases were all nitrogen at settings of 30, 60, 65, and 6 psi, respectively. The LC-MS/MS turbo ion-spray interface was operated at a temperature of 500°C, and the ion-spray voltage was 5,000 V. The monitored transitions were m/z 451.1/411.2, 451.1/368.1, 561.2/441.2, and 470.4/443.1 for compounds 1, 2, 3, and NVP-BEZ235, respectively. The major compound-specific parameter was collision energy at 40, 52, 42, and 48 eV, and the declustering potentials were 100, 101, 97, and 100 V for compounds 1, 2, 3, and NVP-BEZ235, respectively.
Statistical analysis
All results are presented as mean ± SD. Differences between treated groups and the vehicle-control group were analyzed using one-way analysis of variance. The level of significance was established at P<0.05.
Results
Discovery of the major metabolite of compound 1
PK/PD study of compound 1
During preclinical development of the lead compound, ie, compound 1, the PK/PD study was conducted in a BALB/c nude mouse xenograft model with the PC3 cell line. Mice were dosed with compound 1 at 15, 30, and 60 mg/kg, together with NVP-BEZ235 at 30 mg/kg. Compared with the vehicle group, the TGI of compound 1 increased from 37.3% to 82.1% as doses ascended from 15 mg/kg to 60 mg/kg; a dose–effect relationship of compound 1 was clearly observed. In the same experiment, NVP-BEZ235 also showed a similar effect, as illustrated in Figure 2. At 30 mg/kg, the TGI of NVP-BEZ235 was 67.6% vs 59.7% for compound 1.
Combined with early research data, compound 1 and NVP-BEZ235 had comparable in vitro potency against PI3K and mTOR. However, with similar in vivo efficacy at 30 mg/kg, drug concentrations in plasma and tumor tissue of compound 1 and NVP-BEZ235 differed largely, as shown in Table 1. At 30 mg/kg, concentrations of compound 1 in plasma and tumor tissue were 111 ng·h/mL and 5.33±1.97 ng/g, respectively. Even at 60 mg/kg with an inhibition rate of 82.1%, concentrations of compound 1 in plasma and tumor tissue were 328 ng·h/mL and 15.3±4.29 ng/g, respectively. Concentrations of both doses were much lower than NVP-BEZ235 in plasma (4,779 ng·h/mL) and tumor tissue (310±61.8 ng/g). The discordance in exposures of the two compounds indicated the possibility of the contribution of another factor to efficacy besides compound 1.
Identification and quantification of the major metabolite
Compound 1 blood samples from the efficacy study in nude mice were reanalyzed by LC-MS/MS, and a major metabolite was subsequently identified. After enrichment and purification of the major metabolite from the plasma samples, the chemical structure was confirmed by nuclear magnetic resonance (Figure 1) and named compound 2.
To study fully the in vivo metabolite profile of compound 1, a separate PK study was conducted in BALB/c nude mice by oral dosing at 60 mg/kg. The concentration of compound 1 and its major metabolite (compound 2) in the plasma samples was analyzed and quantified using authentic compounds 1 and 2 as reference standards. The PK parameters of compounds 1 and 2 are listed in Table 2, and the PK profile is shown in Figure 3. As can be seen in Table 2, the area under the curve (AUC) of compound 2 was 3,404 ng·h/mL, which was approximately 50 times higher than compound 1. The maximum plasma concentration of compound 2 was 448 ng/mL, approximately six times higher than compound 1.
Characterization of compound 2
Enzyme and cellular inhibition assays
Since the in vivo pharmacodynamic (PD) effect could not be explained by exposure to compound 1 and since in vivo exposure to its major metabolite, compound 2, was much higher than the parent compound, enzyme activities against PI3Kα and mTOR and antiproliferation of different cell lines of compound 2 were tested. The results are presented in Table 3. The IC50 of compound 2 against PI3Kα was 7.8 nM, which was equivalent to compound 1 and lower than NVP-BEZ235. The IC50 of compound 2 against mTOR was 2.5 nM, which was slightly higher than NVP-BEZ235, but much lower than compound 1. Overall, the enzymatic potency of compound 2 against PI3Kα/mTOR was better than compound 1 and equivalent to NVP-BEZ235. Comparison of the antiproliferative effect on different carcinoma cell lines among three compounds showed compound 2 had strong inhibition on selected tumor-cell lines, with IC50 values equal or less than 100 nM, which was more potent than compound 1. However, the IC50 values of NVP-BEZ235 on these cell lines were lower than compound 2 (Table 3).
Ratio of compound 1 transformed to compound 2
Compound 1 was incubated with mouse, rat, dog, and human liver microsomes. At six time points, concentrations of compound 1 and compound 2 were analyzed. Results are summarized in Table 4 and data at 60 minutes plotted in Figure 4.
As incubation time increased, the concentration of compound 1 in mouse liver microsomes decreased, while the concentration of compound 2 increased. At 60 minutes, the concentration of compound 2 was nearly ten times higher than compound 1 (180.5 nM vs 18.95 nM). The in vitro metabolic profiles of compound 1 in mouse liver microsomes corresponded to those obtained from in vivo xenograft models. The data indicated that in the mouse liver microsomes, compound 1 metabolized rapidly to the dominant metabolite – compound 2.
However, in the rat, dog, and human liver microsomes, no compound 2 was detected up to 30 minutes. Furthermore, only 2.5 nM and 5 nM of compound 2 in the liver microsomes of rats and humans were detected, respectively, at the 60-minute time point. It was concluded that compound 1 was metabolized into compound 2 dominantly only in mice and not in rats, dogs, or humans.
Oral bioavailability of compound 2 in rats
Compound 2 was dosed orally in rats using four different formulations with doses of 5 mg/kg or 4 mg/kg. As shown in Table 5, with simple suspensions A and B, the bioavailability of compound 2 was very low: ~1%–2%. To improve the bioavailability of compound 2, amorphous solid-dispersion formulation and solvent-solubilized solution were developed and tested. Bioavailability increased to 8.45% and 14%, respectively, but was still insufficient to develop the oral dosage form later.
Prodrug strategy of compound 2
To improve further bioavailability, compound 3, the prodrug of compound 2, was synthesized and characterized.
Ratio of compound 3 transformed to compound 2
In an attempt to improve the solubility of compound 2, a phosphate ester prodrug (compound 3) was synthesized. The structure is shown in Figure 1. The PK study of compound 3 was performed in rats. Both the prodrug and the parent drug were monitored simultaneously. As shown in Table 6, after intravenous bolus dosing, plasma concentrations of compound 3 and compound 2 were 719±226 ng/mL and 2,383.3±255.4 ng/mL, respectively, at the first time point (0.083 hours), and the ratio of compound 2 to compound 3 was 3.3:1. At the second time point (0.25 hours), the concentration of compound 2 was 2,230±212.8 ng/mL, which was much higher than that of compound 3, and the ratio of compound 2 to compound 3 was 46.9:1. At 0.5 hours, compound 3 totally disappeared, while the concentration of compound 2 decreased slowly with time. It was concluded that compound 3 can be converted to compound 2 quickly and completely in vivo. After oral administration of compound 3 in rats, however, it was undetectable, and only compound 2 was detected in rat plasma (data not shown).
Rat PK study of compound 2 via dosing compound 3
The PK parameters of compound 2 after dosing compound 3 in rats are summarized in Table 7. The clearance of compound 2 in rats was 0.23±0.03 L/h/kg, the AUC0–t of intravenous and oral routes was 8,687±1,209 ng·h/mL and 1,695±1,033 ng·h/mL, respectively, and the bioavailability of compound 2 in rats was 12.2%±9.4%, which was the same as compound 2 with formulation 4 (Table 5).
Discussion
During the process of profiling a clinical lead candidate, it was found that the drug concentration of compound 1 was not correlated with its PD effect in the nude-mouse xenograft model. Compound 1 had equivalent in vitro activity on PI3Kα and mTOR to NVP-BEZ235. However, at a similar tumor-inhibition rate in the nude-mouse xenograft model, drug exposure of compound 1 was approximately 2.3% of NVP-BEZ235. The question arises as to why the in vivo efficacy in the xenograft model did not align with the exposure to compound 1. We speculated that there was some “substance” contributing the PD effect to compound 1, which was not detected at the time.
In order to answer this question, we reanalyzed plasma samples of the nude-mouse xenograft model, and a major metabolite (compound 2) was identified. In subsequent studies, compound 2 was characterized, synthesized, and quantified in the plasma samples of nude mice. These results were reconfirmed by another independent PK study in mice. After dosing compound 1, the AUC of compound 2 was 50 times higher than that of its parent, compound 1, and the PK/PD results revealed a good correlation with compound 2 instead of compound 1.
In the subsequent experiment, the enzyme and cellular inhibitory activities of compound 2 were tested. Compound 2 had better in vitro activity than compound 1. Combined with the fact that the exposure of compound 2 was much higher than compound 1 in nude mice, it can be concluded that the in vivo antitumor activities observed in the mouse xenograft model were due to compound 2 rather than compound 1.
One thing to note is that compound 1 was metabolized into compound 2 dominantly only in mice, but not in rats, dogs, or humans. Since compound 2 is responsible for the antitumor efficacy in the mouse xenograft model, but compound 1 is not metabolized into compound 2 in humans, the question arises: How can the efficacy observed in the xenograft models be translated into clinical practice? Would compound 1 have enough clinical response if it were not metabolized into compound 2 in humans?
In consideration of the metabolic difference of compound 1 between mice and other species and the unavailability of a rat xenograft model, further development of compound 1 was suspended. Moreover, due to the highly encouraging in vitro and in vivo data of compound 2, we decided to profile compound 2, the metabolite of compound 1, as a clinical lead candidate.
Solid oral dosage is a preferred route of administration for medications, and thus the oral bioavailability of compound 2 is a key parameter in its drug-like properties. Rat PK studies of compound 2 were conducted, and indicated that its bioavailability was much lower than 20% in rats and that it was not a good candidate to be developed into an oral drug.
For oral drugs, bioavailability is impacted by absorption and metabolism. As shown in Table 7, the clearance of compound 2 was 0.23±0.03 L/h/kg in rats, and the hepatic extraction ratio was only 5.4% based on the hepatic blood flow of 70 mL/min/kg in rats. Such a low hepatic extraction ratio value indicated that the poor bioavailability of compound 2 was not due to fast metabolism.
Solubility and permeability are the two major factors that influence drug absorption. Therefore, the physicochemical properties of compound 2 were tested. The solubility of compound 2 in pH 7 buffer was less than 1 μg/mL, indicating compound 2 would not dissolve well in the gastrointestinal tract, or would precipitate out with increasing pH values when moving down the gastrointestinal tract. Therefore, the solubility may be the reason for the poor bioavailability. A permeability study across the Caco-2 monolayer showed that compound 2 had low permeability (PappAB ≤2.5×10−6 cm/s) and high efflux ratio of 22.7. Compound 2 can be classified as a Biopharmaceutic Classification System-Class Four compound, and poor solubility and permeability would be the reasons for the low bioavailability.
Prodrug development is a common strategy to improve the solubility of an active ingredient.19,20 Compound 3, a phosphate ester of compound 2, was synthesized. The solubility of the prodrug in pH 7 buffer was improved 5,000-fold compared to compound 2. Furthermore, PK studies through intravenous and oral routes indicated that compound 3 converted into compound 2 immediately and completely, which reduced the complexity in developing this prodrug.
However, the PK study of compound 3 showed that systemic exposure of compound 2 was not improved and the calculated bioavailability of compound 2 in rats was about 12.2%±9.4%. Therefore, oral administration of the prodrug compound 3 may not be a valid option. Phosphate ester is hydrolyzed by alkaline phosphatases,21,22 which are present in all tissues throughout the body and particularly enriched in the liver, bile duct, intestinal mucosa, kidney, bone, and placenta. A phosphate ester prodrug introduced intravenously is metabolized mainly by the enzymes in the blood and liver. When orally dosed, compound 3 was metabolized to compound 2 by the alkaline phosphatases in intestinal mucosa first. Due to poor permeability and high efflux of compound 2, this explains why the prodrug did not improve the exposure of compound 2 by oral administration. Based on these results, further development of compound 3 will focus on the parenteral pathway.
In summary, the process of discovering and developing a dual PI3K–mTOR inhibitor was not straightforward. Compound 2, a metabolite of compound 1, was discovered firstly in plasma samples due to a lack of PK/PD correlation of compound 1 in the mouse xenograft model. Although compound 1 was identified as the clinical lead candidate, its candidacy was suspended because of greatly different metabolic profiles across species. On the basis of better inhibitory activities in enzymatic and cellular assays, compound 2, the active metabolite of compound 1, was explored further for possibilities of developing it as a clinical lead candidate. However, the poor solubility and permeability of compound 2 made this difficult to pursue further. Fortunately, the phosphate ester prodrug, compound 3, resolved the solubility issue. Although the low bioavailability of compound 3 is problematic for its oral dosing, it has adequate solubility for intravenous formulation. Therefore, the development of a parenteral dosage form via the prodrug strategy became attractive, especially for immuno-oncology therapy.23,24 The combination of PD-1/PD-L1 antibody with traditional small molecules has been confirmed to be beneficial to patients.25 It is thus desirable to develop an intravenous medication of a novel dual PI3K–mTOR inhibitor for cancer therapy. As such, further work will be published in the near future.
Acknowledgments
The authors sincerely acknowledge the previous work from Dr Jianjun Xue, Dr Peng Peng, Dr Yan Zhang, Dr Chutian Shu, and Dr Frank Wu.
Disclosure
Yan Zhou, Genyan Zhang, Feng Wang, Jin Wang, Yanwei Ding, Xinyu Li, Chongtie Shi, Jiakui Li and Chengkon Shih all work for Xuanzhu Pharma, Jinan, China. The authors report no other conflicts of interest in this work.
References
Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18(3):349–351. | ||
Gomez-Pinillos A, Ferrari AC. mTOR signaling pathway and mTOR inhibitors in cancer therapy. Hematol Oncol Clin North Am. 2012;26(3):483–505. | ||
Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. | ||
van der Broek R, Mohan S, Eytan DF, Chen Z, van Waes C. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross-talk, and therapies. Oral Dis. 2015;21(7):815–825. | ||
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinase as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. | ||
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | ||
Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. | ||
Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–523. | ||
Janku F, Wheler JJ, Westin SN, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30(8):777–782. | ||
Netland IA, Førde HE, Sleire L, et al. Dactolisib (NVP-BEZ235) toxicity in murine brain tumour models. BMC Cancer. 2016;16:657. | ||
Wen PY, Omuro A, Ahluwalia MS, et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015;17(9):1275–1283. | ||
Dolly SO, Wagner AJ, Bendell JC, et al. Phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2016;22(12):2874–2884. | ||
Chia S, Gandhi S, Joy AA, et al. Novel agents and associated toxicities of inhibitors of the PI3K/Akt/mTOR pathway for the treatment of breast cancer. Curr Oncol. 2015;22(1):33–48. | ||
Wong MH, Xue A, Baxter RC, Pavlakis N, Smith RC. Upstream and downstream co-inhibition of mitogen-activated protein kinase and PI3K/Akt/mTOR pathways in pancreatic ductal adenocarcinoma. Neoplasia. 2016;18(7):425–435. | ||
Simioni C, Ultimo S, Martelli AM, et al. Synergistic effects of selective inhibitors targeting the PI3K/AKT/mTOR pathway or NUP214-ABL1 fusion protein in human acute lymphoblastic leukemia. Oncotarget. 2016;7(48):79842–79853. | ||
Wu F, Shu CT, inventors; Xuanzhu Pharma, assignee. PI3K and/or mTOR inhibitor. World Intellectual Property Organization patent WO2014040373. 2013 Sep 12. | ||
Wu F, Zhang Y, inventors; Xuanzhu Pharma, assignee. Three-ring PI3K and/or mTOR inhibitor. World Intellectual Property Organization patent WO2013071698. 2012 Nov 16. | ||
Wu F, Xue JJ, Li L, inventors; Xuanzhu Pharma, assignee. PI3K and/or mTOR inhibitor prodrug. World Intellectual Property Organization patent WO2014154026. 2014 Mar 28. | ||
Rautio J, Kumpulainen H, Heimbach T, et al. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7(3):255–270. | ||
Browne TR, Kugler AR, Eldon MA. Pharmacology and pharmacokinetics of fosphenytoin. Neurology. 1996;46(6 Suppl 1):S3–S7. | ||
Stella VJ, Nti-Addae KW. Prodrug strategies to overcome poor water solubility. Adv Drug Deliv Rev. 2007;59(7):677–694. | ||
Varia SA, Schuller S, Sloan KB, Stella VJ. Phenytoin prodrugs III: water-soluble prodrugs for oral and/or parenteral use. J Pharm Sci. 1984;73(8):1068–1073. | ||
Ott PA, Adams S. Small-molecule protein kinase inhibitors and their effects on the immune system: implications for cancer treatment. Immunotherapy. 2011;3(2):213–227. | ||
Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov. 2015;14(9):603–622. | ||
Ribas A, Butler M, Lutzky J, et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J Clin Oncol. 2015;33(15 Suppl 7):S3003. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.