")
Back to Journals » Infection and Drug Resistance » Volume 16
Detection of Pathogens and Antimicrobial Resistance Genes in Ventilator-Associated Pneumonia by Metagenomic Next-Generation Sequencing Approach
Authors Chen T , Zhang L, Huang W , Zong H, Li Q, Zheng Y, Lv Q, Kong D, Ren Y, Jiang Y, Li Y, Liu P
Received 22 November 2022
Accepted for publication 26 January 2023
Published 15 February 2023 Volume 2023:16 Pages 923—936
DOI https://doi.org/10.2147/IDR.S397755
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Ting Chen,1,2,* Lei Zhang,3,* Wenhua Huang,3,* Huijun Zong,1,2 Qian Li,3 Yuling Zheng,3 Qingyu Lv,3 Decong Kong,3 Yuhao Ren,3 Yongqiang Jiang,3 Yan Li,1,2 Peng Liu3
1The PLA 307 Clinical College of Anhui Medical University, The Fifth Clinical Medical College of Anhui Medical University, Hefei, People’s Republic of China; 2Department of Critical Care Medicine, The Fifth Medical Center, Chinese PLA General Hospital, Beijing, 100071, People’s Republic of China; 3State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology, Academy of Military Medical Sciences, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan Li, The PLA 307 Clinical College of Anhui Medical University, The Fifth Clinical Medical College of Anhui Medical University, Hefei, 230032, People’s Republic of China, Email [email protected] Peng Liu, Beijing Institute of Microbiology and Epidemiology, Dongdajie Road 20, Beijing, 100071, People’s Republic of China, Tel +86-010-66948487, Email [email protected]
Background: The early identification of pathogens and their antibiotic resistance are essential for the management and treatment of patients affected by ventilator-associated pneumonia (VAP). However, microbiological culture may be time-consuming and has a limited culturability of many potential pathogens. In this study, we developed a rapid nanopore-based metagenomic next-generation sequencing (mNGS) diagnostic assay for detection of VAP pathogens and antimicrobial resistance genes (ARGs).
Patients and Methods: Endotracheal aspirate (ETA) samples from 63 patients with suspected VAP were collected between November 2021 and July 2022. Receiver operating characteristic (ROC) curves were established to compare the pathogen identification performance of the target pathogen reads, reads percent of microbes (RPM) and relative abundance (RA). The evaluation of the accuracy of mNGS was performed comparing with the gold standard and the composite standard, respectively. Then, the ARGs were analyzed by mNGS.
Results: ROC curves showed that RA has the highest diagnostic value and the corresponding threshold was 9.93%. The sensitivity and specificity of mNGS test were 91.3% and 78.3%, respectively, based on the gold standard, while the sensitivity and specificity of mNGS test were 97.4% and 100%, respectively, based on the composite standard. A total of 13 patients were virus-positive based on mNGS results, while the coinfection rate increased from 27% to 46% compared to the rate obtained based on clinical findings. The mNGS test also performed well at predicting antimicrobial resistance phenotypes. Patients with a late-onset VAP had a significantly greater proportion of ARGs in their respiratory microbiome compared to those with early-onset VAP (P = 0.041). Moreover, the median turnaround time of mNGS was 4.43 h, while routine culture was 72.00 h.
Conclusion: In this study, we developed a workflow that can accurately detect VAP pathogens and enable prediction of antimicrobial resistance phenotypes within 5 h of sample receipt by mNGS.
Keywords: mechanical ventilation, endotracheal aspirate, pathogen diagnosis, antibiotic resistance, NGS
Introduction
Patients admitted to the intensive care unit (ICU) often need to be treated with mechanical ventilation to overcome the respiratory disorders and failure, however, this type of treatment can cause several medical complications, such as displacement of tracheal intubation, septic shock, and ventilator-associated pneumonia (VAP).1,2 Specifically, VAP represents a pneumonia that occurs later than 48 h post-mechanical ventilation in patients with tracheal intubation or tracheotomy, or within 48 h after extubation.3 VAP is the most common and fatal nosocomial infection mostly diagnosed within the critical care settings.4 Indeed, the incidence of VAP in ICU patients undergoing mechanical ventilation ranged from 9% to 69%, while the average case fatality rate ranged between 20% and 50%, with a peak up to 38.9–60.0%, in case of multi-drug resistant bacteria.3,5 Therefore, extremely necessary is the correct diagnosis of the pathogens involved in the VAP patients in order to test their antibiotic susceptibility and identifying the correct treatment, to significantly improve the survival rate, and reduce drug-related side effects and medical costs.6
VAP is diagnosed by the simultaneous presence of the specific criteria: clinical signs, positive microbiology cultures from lower respiratory tract specimens, and new or progressive radiographic infiltrates.7 However, the microbiological culture may sometimes does not lead to the pathogen detection, in case the pathogens are present in low concentration or in case of fastidious bacteria.8 This long time gap, which is between the initial clinical diagnosis and the identification of the correct antibiotic to use, often leads to a variety of empirical antibiotic treatments.9 Molecular detection methods are culture-independent, and therefore they can allow to obtain a correct diagnosis in a timely manner. In this regard, designated pathogens and antimicrobial resistance genes (ARGs), such as mecA, the gene responsible for methicillin resistance in Staphylococcus aureus can be quickly detected by polymerase chain reaction (PCR), but it is not entirely comprehensive.10 In addition, the metagenomic next-generation sequencing (mNGS)-based approach can be considered as an ideal method for detection of all bacteria, fungi and viruses present in a specimen, due to pan-microbial coverage.11 One of the most common NGS platforms, the Illumina ones, show a high sensitivity, but they require a complex preparation process and a running time of more than 24 hours, with a result obtained only at the end of the sequencing run.12 Moreover, Illumina platforms are short reads-based sequencing methods, the taxonomical classification at the genus and species level is less efficient, which may lead to misdiagnosis.13
In contrast, the real-time analysis and ultra-long reads (>100kb)-based approaches, such as the nanopore sequencing is more suitable for clinical laboratories.14,15 In this regard, the feasibility of the nanopore sequencing application for clinical diagnosis has been demonstrated in clinical cases of meningitis,16 peritonitis,12 urinary tract infection,17 lower respiratory infection,18 and sepsis.19 However, due to several combinations of the pathogens isolated in different clinical cases, suitable mNGS approaches for comprehensive pathogens detection and drug resistance prediction of VAP in ICU are still limited.20,21 In addition, the diagnostic criteria of mNGS are not uniform, and it is not clear which criteria is the most accurate for the diagnosis of VAP pathogens in ICU.18,22,23 In this study, a nanopore-based mNGS workflow for a comprehensive detection of VAP pathogens and ARGs to determine the applicability of this method for a correct clinical diagnosis in terms of sensitivity, specificity, and prompt diagnostic results.
Materials and Methods
Participants and Study Design
Between November 2021 and July 2022, endotracheal aspirate (ETA) samples were collected from 74 patients with a clinical suspect of VAP admitted to the ICU of the Fifth Medical Center of Chinese PLA General Hospital, Beijing, China. Based on clinical criteria, a clinical suspect of VAP was defined as follows: (1) duration of the mechanical ventilation treatment performed with tracheal intubation or tracheotomy >48 h, (2) chest X-ray or CT (computed tomography) demonstrating new or progressive infiltration, consolidation or ground glass shadow, associated with at least one of following four clinical criteria: (1) fever >38°C; (2) cough, expectoration and/or dyspnea; (3) purulent airway secretion; (4) peripheral white blood count >10 × 109/L or <4 × 109/L. (American Thoracic Society, 2005)24 After that, only the samples that meet the following criteria were included: (i) consistency with the clinical standards of suspected VAP; (ii) presence of comprehensive information regarding anamnesis, signalment, and clinical data; (iii) age ≥18 years old. On the other side, the following exclusion criteria were followed: (i) presence of other intercurrent infections; (ii) pregnancy and lactation. Based on the inclusion and exclusion criteria, 63 patients were finally enrolled in this study (Figure 1). The clinical data of patients were collected from the electronic medical records, avoiding the record of their identity. Information regarding tests for clinical respiratory viruses was also recorded for each patient. ETA samples were collected within 12 h after the suspect of diagnosis. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the Fifth Medical Center of Chinese PLA General Hospital [ky-2019-1-4], and all samples were obtained following the patients’ consents.
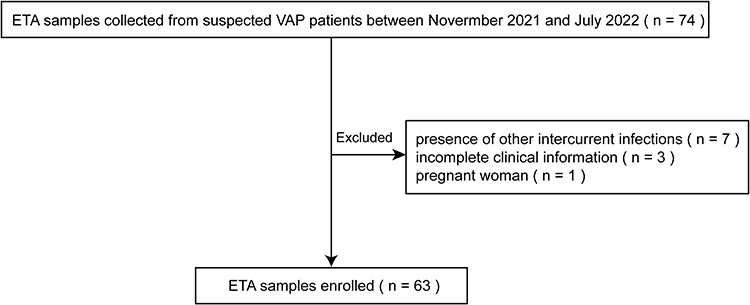 |
Figure 1 Schematic of samples inclusion and exclusion. |
Sampling, Processing, and DNA Extraction
Each patient’s sample was aliquoted and placed in two sterile tubes (each 1–2mL). The first one was sent to the clinical laboratory for microbiological culture and antimicrobial susceptibility testing (AST), and the second one was sequenced by mNGS and stored at 4°C for up to 72 h before processing. The bacterial culture was performed streaking 10 µL of samples onto blood agar and MacConkey agar plates (bioMérieux, Marcy-l’ Etoile, France), and then incubated in aerobic condition at 37°C for 48 h. The fungal culture was performed incubating the samples in aerobic condition at 27°C for 7 days using Sabouraud agar plates (Moltox, USA). The microorganisms were identified using the VITEK 2 automated system (bioMérieux, Marcy-l’ Etoile, France). Accordingly, with the standard clinical practices, culture-negative samples were reported as “normal respiratory flora” or as “no organismal growth” if no microorganisms were isolated. AST was performed following the guidelines of Clinical and Laboratory Standards Institute (CLSI).25
The mNGS testing was performed as follows (Figure 2): 300 µL sample was mixed with sterile Phosphate Buffer Saline (PBS) in a 1:3 ratio using 1.5 mL Eppendorf tubes (Eppendorf) and centrifuged for 5 min at 20,000×g for enrichment. After discarding 1 mL supernatant, 5 µL of lytic enzyme solution (Qiagen Inc., Hilden, Germany) and 10 µL of MetaPolyzyme (Sigma Aldrich, USA; reconstituted in 750 µL PBS) were added to the samples and gently mixed. After that, the samples were incubated for 1 h at 37°C in the shaker to lyse microbial cells, followed by DNA extraction using an IndiSpin Pathogen Kit (Indical Bioscience). Sterile deionized water was extracted as well and it was used as negative control. DNA concentrations were assessed using a Qubit 4.0 fluorometer with the dsDNA HS Assay kit (Thermo Fisher Scientific).
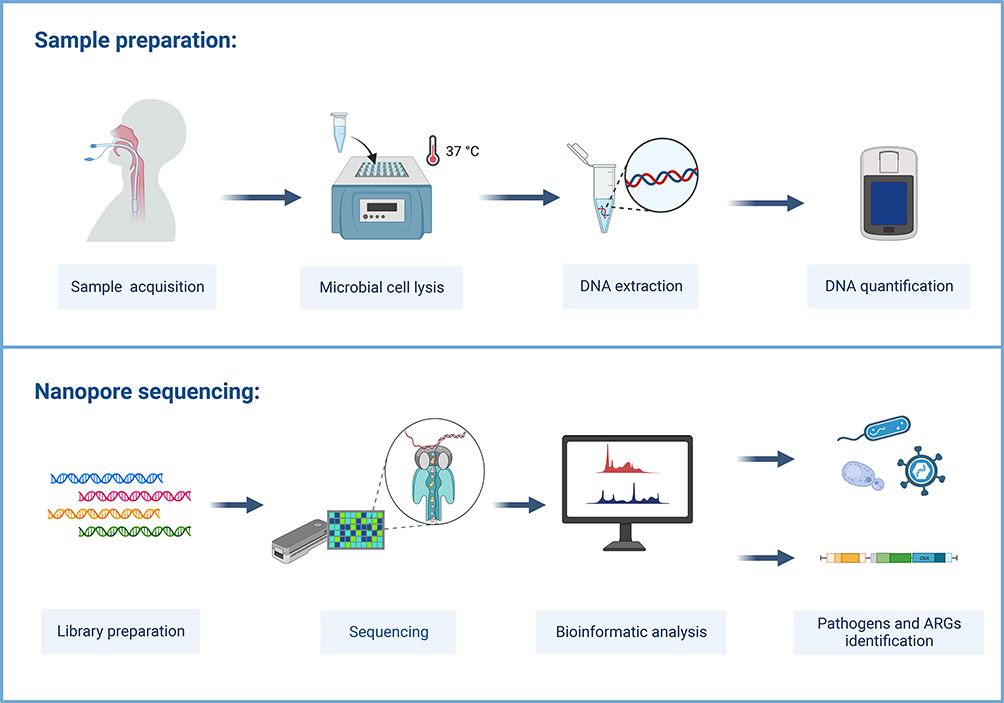 |
Figure 2 Schematic representation of the MinION-based mNGS workflow. |
Library Preparation and Sequencing
All the samples were sequenced using the MinION platform (Oxford Nanopore Technology (ONT)). All the samples, including the ones showing a low DNA concentration, were included in the workflow, to obtain an accurate representation of all the microorganisms that can be detected in lower respiratory tract specimens collected from clinical cases, using a metagenomic approach. According to the manufacturer’s instruction, library preparation was performed using the PCR Barcoding Kit (SQK-PBK004, ONT), using 4-min of DNA extension and 15 cycles within the PCR amplification step. A total of 75 µL of library DNA was loaded into the R9.4.1 flow cell (FLO-MIN106) on MinION device according to the manufacturer’s instruction. ONT MinKNOW GUI software (version 4.2.8) was used to collect raw sequencing data. The sequencing run was stopped when approximately 50k of total read counts for each sample was accumulated.
Bioinformatic Analysis
The raw sequencing data were processed using an automatic bioinformatics pipeline composed of a set of fixed external software (ont-Guppy, bwa, SAMtools, BLASTn). The processing step consisted of (1) trimming of adapters using ont-Guppy; (2) subtraction of human host sequences once mapped to the human reference genome (GRCh38, https://www.ncbi.nlm.nih.gov/data-hub/assembly/GCF_000001405.39/) using Burrows-Wheeler alignment with BWA-MEM algorithm; (3) the SAM files as output were indexed and sorted with SAMtools (version 1.7) to generate nonhuman reads; (4) all the nonhuman reads were classified by simultaneous alignment to RefSeq microbial genome databases (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq), which included viruses, bacteria, fungi, and parasites sequences using BLASTn (version 2.10.1); (5) the final species classification results were finally outputted as csv files following the process by two custom Python scripts and Linux commands; (6) the ARGS were detected using ABRicate (version 0.8, https://github.com/tseemann/abricate) based on Comprehensive Antibiotic Research Database (CARD), using the input of non-human reads and with the following parameters as follows: “-minid 85 -mincov 85 -csv”. The automatic bioinformatics pipeline is available at https://github.com/gitzl222/APDNS/.
Positive Standards of mNGS Testing
To determine the best threshold to be used for the detection of VAP pathogens, excluding sample or environmental contaminant microorganisms, threshold standards were established. Specifically, for the detection of bacterial and fungal pathogens, we established receiver operating characteristic (ROC) curves following the microbiological culture results in order to compare three main indicators in mNGS, including the target pathogen reads (reads), reads percent of microbes (RPM, defined as the percentage of target pathogen reads in the remaining total reads following the barcode trimming)26 and relative abundance (RA, defined as the percentage of target pathogen reads in the total microbe reads). Later, the indicator with the highest diagnostic value was selected and the threshold standard was determined according to the best Youden index. Positive standards of bacterial and fungal pathogens were as the ones that exceed the threshold standard, while the microbes above the threshold, but not considered pathogens, were listed in Table S1. As for detection of viruses, positive standard was defined when 1 or more reads were detected, while the coinfection was defined when more than one pathogen was detected.
Composite Standard for Pathogen Diagnosis
The composite standard for pathogen diagnosis was defined as a combination of clinical tests (culture, staining, immunology-related tests), confirmatory qPCR test and clinical adjudication by doctors. When the mNGS results were inconsistent with culture results, these pathogens were further validated by qPCR test. All the primers and probes for qPCR test and detailed steps were reported in Table S2.
Accuracy Evaluation of Pathogen Detection by MinION-Based mNGS
To perform a statistical analysis on the diagnostic performance, the mNGS results were compared using two criteria: (1) the gold standard of microbiological culture, and (2) the composite standard. A specific scoring algorithm was employed for detection of bacterial and fungal pathogens. For the gold standard culture method, in case of consistency between culture results and mNGS results, the samples were identified as true positive/true negative (TP/TN); otherwise, they were identified as false positive/false negative (FP/FN). For the composite standard, the results of inconsistency between sequencing and culture were further verified by qPCR test. In case of the mNGS results were consistent with the verification results and clinical adjudication, they were identified as TP/TN; otherwise, they were identified as FP/FN. In all two standards, a sample scored a maximum of 1 point, and when multiple pathogens were determined to be positive, the score was divided into several equal points according to the number of pathogens.
Statistical Analysis
The data were analyzed using SPSS 25.0 software. The quantitative data with a normal distribution were expressed as mean ± standard deviation (SD) values using t-test, whereas quantitative data with a non-normal distribution were expressed as median [interquartile range (IQR)] using Mann–Whitney U-test. Categorical data were expressed as numbers (percentage) using chi-squared test. A P-value <0.05 was considered significant. The ROC curves were drawn by SPSS 25.0 software. 2×2 contingency tables were used to calculate sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV).
Results
Patient Characteristics and Microorganisms Isolated
In this study, 63 patients were divided into two groups, a microbiological culture-positive group (n = 40) and a microbiological culture-negative group (n = 23). Culture-positive and negative patients had similar distribution of gender, comorbidities and clinical laboratory results, while the age, length of the mechanical ventilation treatment before sampling and ICU mortality were significantly higher in the first group than in the negative one (P ≤ 0.01) (Table 1). A total of 57 pathogens were isolated, including 53 bacteria and 4 fungi. Fifty-three strains of bacteria were 17 Acinetobacter baumannii (32.1%), 14 Pseudomonas aeruginosa (26.4%), 8 Klebsiella pneumoniae (15.1%), 2 Stenotrophomonas maltophilia (3.8%), 2 Staphylococcus aureus (3.8%), 2 Corynebacterium striatum (3.8%), 2 Enterococcus faecium (3.8%), 2 Proteus mirabilis (3.8%), 1 Burkholderia contaminans (1.9%), 1 Burkholderia gladioli (1.9%), 1 Staphylococcus haemolyticus (1.9%), and 1 Morganella morganii (1.9%), while 4 strains of fungi were 3 Candida albicans (75%), and 1 Aspergillus fumigatus (25%). The AST analysis was performed for 51 culture-confirmed bacterial pathogens (except 2 Corynebacterium striatum) isolated from 37 patients. To this regard, we found that 42 (82.4%) were resistant to penicillin, 39 (76.5%) to cephalosporin, 38 (74.5%) to fluoroquinolone, 33 (64.7%) to aminoglycoside, 32 (62.7%) to carbapenem, 26 (51.0%) to sulfonamide, 22 (43.1%) to tetracycline, 9 (17.6%) to monobactam, 8 (15.7%) to cephamycin, 4 (7.8%) to macrolide, 2 (3.9%) to lincosamide, and 1 (2.0%) to polypeptide. Detailed results of AST were shown in Table S3.
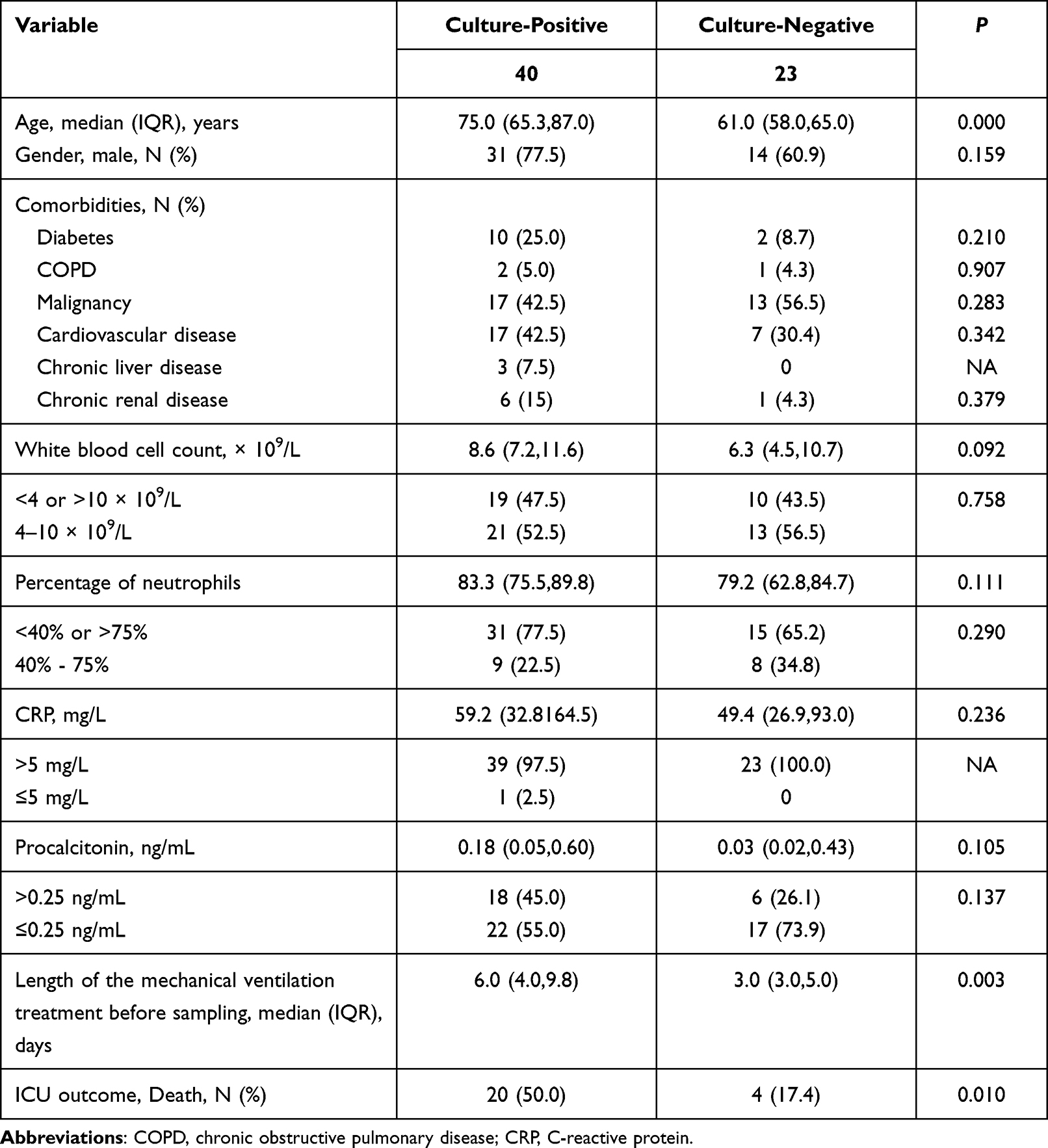 |
Table 1 The Clinical Characteristics of Patients in Microbiological Culture-Positive and Culture-Negative Group |
Diagnostic Threshold Establishment and Evaluation of the Accuracy for MinION-Based mNGS
ROC curves of the reads, RPM and RA were established using the culture results as the gold standard. As shown in Figure 3A, the area under the curve (AUC) of the RA represented the maximum (AUC = 0.847), which has the highest diagnostic value, while the RPM (AUC = 0.740) was the second, and the AUC of the reads was the minimum (AUC = 0.686) with the lowest diagnostic value obtained. Therefore, the RA was selected as the best indicator to detect bacterial and fungal pathogens in VAP cases by mNGS. Moreover, the corresponding threshold value was 9.93% according to the best Youden index. A detailed description of the bacteria and fungi detected was shown in Table S4.
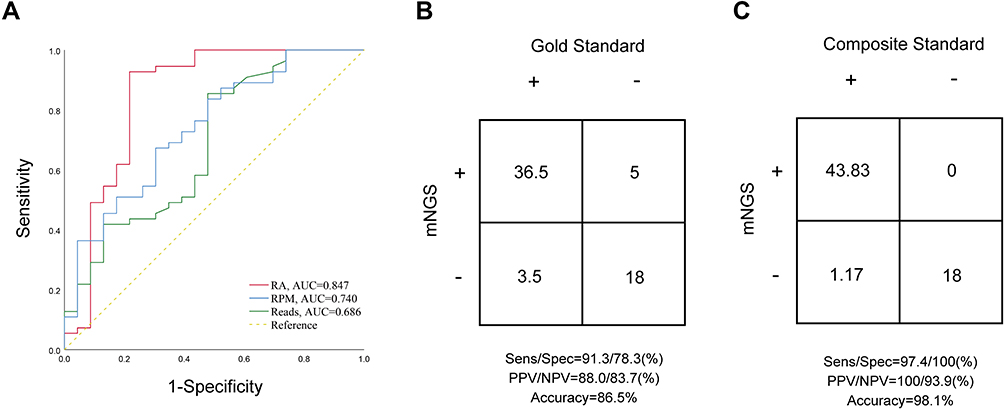 |
Figure 3 (A) Evaluation of the performance of three indicators (reads, RPM and RA) to detect VAP pathogens using ROC curves. (B and C) Accuracy evaluation by 2×2 contingency tables based on the gold standard culture test and composite standard, respectively. |
Based on the microbiological culture results, the 40 culture-positive samples were also found positive for 97.5% of times (39/40) mNGS, with a full consistency of the pathogen detected in the 87.2% of times (34/39), while partially consistent results were obtained in the remaining 5 samples, indicating at least 1 pathogen in the context of polymicrobial cultures was found. And only 1 sample (2.5%) showed inconsistent results. Finally, mNGS results scored 36.5 out of 40 and showed a sensitivity of 91.3% and a PPV of 88.0% (Figure 3B). In 23 culture-negative samples, 18 (78.3%) mNGS results were consistent with culture results while in 5 samples, positive pathogens were detected, resulting in a specificity of 78.3% and a NPV of 83.7% (Figure 3B). The false-positive and false-negative results based on the gold standard were shown in Table S5.
Based on the composite standard, the mNGS results were confirmed for the samples that show inconsistency in the culture results. In all culture-positive samples, only 6 pathogens detected by culture were lost in mNGS. Among them, 3 pathogens also could not be detected by qPCR tests, while 3 pathogens did not reach the thresholds of the mNGS. For the 5 FP samples based on gold standard, the mNGS results were further validated by qPCR test and clinical adjudication, increasing the scores by 5.0. Therefore, the sensitivity and specificity were 97.4% and 100%, respectively, and the PPV and NPV were 100% and 93.9%, respectively (Figure 3C). The false-positive and false-negative results based on the composite standard were shown in Table S5.
Detection of DNA Viruses by MinION-Based mNGS
Viral pathogens were also detected in our samples by mNGS. A total of 11 out of 40 culture positive samples were identified as virus positive by mNGS. The virus identified included 4 Epstein-Barr virus (EBV) and 7 Herpes simplex virus 1 (HSV-1). All the mNGS results were further confirmed by qPCR test. In addition, 2 virus positive samples were found within the 23 culture negative samples, specifically one sample (N11) was HSV-1 positive, while the second (N18) demonstrated to be coinfected with EBV and HSV-1. Following the qPCR validation, sample N18 passed the verification, but the CT value of sample N11 did not reach the threshold of qPCR test, likely due to low viral concentration (Table S6).
Detection of Coinfections by MinION-Based mNGS
In a total of 29 out of 63 patients (46.0%), coinfection was detected using mNGS, increasing the detection rate of coinfection by 19% (12/63) compared to clinical laboratory findings. It is worth noting that 20.6%, 3.2%, 22.2% patients were diagnosed as bacterial-viral, bacterial-fungal, and bacterial-bacterial coinfections, respectively, showing an increase in the coinfection detection rate, especially for bacterial-viral coinfection (Figure 4).
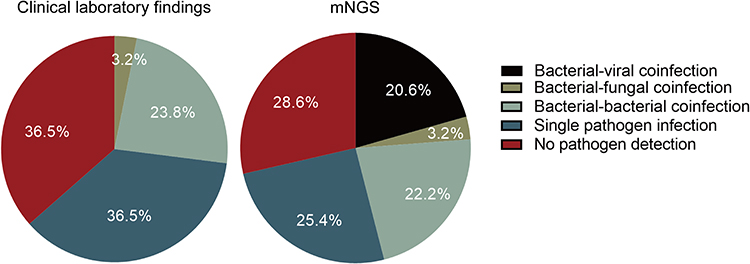 |
Figure 4 Comparison of the pathogen detection rates between clinical laboratory findings and mNGS results. |
Detection of ARGs by MinION-Based mNGS
To evaluate the ability of mNGS in prediction of the antibiotic resistance, the ARGs of bacteria were investigated in all the 45 mNGS-positive samples. A total of 52 ARGs were detected in 28 samples, while 17 samples showing no mNGS reads that mapped to known ARGs. Most of ARGs were related to multi-drug resistance (48.1%, eg, abe, acr), aminoglycoside (11.5%, eg, AAC, APH), and fluoroquinolone (9.6%, eg, AbaQ, CrpP). The most prevalent (39.3%,11/28) ARG was OXA, which is a β-lactamase gene. The ARGs detected in each sample and the corresponding type of antimicrobial agents are shown in Figure 5.
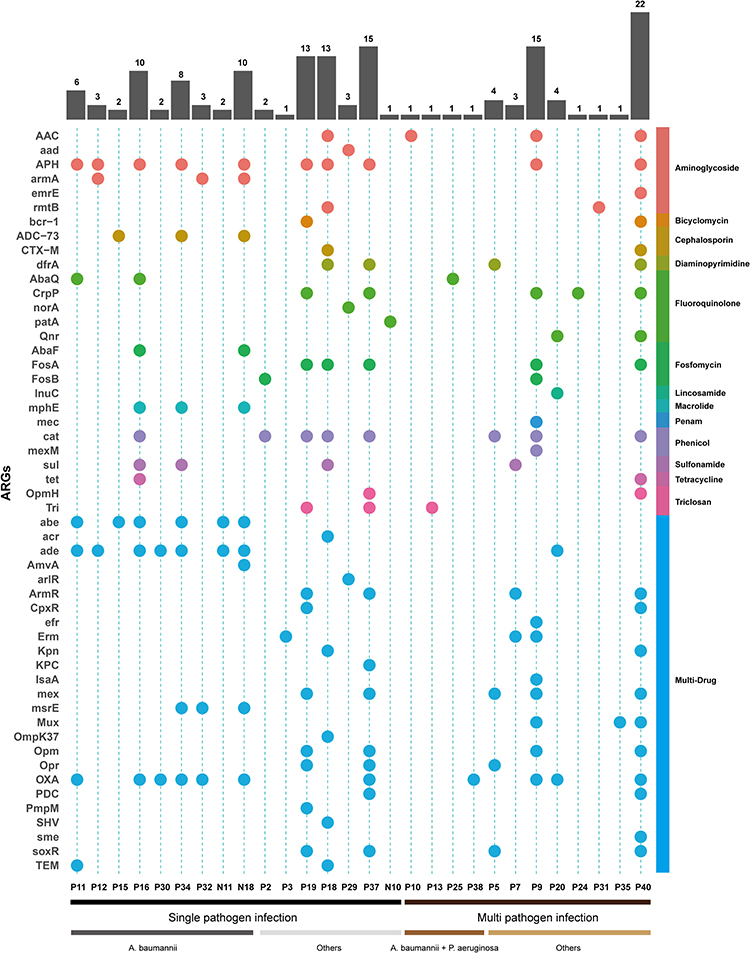 |
Figure 5 ARGs profile in the mNGS-positive samples. The heatmap strip at the right with different colors corresponds to different antimicrobial class. The heatmap strip at the bottom represents different pathogen species. The bar chart indicates the number of ARGs per sample. |
We also investigated the consistency between the results of ARGs and clinical AST (excluded 3 culture-negative samples without AST results: N10, N11, N18). As shown in Table 2, there was a high consistency between ARGs detected from gram-negative pathogens and aminoglycoside and β-lactam phenotypes. It was observed that resistance to aminoglycoside antibiotics was shown in 11 of 13 samples in which aminoglycoside ARGs were detected, with APH being the most common gene, followed by AAC. The enzymes encoded by ade, Opr, ArmR and Kpn belong to multidrug efflux complex, which can be responsible for the resistance to carbapenem, cephalosporin and penicillin, while class D β-lactamases encoded by OXA can hydrolyze cephalosporin and penicillin. The detected ARGs were consistent with the AST results to β-lactam antibiotics in 13 of all 15 samples. Most of the carbapenem resistance was due to the presence of ade gene in A.baumannii, while Opr or ArmR in P. aeruginosa. In general, the two methods produced fully concordant results in 7 (28.0%) samples and partially consistent results in the remaining 18 (72.0%) samples, with the antibiotic sulfonamide as the most frequently missed in ARGs screening of the mNGS data.
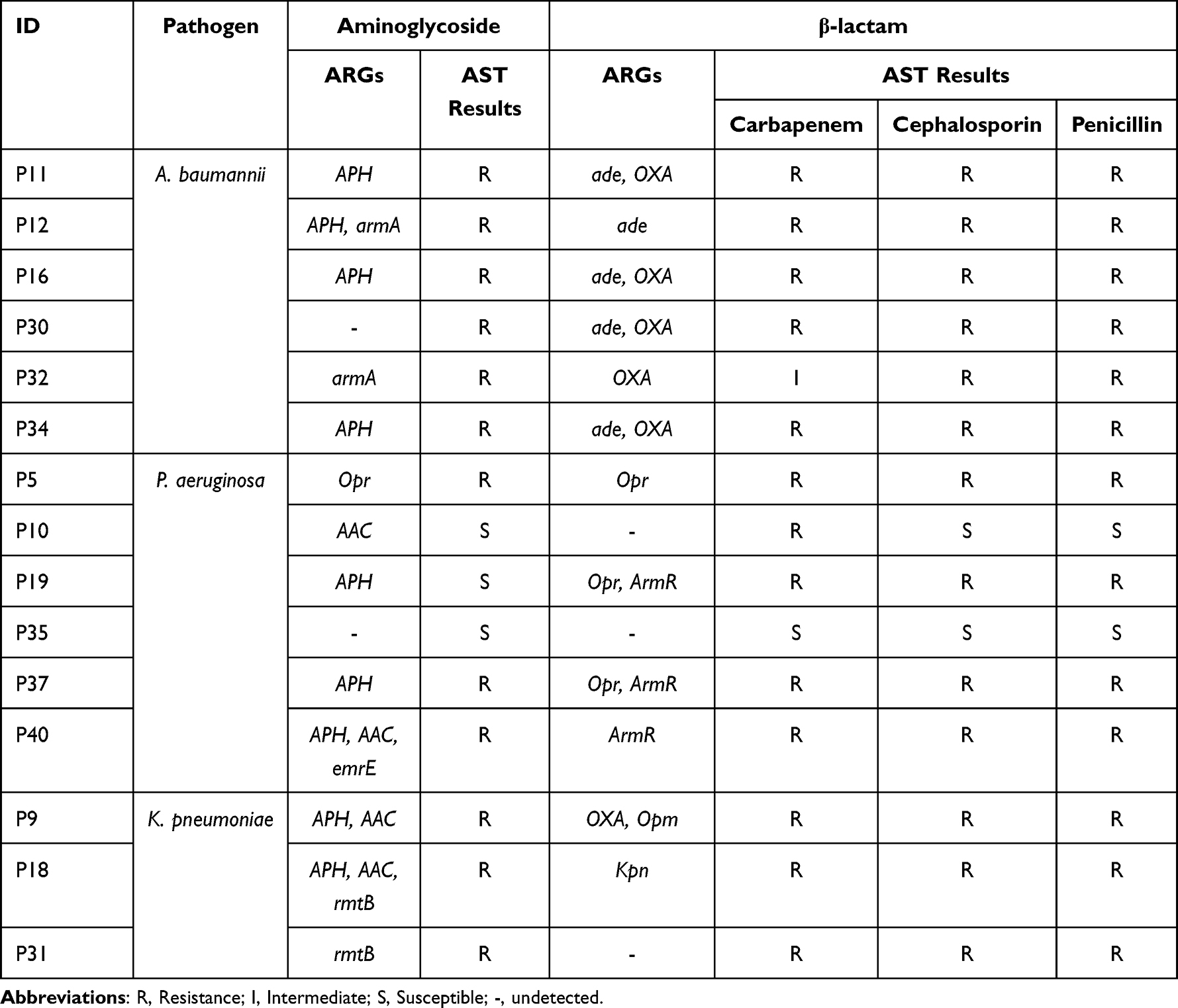 |
Table 2 ARGs Detected from Gram-Negative Pathogens by mNGS and the Corresponding AST Results |
Next, we divided the samples of these 28 patients into two groups according to the mechanical ventilation exposure (≤4 d or ≥5 d), to evaluate the association between mechanical ventilation exposure and ARGs in the respiratory microbiome. Following the comparison of the sequencing depth of two groups, we found that the total number of reads generated by mNGS showed no significant difference between the two groups (P > 0.05, Figure 6A). Based on the same depth of sequencing, patients with late-onset (mechanical ventilation exposure ≥5 d) VAP had a significantly greater proportion of ARGs in their respiratory microbiome compared to those with early-onset (mechanical ventilation exposure ≤4 d) VAP (P = 0.041, Figure 6B).
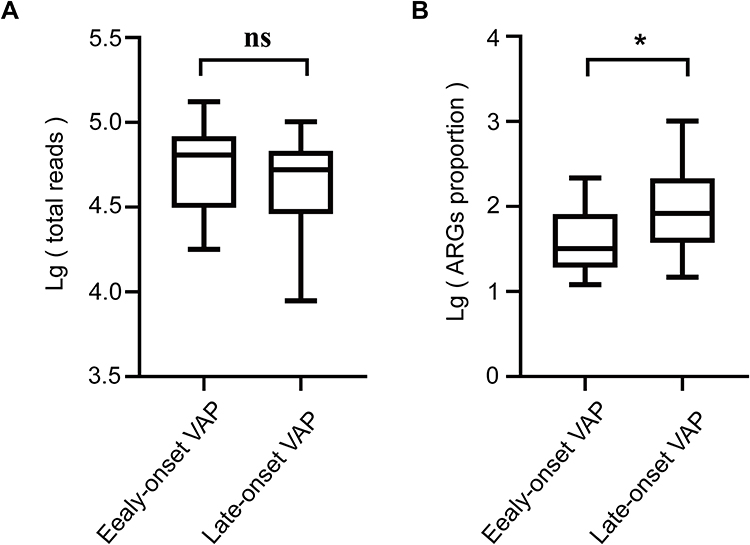 |
Figure 6 Boxplots demonstrating the statistical analyses for ARGs detected from different types of VAP clinical cases. Median values are indicated by the line within the boxplot. The box extends from the 25th to 75th percentile, and whiskers indicate the minimum and maximum values. *P < 0.05; ns, nonsignificant. (A) Lg (total reads). (B) Lg (ARGs proportion). |
Comparison of Turnaround Time Between mNGS and Routine Culture
To compare the performance and the promptness of the results between mNGS and routine culture methods, the turnaround times of all samples between the two methods were compared. The time needed for a sample to be processed and be loaded into the flow cell was 3 h, followed by a real time analysis of data during the sequencing process, being the analysis begin when the first fastq file was output (median time was 7 min, Table S7). The median sequencing duration was therefore only 1.35 h, followed by a 5 min analysis using an automatic bioinformatics pipeline. To conclude, the total median turnaround time to obtain results was 4.43 h for mNGS, and up to 72.00 h for routine culture (Figure 7).
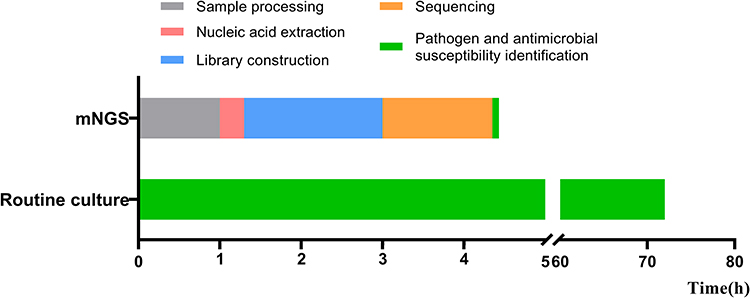 |
Figure 7 Turnaround time: mNGS vs routine microbial culture. |
Discussion
In this study, a rapid nanopore-based mNGS diagnostic assay was developed for a fast detection of VAP pathogens and ARGs by 5 h after sample receiving. In addition, the performance of three different indicators to detect bacterial and fungal pathogens were evaluated, along with the hypothesis that the RA resulted in the highest diagnostic value that was consistent with clinical microbiological culture. Indeed, following the confirmation that the assay could be useful to establish the correct therapy, the main beneficial consequences are that patients might have their empiric therapy correctly de-escalated, while uninfected patients might substantially decrease their antibiotic exposure gradually decreasing its administration from the diagnosis.
The research from this study showed that the RA was the best mNGS indicator for detection of VAP pathogens. Kitsios GD et al27 also used machine-learning algorithms to identify the RA through sequencing as the most informative predictor of culture positivity. In contrast, no significant results were obtained from the reads and RPM parameters, mainly due to the influence of the sequencing depth on the reads, while the RPM was influenced by the host genome proportion in the sample.
More pathogens were identified by mNGS compared with microbiological culture, confirming a better detection performance of mNGS methodology. According to the final composite standard, the diagnostic accuracy of mNGS significantly increased from 86.5% to 98.1% compared with the culture method. For the 6 FN pathogens based on culture results, 3 results were paradoxical with the clinical adjudication, while the remaining pathogens were detected by mNGS but remove from the analysis because the set threshold for mNGS was not achieved. Based on the composite standard, only 3 pathogens were real FN. Additionally, in 2 culture-negative samples, Mycobacterium tuberculosis and Legionella pneumophila were detected by mNGS, respectively. In this regard, the culture method is considered the gold standard for the identification of Mycobacterium tuberculosis and Legionella pneumophila, however, the method requires a time of 2-weeks and the growth of them can be inhibited by the presence of other bacteria.28–30 In addition, since the clinical signs and radiological findings of VAP are nonspecific, the immunology-related test and qPCR methods are often not included in the diagnostic workflow on time. It is really clear that mNGS shows more advantages than traditional methods in the identification of fastidious and nonspecific pathogens.
Traditionally, respiratory viruses have been given minimal attention as a cause of hospital-acquired pneumonia; however, research study showed that respiratory viral infection was associated with mortality rates comparable to the ones associated with bacterial infection.31 HSV even can be responsible for viral reactivation pneumonia in mechanically ventilated patients.7 Based on the clinical virus test results, only 5 out of 63 patients were subjected to respiratory virus test, but none of them resulted positive (Table S6). Viral pathogens were found in 13 (20.6%) patients by mNGS, indicating that mNGS has a great advantage over traditional methods in virus detection, consistently with previous study.32
In addition, the ability of mNGS to simultaneously identify bacteria, fungi, and viruses was also demonstrated in this study. By comparing the results of clinical laboratory findings and mNGS, the coinfection rate in VAP cases increased from 27% to 46%, especially regarding bacterial-viral coinfection. Several reports showed that coinfection can be associated with poorer outcomes in VAP.33,34 Therefore, the improvement in coinfections detection by mNGS may provide more references to clinicians for a better diagnosis and treatment.
The ability of genome sequencing to predict antimicrobial resistance has been extensively established,35,36 but studies assessing the efficiency of this methodology in direct respiratory specimens are still limited,37,38 likely due to the low pathogen concentration in clinical body fluids.39 This problem can be potentially overcome by increasing sequencing depth.40 On the other hand, the long reads-based nanopore sequencing allows for a more reliable assembly through repetitive regions, and allow the ARGs detection by mNGS directly from specimens.41 In our study, the ARGs detected by mNGS were highly consistent with the aminoglycoside and β-lactam resistance phenotypes. Overall, the results of AST and ARGs were fully consistent in 28% samples, with a partial consistency in the rest samples. Those findings may be due to as follows: (1) Although resistance mechanisms associated with specific ARGs can be detected and acquired easily, the mNGS may be inadequate in detecting unknown resistance mechanisms;42 (2) ARG is not always correlated with the protein expression and may lead to discordant genotype/phenotype results.43 A previous study demonstrated that the early-onset VAP is more likely to be caused by antibiotic-susceptible bacteria, while late-onset VAP is usually caused by multi-drug resistant pathogens associated with increased morbidity and mortality.44 Similarly, our findings showed that there was an association between mechanical ventilation exposure and the ARGs burden in VAP, demonstrating the feasibility of mNGS for monitoring the antimicrobial resistance and its epidemiological trends.
As reported, a delay in the initiation of appropriate antibiotic treatment is associated with an increased mortality in severe pneumonia cases.45 However, 2–4 days of turnaround time to obtain culture results often do not allow to establish a prompt treatment.46 In contrast, the total turnaround time of nanopore-based mNGS was within 5 hours after sample submission, which might lead to a faster choice of the appropriate antibiotic before the disease will become irreversible. In addition, the respective libraries were pooled and multiplex sequencing was performed to improve cost-effectiveness, making the sequencing cost drop to $92.17 Although the cost for a microbial culture is $60, more than one test and/or combinations with other ones (such as staining, immunology-related tests and/or PCR test) are often needed, the sequencing-based method developed here is more feasible and cost-effective.
Our study includes certain limitations. First, this is a study based on a single center design, and further research in additional hospitals is required. Second, all the samples had only undergone DNA sequencing but not RNA sequencing, which may have contributed to some of the false-negative results. Third, ETA samples contain greater proportion of host genome DNA, and different depths of sequencing for pathogens, limiting the detection of ARGs. Fourth, the mNGS cost still seems to be high for the rural area of developing countries; therefore, further process optimization is required to reduce cost.
Conclusion
In summary, a nanopore-based mNGS workflow was developed to identify VAP-associated pathogens and ARGs. The pathogen diagnostic accuracy was of 98.1% based on composite standard and the turnaround time was less than 5 h, proving that our method may be beneficial and feasible. In addition, we also demonstrated the efficacy of mNGS in ARGs detection and their epidemiological surveillance using a direct sequencing method of ETA samples from patients affected by VAP. Our study thus showed that mNGS can be beneficially used as a tool for VAP etiological diagnosis in clinical settings.
Data Sharing Statement
The datasets generated for this study can be found in the NCBI BioProject database (BioProject ID: PRJNA893091).
Ethics Approval and Informed Consent
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the Fifth Medical Center of Chinese PLA General Hospital [ky-2019-1-4], and all samples were obtained following the patients’ consents.
Acknowledgments
This study was funded by National Natural Science Foundation of China (82002115 and 81571959).
Disclosure
Ting Chen, Wenhua Huang, Huijun Zong, Qingyu Lv, Yongqiang Jiang, Yan Li, and Peng Liu report grants from National Natural Science Foundation of China, during the conduct of the study. The authors report no conflicts of interest in this work.
References
1. Dai W, Lin Y, Yang X, Huang P, Xia L, Ma J. Meta-analysis of the efficacy and safety of chlorhexidine for ventilator-associated pneumonia prevention in mechanically ventilated patients. Evid Based Complement Alternat Med. 2022;2022:5311034. doi:10.1155/2022/5311034
2. Alecrim RX, Taminato M, Belasco A, Longo MCB, Kusahara DM, Fram D. Strategies for preventing ventilator-associated pneumonia: an integrative review. Revista Brasileira de Enfermagem. 2019;72(2):521–530. doi:10.1590/0034-7167-2018-0473
3. Kalil AC, Metersky ML, Klompas M, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 Clinical Practice Guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):e61–e111. doi:10.1093/cid/ciw353
4. Modi AR, Kovacs CS. Hospital-acquired and ventilator-associated pneumonia: diagnosis, management, and prevention. Cleve Clin J Med. 2020;87(10):633–639. doi:10.3949/ccjm.87a.19117
5. Ranjan N, Chaudhary U, Chaudhry D, Ranjan KP. Ventilator-associated pneumonia in a tertiary care intensive care unit: analysis of incidence, risk factors and mortality. Indian J Crit Care Med. 2014;18(4):200–204. doi:10.4103/0972-5229.130570
6. Torres A, Ferrer M, Badia JR. Treatment guidelines and outcomes of hospital-acquired and ventilator-associated pneumonia. Clin Infect Dis. 2010;51(Suppl 1):S48–53. doi:10.1086/653049
7. Papazian L, Klompas M, Luyt CE. Ventilator-associated pneumonia in adults: a narrative review. Intensive Care Med. 2020;46(5):888–906. doi:10.1007/s00134-020-05980-0
8. Overmann J, Abt B, Sikorski J. Present and future of culturing bacteria. Annu Rev Microbiol. 2017;71:711–730. doi:10.1146/annurev-micro-090816-093449
9. Vaughn VM, Flanders SA, Snyder A, et al. Excess antibiotic treatment duration and adverse events in patients hospitalized with pneumonia: a multihospital cohort study. Ann Intern Med. 2019;171(3):153–163. doi:10.7326/m18-3640
10. Thomas LC, Gidding HF, Ginn AN, Olma T, Iredell J. Development of a real-time Staphylococcus aureus and MRSA (SAM-) PCR for routine blood culture. J Microbiol Methods. 2007;68(2):296–302. doi:10.1016/j.mimet.2006.09.003
11. Miao Q, Ma Y, Wang Q, et al. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin Infect Dis. 2018;67(suppl_2):S231–s240. doi:10.1093/cid/ciy693
12. Goelz H, Wetzel S, Mehrbarzin N, Utzolino S, Häcker G, Badr MT. Next- and third-generation sequencing outperforms culture-based methods in the diagnosis of ascitic fluid bacterial infections of ICU patients. Cells. 2021;10(11):3226. doi:10.3390/cells10113226
13. Johnson JS, Spakowicz DJ, Hong B-Y, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10(1):5029. doi:10.1038/s41467-019-13036-1
14. Grumaz C, Hoffmann A, Vainshtein Y, et al. Rapid next-generation sequencing–based diagnostics of bacteremia in septic patients. J Mol Diagn. 2020;22(3):405–418. doi:10.1016/j.jmoldx.2019.12.006
15. Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20(6):341–355. doi:10.1038/s41576-019-0113-7
16. Moon J, Kim N, Kim T-J, et al. Rapid diagnosis of bacterial meningitis by nanopore 16S amplicon sequencing: a pilot study. Int J Med Microbiol. 2019;309(6):151338. doi:10.1016/j.ijmm.2019.151338
17. Zhang L, Huang W, Zhang S, et al. Rapid detection of bacterial pathogens and antimicrobial resistance genes in clinical urine samples with urinary tract infection by metagenomic nanopore sequencing. Front Microbiol. 2022;13:858777. doi:10.3389/fmicb.2022.858777
18. Charalampous T, Kay GL, Richardson H, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol. 2019;37(7):783–792. doi:10.1038/s41587-019-0156-5
19. Irwin AD, Coin LJM, Harris PNA, et al. Optimising treatment outcomes for children and adults through rapid genome sequencing of sepsis pathogens. A study protocol for a prospective, multi-centre trial (DIRECT). Front Cell Infect Microbiol. 2021;11:667680. doi:10.3389/fcimb.2021.667680
20. Wu N, Ranjan P, Tao C, et al. Rapid identification of pathogens associated with ventilator-associated pneumonia by Nanopore sequencing. Respir Res. 2021;22(1):310. doi:10.1186/s12931-021-01909-3
21. Chan WS, Au CH, Leung SM, et al. Potential utility of targeted Nanopore sequencing for improving etiologic diagnosis of bacterial and fungal respiratory infection. Diagn Pathol. 2020;15(1):41. doi:10.1186/s13000-020-00960-w
22. Baldan R, Cliff PR, Burns S, et al. Development and evaluation of a nanopore 16S rRNA gene sequencing service for same day targeted treatment of bacterial respiratory infection in the intensive care unit. J Infect. 2021;83(2):167–174. doi:10.1016/j.jinf.2021.06.014
23. Li H, Gao H, Meng H, et al. Detection of pulmonary infectious pathogens from lung biopsy tissues by metagenomic next-generation sequencing. Front Cell Infect Microbiol. 2018;8:205. doi:10.3389/fcimb.2018.00205
24. Niederman MS. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388–416. doi:10.1164/rccm.200405-644ST
25. Humphries R, Bobenchik AM, Hindler JA, Schuetz AN, McAdam AJ. Overview of Changes to the Clinical and Laboratory Standards Institute Performance Standards for Antimicrobial Susceptibility Testing, M100, 31st Edition. J Clin Microbiol. 2021;59(12):e0021321. doi:10.1128/jcm.00213-21
26. Gu W, Deng X, Lee M, et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med. 2021;27(1):115–124. doi:10.1038/s41591-020-1105-z
27. Kitsios GD, Fitch A, Manatakis DV, et al. Respiratory microbiome profiling for etiologic diagnosis of pneumonia in mechanically ventilated patients. Front Microbiol. 2018;9:1413. doi:10.3389/fmicb.2018.01413
28. Eble D, Gehrig V, Schubert‐Ullrich P, Köppel R, Füchslin HP. Comparison of the culture method with multiplex PCR for the confirmation of Legionella spp. and Legionella pneumophila. J Appl Microbiol. 2021;131(5):2600–2609. doi:10.1111/jam.15103
29. Falzone L, Gattuso G, Lombardo C, et al. Droplet digital PCR for the detection and monitoring of Legionella pneumophila. Int J Mol Med. 2020;46(5):1777–1782. doi:10.3892/ijmm.2020.4724
30. Machado D, Couto I, Viveiros M. Advances in the molecular diagnosis of tuberculosis: from probes to genomes. Infect Genet Evol. 2019;72:93–112. doi:10.1016/j.meegid.2018.11.021
31. Hong HL, Hong SB, Ko GB, et al. Viral infection is not uncommon in adult patients with severe hospital-acquired pneumonia. PLoS One. 2014;9(4):e95865. doi:10.1371/journal.pone.0095865
32. Lu H, Ma L, Zhang H, et al. The comparison of metagenomic next-generation sequencing with conventional microbiological tests for identification of pathogens and antibiotic resistance genes in infectious diseases. Infect Drug Resist. 2022;15:6115–6128. doi:10.2147/idr.S370964
33. Cillóniz C, Ewig S, Ferrer M, et al. Community-acquired polymicrobial pneumonia in the intensive care unit: aetiology and prognosis. Crit Care. 2011;15(5):R209. doi:10.1186/cc10444
34. Jamieson AM, Pasman L, Yu S, et al. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340(6137):1230–1234. doi:10.1126/science.1233632
35. Hendriksen RS, Bortolaia V, Tate H, Tyson GH, Aarestrup FM, McDermott PF. Using genomics to track global antimicrobial resistance. Front Public Health. 2019;7:242. doi:10.3389/fpubh.2019.00242
36. Mahfouz N, Ferreira I, Beisken S, von Haeseler A, Posch AE. Large-scale assessment of antimicrobial resistance marker databases for genetic phenotype prediction: a systematic review. J Antimicrob Chemother. 2020;75(11):3099–3108. doi:10.1093/jac/dkaa257
37. Yang L, Haidar G, Zia H, et al. Metagenomic identification of severe pneumonia pathogens in mechanically-ventilated patients: a feasibility and clinical validity study. Respir Res. 2019;20(1):265. doi:10.1186/s12931-019-1218-4
38. Charalampous T, Alcolea-Medina A, Snell LB, et al. Evaluating the potential for respiratory metagenomics to improve treatment of secondary infection and detection of nosocomial transmission on expanded COVID-19 intensive care units. Genome Med. 2021;13(1):182. doi:10.1186/s13073-021-00991-y
39. Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, Zengler K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42. doi:10.1186/s40168-018-0426-3
40. Siddiqui H, Nederbragt AJ, Lagesen K, Jeansson SL, Jakobsen KS. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol. 2011;11:244. doi:10.1186/1471-2180-11-244
41. Tamma PD, Fan Y, Bergman Y, et al. Applying rapid whole-genome sequencing to predict phenotypic antimicrobial susceptibility testing results among carbapenem-resistant klebsiella pneumoniae clinical isolates. Antimicrob Agents Chemother. 2019;63(1):Jan. doi:10.1128/aac.01923-18
42. Mitchell SL, Simner PJ. Next-generation sequencing in clinical microbiology: are we there yet? Clin Lab Med. 2019;39(3):405–418. doi:10.1016/j.cll.2019.05.003
43. Yee R, Simner PJ. Next-generation sequencing approaches to predicting antimicrobial susceptibility testing results. Clin Lab Med. 2022;42(4):557–572. doi:10.1016/j.cll.2022.09.011
44. Patil HV, Patil VC. Incidence, bacteriology, and clinical outcome of ventilator-associated pneumonia at tertiary care hospital. J Nat Sci Biol Med. 2017;8(1):46–55. doi:10.4103/0976-9668.198360
45. Heath CH, Grove DI, Looke DF. Delay in appropriate therapy of Legionella pneumonia associated with increased mortality. Eur J Clin Microbiol Infect Dis. 1996;15(4):286–290. doi:10.1007/bf01695659
46. Jain S, Self WH, Wunderink RG, et al. Community-acquired pneumonia requiring hospitalization among U.S. adults. N Engl J Med. 2015;373(5):415–427. doi:10.1056/NEJMoa1500245
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.