")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Design, synthesis, and characterization of (1-(4-aryl)-1H-1,2,3-triazol-4-yl)methyl, substituted phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates against Mycobacterium tuberculosis
Authors Venugopala K , Rao D, Bhandary S , Pillay M, Chopra D , Aldhubiab B, Attimarad M , Alwassil O, Harsha S , Mlisana K
Received 3 April 2016
Accepted for publication 19 May 2016
Published 25 August 2016 Volume 2016:10 Pages 2681—2690
DOI https://doi.org/10.2147/DDDT.S109760
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Katharigatta N Venugopala,1,2 G B Dharma Rao,3 Subhrajyoti Bhandary,3 Melendhran Pillay,4 Deepak Chopra,3 Bandar E Aldhubiab,1 Mahesh Attimarad,1 Osama Ibrahim Alwassil,1 Sree Harsha,1 Koleka Mlisana4
1Department of Pharmaceutical Sciences, College of Clinical Pharmacy, King Faisal University, Al-Ahsa, Kingdom of Saudi Arabia; 2Department of Biotechnology and Food Technology, Durban University of Technology, Durban, South Africa; 3Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal, India; 4Department of Microbiology, National Health Laboratory Services, KZN Academic Complex, Inkosi Albert Luthuli Central Hospital, Durban, South Africa
Abstract: The novel (1-(4-aryl)-1H-1,2,3-triazol-4-yl)methyl, substituted phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate derivatives were synthesized by the click reaction of the dihydropyrimidinones, bearing a terminal alkynyl group, with various substituted aryl azides at room temperature using a catalytic amount of Cu(OAc)2 and sodium ascorbate in a 1:2 ratio of acetone and water as a solvent. The newly synthesized compounds were characterized by a number of spectroscopic techniques, such as infrared, liquid chromatography-mass spectrometry, 1H, and 13C nuclear magnetic resonance along with single crystal X-ray diffraction. The current procedure for the synthesis of 1,2,3-triazole hybrids with dihydropyrimidinones is appropriate for the synthesis of a library of analogs 7a-l and the method accessible here is operationally simple and has excellent yields. The title compounds 7a-l were evaluated for their in vitro antitubercular activity against H37RV and multidrug-resistant strains of Mycobacterium tuberculosis by resazurin microplate assay plate method and it was found that compound 7d was promising against H37RV and multidrug-resistant strains of M. tuberculosis at 10 and 15 µg/mL, respectively.
Keywords: 1,2,3-triazole, dihydropyrimidinone, click chemistry, antitubercular drug discovery, synthesis
Introduction
The pyrimidine system is an important pharmacophore with abundant occurrence in nature. Natural and synthetic dihydropyrimidine derivatives have a wide range of pharmacological actions, such as anticancer,1 antiviral,2,3 antihypertensive,4 calcium channel blocking,5 antitubercular,6 antimicrobial,7,8 anti-inflammatory,9,10 and larvicidal and insecticide actions.11,12 1,2,3-Triazoles, as a vital class of N-heterocyclic compounds, due to their unique chemical and structural properties, have received a great deal of attention over the past few decades and found broad application in medicinal chemistry13 and particularly as anticancer,14 antimicrobial,15 antitubercular,16 anti-HIV,17 and antifungal agents.18 On the other hand, this special class of scaffolds has also found relevance in objective oriented synthesis,19 bioconjugation,20 materials and surface science,21 combinatorial chemistry,22 and medicinal chemistry.23 Moreover, 1,2,3-triazole can mimic natural peptides and heterocycles in geometrical shape and interaction function.24 1,2,3-Triazoles could be easily constructed by click chemistry reaction25 and which yielded small molecules with special properties, such as moderate dipole character, hydrogen bonding capability, rigidity, and stability.26 Heterocyclic27–29 fluorine-containing compounds have been shown to exhibit promising anti-tuberculosis (anti-TB) activity,30 including 1,2,3-triazole analogs, for their promising anti-TB activity.31 Keeping this in mind and considering the pharmacological significance of dihydropyrimidine and 1,2,3-triazole pharmacophores, in the present investigation it was decided to design and synthesize a series of novel 1,2,3-triazole hybrid with dihydropyrimidinone (DHPM) scaffolds in accordance with Lipinski rule except compound 7d.32 The title compounds, (1-(4-aryl)-1H-1,2,3-triazol-4-yl)methyl, substitutedphenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates 7a-l, have been tested for safety studies by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay33 following in vitro antitubercular activity against H37RV and multidrug-resistant strains of Mycobacterium tuberculosis (MDR-MTB) by resazurin microplate assay plate method.
In this communication and in continuation of our work on the development of pharmacologically active heterocyclic compounds6,34,35 and screening of heterocyclic compounds for properties of polymorphism,36–38 we have synthesized 1,2,3-triazole hybrid with DHPMs using aryl azide as well as DHPMs having a terminal alkynyl group, which was synthesized by the four component Biginelli-like cyclocondensation reaction (tert-butyl β-ketoester, propargyl alcohol, aryl aldehyde, and urea) along with catalytic amount of Cu(OAc)2 and sodium ascorbate in a 1:2 ratio of acetone and water as a solvent at room temperature as shown in Figure 1.
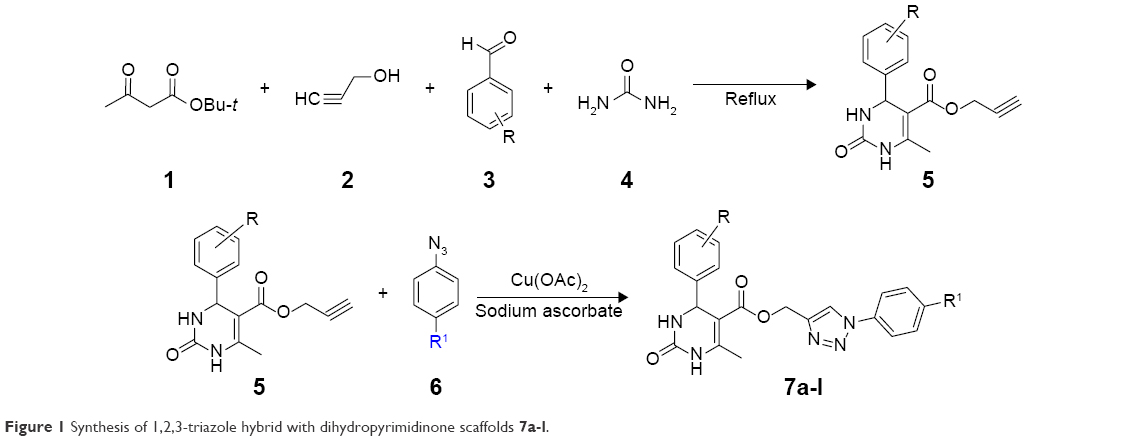 | Figure 1 Synthesis of 1,2,3-triazole hybrid with dihydropyrimidinone scaffolds 7a-l. |
Materials and methods
Chemistry
All the chemicals were purchased from Sigma-Aldrich Corporation (St Louis, MO, USA; analytical grade) and used without further purification. Fourier transform infrared (FTIR) spectra were registered on a Bruker Corporation (Billerica, MA, USA) IFS 55 equinox Fourier transform IR spectrophotometer as KBr discs. 1H- and 13C-nuclear magnetic resonance (NMR) spectra were recorded using a Bruker 400 or 500 MHz spectrometer in the solvents indicated (referenced to the residual 1H signals in the deuterated solvents) using tetramethylsilane (TMS) as an internal standard. Chemical shifts are reported in ppm (δ scale) and coupling constant (J) values given in hertz (Hz). The splitting pattern is abbreviated as follows: s, singlet; d, doublet; and m, multiplet. Thin layer chromatography (TLC) analysis of reaction mixtures was performed on Merck (Merck Serono, Darmstadt, Germany) aluminum plates coated with silica gel (60 F254). Compounds were visualized by ultraviolet irradiation at 254 and 366 nm. Merck silica gel (60–120 mesh) was used for column chromatography.
Spectra of the compounds are available as Supplementary Materials.
General procedure for the synthesis of (1-(4-aryl)-1H-1,2,3-triazol-4-yl)methyl, substituted phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates (7a-l)
A 25 mL round bottom flask equipped with a condenser was charged with substituted aryl azides (1.0 mmol) (6) as well as DHPMs having terminal alkynyl group (1.0 mmol) (5). Compound 5 was synthesized by the four components Biginelli-like cyclocondensation reaction of tert-butyl β-ketoester (1.0 mmol) (1), propargyl alcohol (1.2 mmol) (2), substituted aryl aldehyde (1.0 mmol) (3), and urea (1.2 mmol) (4) by a reflux method.39 The entire reaction mixture was allowed to stir for 3 hours at room temperature along with a catalytic amount of Cu(OAc)2 (0.1 mmol) and sodium ascorbate (0.2 mmol) in a 1:2 ratio of acetone and water (2 mL) as a solvent till the reaction was complete. The progress of the reaction was monitored on TLC (4:6 of hexane and ethyl acetate). After completion of the reaction as indicated on TLC, the contents were concentrated under reduced pressure to remove excess of the acetone and the crude reaction mixture was extracted with ethyl acetate and water. The combined organic extract, after drying over anhydrous sodium sulfate, was again concentrated under reduced pressure to obtain the crude product. For analytically pure products, the final solid mass was purified by column chromatography using the hexane/ethyl acetate (4:6) as the eluent to give the pure products 7a-l at 77%–92% yield. Physicochemical characteristics of the title compounds are tabulated in Table 1.
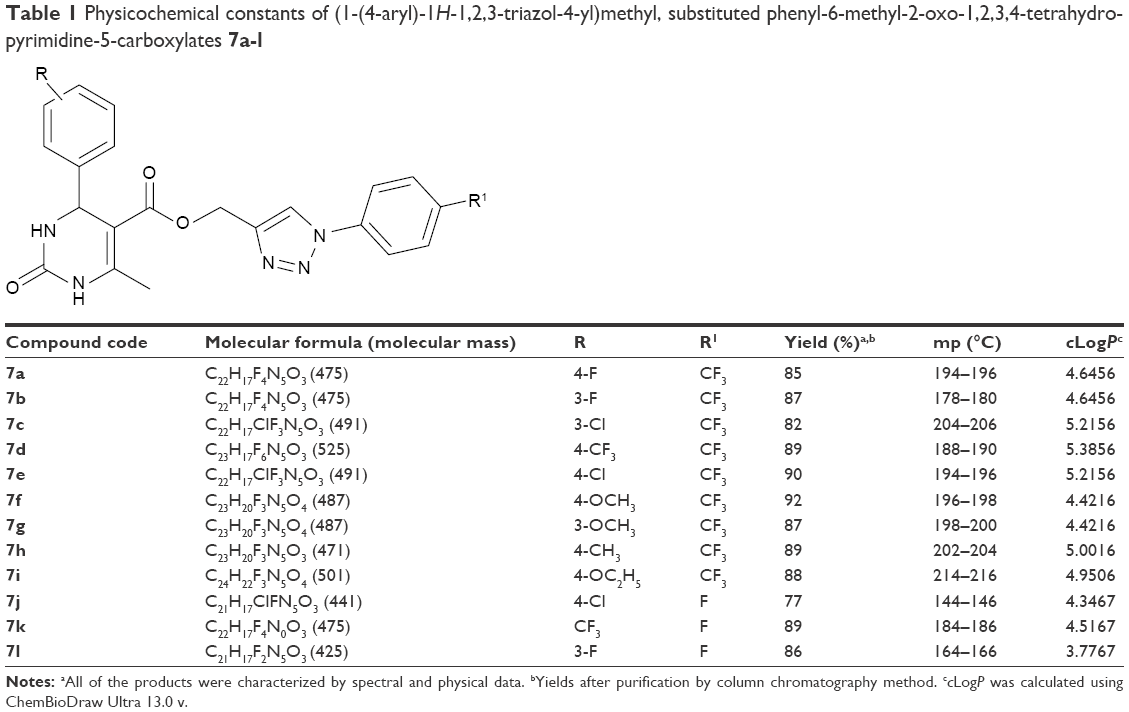 | Table 1 Physicochemical constants of (1-(4-aryl)-1H-1,2,3-triazol-4-yl)methyl, substituted phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates 7a-l |
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(4-fluorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7a)
IR (KBr) ν/cm−1 3,333, 2,946, 2,370, 1,670, 1,332, 844, 785, 753. 1H NMR (500 MHz, dimethyl sulfoxide [DMSO]) δ 9.29 (s, 1H), 8.72 (s, 1H), 8.12 (d, J=8.5 Hz, 2H), 7.99 (d, J=8.6 Hz, 2H), 7.77 (s, 1H), 7.24–7.21 (m, 2H), 7.06 (t, J=8.8 Hz, 2H), 5.21–5.17 (m, 3H), 2.27 (s, 3H). 13C NMR (126 MHz, DMSO) δ 165.3, 162.7, 160.7, 152.3, 150.04, 144.2, 141.3, 141.2, 139.6, 128.7, 128.6, 127.6, 123.2, 121.0, 115.6, 115.4, 99.0, 56.6, 53.7, 18.3. Liquid chromatography-mass spectrometry (LCMS): 475.2.
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(3-fluorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7b)
IR (KBr) ν/cm−1 3,367, 2,979, 2,345, 1,700, 1,636, 1,329, 873, 845, 757. 1H NMR (400 MHz, DMSO) δ 9.31 (s, 1H), 8.80 (s, 1H), 8.10 (d, J=7.9 Hz, 2H), 7.96 (d, J=8.0 Hz, 2H), 7.79 (s, 1H), 7.27 (d, J=6.6 Hz, 1H), 7.06–6.87 (m, 3H), 5.27–5.10 (m, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.2, 163.6, 161.2, 152.3, 150.4, 147.9, 144.2, 139.7, 130.9, 130.8, 127.7, 127.6, 123.2, 122.5, 121.0, 114.6, 114.4, 113.5, 113.3, 98.5, 56.7, 53.8, 18.4. LCMS: 475.4.
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(3-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7c)
IR (KBr) ν/cm−1 3,363, 2,969, 2,349, 1,695, 1,330, 843, 788, 757. 1H NMR (400 MHz, DMSO) δ 9.33 (s, 1H), 8.80 (s, 1H), 8.11 (d, J=8.4 Hz, 2H), 7.96 (d, J=8.5 Hz, 2H), 7.79 (s, 1H), 7.34–7.13 (m, 4H), 5.20–5.13 (m, 3H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.2, 158.0, 152.2, 150.5, 147.5, 146.9, 144.2, 133.3, 130.8, 127.7, 127.6, 126.6, 125.2, 123.2, 121.0, 98.4, 56.7, 53.9, 18.4. LCMS: 491.2 (M+), 493.2 (M +2).
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 6-methyl-2-oxo-4-(4-(trifluoromethyl)phenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7d)
IR (KBr) ν/cm−1 3,412, 2,968, 2,345, 1,700, 1,641, 1,326, 844, 795, 718. 1H NMR (400 MHz, DMSO) δ 9.35 (s, 1H), 8.79 (s, 1H), 8.10 (d, J=8.1 Hz, 2H), 7.95 (d, J=8.2 Hz, 2H), 7.83 (s, 1H), 7.57 (d, J=7.7 Hz, 2H), 7.38 (d, J=7.6 Hz, 2H), 5.25–5.12 (m, 3H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.1, 152.2, 150.5, 144.2, 139.6, 127.6, 127.6, 127.6, 127.5, 125.8, 125.7, 123.2, 120.9, 98.3, 59.3, 56.7, 54.1, 18.4. LCMS: 525.1.
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(4-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7e)
IR (KBr) ν/cm−1 3,365, 2,966, 2,309, 1,709, 1,638, 1,331, 843, 786, 746. 1H NMR (400 MHz, DMSO) δ 9.30 (s, 1H), 8.75 (s, 1H), 8.11 (d, J=8.5 Hz, 2H), 7.96 (d, J=8.6 Hz, 2H), 7.76 (s, 1H), 7.27 (d, J=8.4 Hz, 2H), 7.17 (d, J=8.4 Hz, 2H), 5.31–5.03 (m, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.2, 152.2, 150.2, 144.3, 144.0, 139.7, 132.2, 129.4, 128.7, 128.6, 127.7, 127.6, 123.2, 121.0, 98.6, 56.7, 53.8, 18.4. LCMS: 491.2 (M+), 493.2 (M +2).
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7f)
IR (KBr) ν/cm−1 3,390, 2,964, 2,345, 1,695, 1,638, 1,336, 845, 790, 757. 1H NMR (400 MHz, DMSO) δ 9.21 (s, 1H), 8.68 (s, 1H), 8.10 (d, J=8.4 Hz, 2H), 7.96 (d, J=8.6 Hz, 2H), 7.66 (s, 1H), 7.08 (d, J=8.6 Hz, 2H), 6.76 (d, J=8.6 Hz, 2H), 5.18–5.08 (m, 3H), 3.59 (s, 3H), 2.23 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.3, 158.8, 152.4, 149.6, 144.4, 139.6, 137.3, 127.8, 127.6, 127.6, 123.1, 121.0, 114.0, 99.3, 56.6, 55.3, 53.7, 31.1, 18.3. LCMS: 487.4.
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(3-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7g)
IR (KBr) ν/cm−1 3,414, 2,964, 2,349, 1,718, 1,642, 1,333, 843, 788, 726. 1H NMR (400 MHz, DMSO) δ 9.25 (s, 1H), 8.74 (s, 1H), 8.10 (d, J=8.5 Hz, 2H), 7.97 (d, J=8.6 Hz, 2H), 7.72 (s, 1H), 7.15 (t, J=7.9 Hz, 1H), 6.82–6.65 (m, 3H), 5.19–5.11 (m, 3H), 3.59 (s, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.3, 159.6, 152.5, 150.0, 146.6, 144.3, 130.0, 127.7, 127.6, 123.2, 121.0, 118.6, 112.8, 112.6, 98.9, 56.7, 55.3, 54.1, 18.3. LCMS: 487.3.
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 6-methyl-2-oxo-4-(p-tolyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7h)
IR (KBr) ν/cm−1 3,367, 2,967, 2,349, 1,710, 1,646, 1,324, 841, 792, 703. 1H NMR (400 MHz, DMSO) δ 9.21 (s, 1H), 8.68 (s, 1H), 8.10 (d, J=8.5 Hz, 2H), 7.97 (d, J=8.6 Hz, 2H), 7.68 (s, 1H), 7.05–6.99 (m, 4H), 5.21–5.09 (m, 3H), 2.23 (s, 3H), 2.12 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.3, 152.4, 149.7, 144.4, 142.2, 139.7, 136.8, 129.2, 127.7, 127.6, 126.6, 125.6, 123.1, 121.0, 99.2, 56.6, 54.0, 20.9, 18.3. LCMS: 471.2.
(1-(4-(Trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(4-ethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7i)
IR (KBr) ν/cm−1 3,323, 2,982, 2,350, 1,696, 1,644, 1,331, 839, 769, 696. 1H NMR (400 MHz, DMSO) δ 9.20 (s, 1H), 8.65 (s, 1H), 8.09 (d, J=8.5 Hz, 2H), 7.96 (d, J=8.6 Hz, 2H), 7.65 (s, 1H), 7.05 (d, J=8.6 Hz, 2H), 6.72 (d, J=8.6 Hz, 2H), 5.26–5.05 (m, 3H), 3.88–3.72 (m, 2H), 2.23 (s, 3H), 1.19 (t, J=7.0 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 165.3, 158.1, 152.4, 149.5, 144.4, 139.6, 137.2, 127.8, 127.6, 127.6, 123.0, 121.0, 114.5, 99.3, 63.3, 56.5, 53.7, 18.3, 14.9. LCMS: 501.6.
(1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(4-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7j)
IR (KBr) ν/cm−1 3,414, 2,960, 2,349, 1,712, 1,640, 1,323, 838, 792, 710. 1H NMR (400 MHz, DMSO) δ 9.36 (s, 1H), 8.63 (s, 1H), 7.93–7.79 (m, 3H), 7.59 (d, J=7.9 Hz, 2H), 7.45–7.38 (m, 4H), 5.27–5.09 (m, 3H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.2, 152.2, 150.5, 149.5, 143.8, 127.5, 125.8, 125.8, 123.2, 122.8, 122.8, 117.3, 117.0, 98.4, 56.8, 54.1, 31.1, 23.1, 18.4. LCMS: 441.2 (M+), 443.2 (M +2).
(1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl 6-methyl-2-oxo-4-(4-(trifluoromethyl)phenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7k)
IR (KBr) ν/cm−1 3,367, 2,960, 2,349, 1,711, 1,638, 1,313, 835, 787, 691. 1H NMR (400 MHz, DMSO) δ 9.30 (s, 1H), 8.58 (s, 1H), 7.90–7.87 (m, 2H), 7.76 (s, 1H), 7.43 (t, J=8.4 Hz, 2H), 7.28 (d, J=8.0 Hz, 2H), 7.18 (d, J=8.0 Hz, 2H), 5.20–5.09 (m, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.2, 152.2, 150.2, 144.0, 143.9, 133.5, 132.2, 128.7, 128.6, 123.1, 123.0, 122.9, 98.6, 56.8, 53.8, 31.1, 18.4. LCMS: 475.3.
(1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl 4-(3-fluorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (7l)
IR (KBr) ν/cm−1 3,324, 3,079, 2,349, 1,663, 1,638, 1,239, 845, 761, 703. 1H NMR (400 MHz, DMSO) δ 9.31 (s, 1H), 8.62 (s, 1H), 7.90–7.87 (m, 2H), 7.79 (s, 1H), 7.43 (t, J=8.7 Hz, 2H), 7.28 (m, 1H), 7.06–6.86 (m, 3H), 5.23–5.10 (m, 3H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.2, 163.7, 161.2, 160.9, 152.3, 150.4, 147.9, 147.9, 143.8, 133.5, 130.9, 130.8, 123.2, 123.0, 122.9, 122.5, 117.3, 117.0, 114.6, 114.4, 113.5, 113.3, 98.5, 56.8, 53.8, 18.4. LCMS: 425.4.
Crystal growth and single crystal X-ray study
Title compound 7g was used to grow single crystals at room temperature (25°C) using benzene as solvent for crystallographic studies. Single crystal data were collected on the Bruker D8 VENTURE diffractometer equipped with CMOS type PHOTON 100 detector using monochromated Mo Kα radiation (λ=0.71073 Å). Unit cell measurement, data collection, integration, scaling, and absorption corrections for the crystal were done using Bruker Apex II software.40 Data reduction was done by Bruker SAINT suite.41 The crystal structure was solved by direct methods using SIR 201442 and refined by the full matrix least squares method using SHELXL 201443 present in the program suite WinGX (version 2014.1, Louis J. Farrugia, Glasgow, Scotland).44 Absorption correction was applied using SADABS.45 All non-hydrogen atoms were refined anisotropically and all hydrogen atoms (except H-atoms bonded to N4 and N5) were positioned geometrically and refined using a riding model with Uiso(H) =1.2Ueq. The H-atoms bonded to N4 and N5 were taken directly from difference Fourier maxima. ORTEP (Oak Ridge Thermal Ellipsoid Plot) was generated using Mercury 3.5.1 Cambridge Crystallographic Data Center (CCDC) program.46 Geometrical calculations were done using PARST47 and PLATON.48 Crystallographic and refinement data of the title compound 7g are tabulated in Table 2.
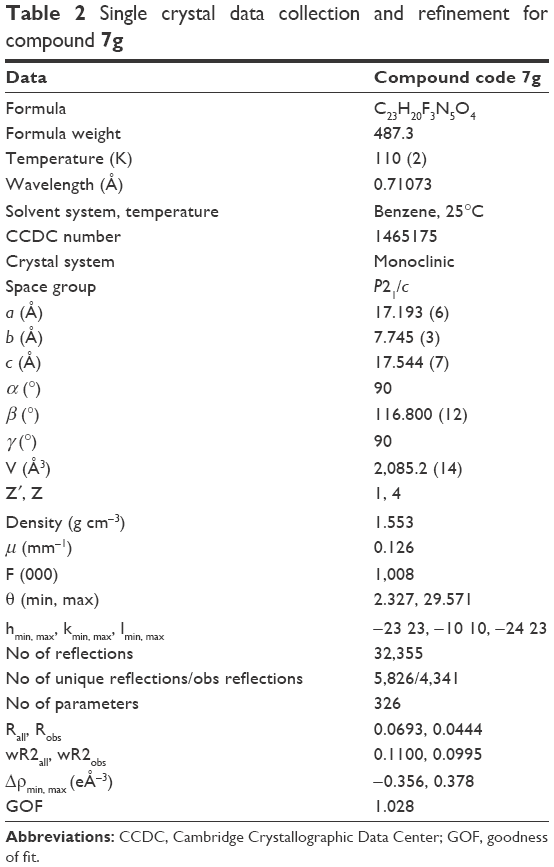 | Table 2 Single crystal data collection and refinement for compound 7g |
Safety studies
The safety of the test compounds 7a-l was evaluated by an MTT assay. The MTT cytotoxicity assay was used to evaluate the cytotoxic effect of the most promising compounds against peripheral blood mononuclear cells according to the protocol described.49 Cells were pipetted (90 μL of cell culture, 1×105 cells/mL) into each well of 96-well microtiter plates, and the outer wells were filled with phosphate-buffered saline in order to prevent the medium from evaporation during incubation. Thereafter, plates were incubated at 37°C for 24 hours. Each well of the plate was then treated with 10 μL of the compounds (1,000–5 μg/mL). In the control wells, the negative control DMSO and media were added. Thereafter, the plates were incubated for 2 days at 37°C in a humidified incubator that contained a 5% CO2 atmosphere. After the incubation time, 20 μL of MTT reagent (5 mg/mL) was further added to individual well. The plate was then incubated for a further 4 hours at 37°C (5% CO2 incubator). The media were then removed after incubation, and an aliquot of 100 μL DMSO was added to each well in order to dissolve the formazan crystals that were formed in metabolically active cells. Thereafter, the plates were incubated for an extra hour. The absorbance of the formazan was evaluated at 590 nm using an ELISA plate reader.
Antitubercular activity
Resazurin microplate assay plate method
The susceptibility of clinical isolates comprising of both fully sensitive and MDR TB isolates were evaluated against test compounds 7a-l by the colorimetric resazurin microplate assay plate method.50 An amount of 100 μL of Middlebrook 7H9 (Becton, Dickinson and Company, New Jersey, USA) broth was aseptically prepared and dispensed in each of the wells of a 96-well flat-bottomed microtiter plate with lids (Lasec, Ndabeni, South Africa). Each of the test compounds 7a-l was weighed out accordingly, dissolved in the appropriate solvent, and filter sterilized using a 0.2 micron polycarbonate filter. Stock solutions of the test samples were aliquoted into cryovials and stored at −20°C. An amount of 100 μL of the test samples was added to each of the well containing Middlebrook 7H9 broth supplemented with 0.1% casitone, 0.5% glycerol, and 10% oleic acid, albumin, dextrose, and catalase. The test samples were then further serially diluted two-fold directly in the broth of the microtiter plate to a desired concentration ranging from 40 to 0.625 μg/mL.
Inoculums from clinical isolates were prepared fresh from Middlebrook 7H11 agar plates by scraping and resuspending loopful of colonies into Middlebrook 7H9 broth containing glass beads. The inoculum turbidity was adjusted to a McFarland number 1 standard and further diluted 1:10 in M7H9 broth prior to addition (100 μL) to each of the test samples and drug-free wells. A growth control and a sterile control were also included for each isolate. Sterile M7H9 broth was added to all perimeter wells to avoid evaporation during the incubation. The plate was covered, sealed in a plastic bag, and incubated at 37°C. After 8 days of incubation, 30 μL of 0.02% working solution of resazurin salt was inoculated into each microtiter well. The plates were then incubated overnight and read the following day. A positive reaction resulted in a color change from blue to pink owing to the reduction of resazurin to rezarufin, which confirmed MTB cell viability/growth and hence drug resistance. The minimum inhibitory concentrations were defined as the minimum drug concentration to inhibit the growth of the organism with no color changes present in the well. The anti-TB results of title compounds 7a-l are tabulated in Table 3.
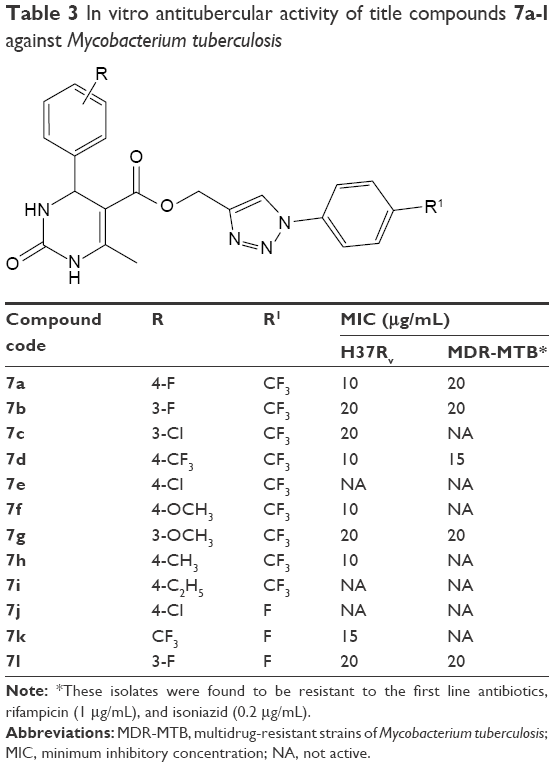 | Table 3 In vitro antitubercular activity of title compounds 7a-l against Mycobacterium tuberculosis |
Results and discussion
Chemistry
As a continuing aspect of our earlier work6,37,51 and after much efforts over the years to develop efficient synthetic procedures for multicomponent reactions under greener conditions, there was a requirement to synthesize a huge library of diversified 1,2,3-triazole hybrids with DHPMs analogs with reduced time, outstanding yields, and excellent biological activities. The DHPMs with terminal alkynyl group were synthesized following a previously reported procedure.52 Aromatic azides were synthesized in high yields from arenediazonium tosylates and sodium azide in water at room temperature. An in situ diazotization followed by azidation in the presence of p-toluenesulfonic acid allows the direct transformation of aromatic amines.53
For our initial studies, 4-trifluoromethyl aryl azide and prop-2-yn-1-yl 4-(4-fluorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate were chosen as model substrates (entry 1; compound 7a, Table 1) with a catalytic amount of Cu(OAc)2 and sodium ascorbate in a 1:2 ratio of acetone and water (2 mL) as a solvent. A whole reaction mixture of 4-trifluoromethyl aryl azide and terminal alkynyl DHPMs was stirred at room temperature (25°C). The starting material was consumed within 3 hours as indicated by TLC analysis. It was observed that when aryl azide, terminal alkynyl DHPMs, Cu(OAc)2, and sodium ascorbate were used in the ratio of 1:1:0.1:0.2 in 2 mL of a mixture of 1:2 ratio of acetone and water as a solvent, they gave the best result. After workup and purification by silica gel column chromatography, the final desired product 1,2,3-triazole hybrid with DHPMs was isolated in 85% yield.
The practicality of optimized reaction conditions was further extended to the synthesis of more functionalized 1,2,3-triazole hybrid DHPM derivatives 7b-l and experiments were performed by making use of a wide range of aryl azides. It was found that in all the cases the reaction occurred smoothly. We have also reacted a variety of DHPMs analogs having both electron-releasing and -withdrawing substituent to synthesize the diversified DHPMs derivatives with 1,2,3-triazole linkage and all the results are appended in Table 1. The partition coefficient of the title compounds was calculated by ChemBioDraw Ultra 13.0v (PerkinElmer Inc., Waltham, MA, USA) and the results were in the range of 3.7767–5.3856. The purity of the compounds was confirmed by high performance liquid chromatography and it was over 99%.
Crystallographic studies
Test compound 7g emerged as one of the promising compounds for anti-TB activity from the series subjected to single crystal X-ray studies. The compound crystallizes in the centrosymmetric monoclinic space group P21/c with one molecule in the asymmetric unit (Z=4). ORTEP is shown in Figure 2. Structural investigation shows that the DHPM ring of the molecule exists in a boat-like conformation due to minimization of the steric repulsion between ester moiety containing a triazole ring with meta methoxy phenyl ring. The para trifluoro substituted benzene ring and triazole unit remain almost in the same molecular plane whereas the methoxy phenyl substituted DHPM unit is almost perpendicular to the plane. Crystal packing is mainly controlled by strong intermolecular N-H…O dimeric motif along the crystallographic axis a and weak C-H…O hydrogen bonding dimers along the axis c (shaded regions in Figure 3A). In addition, molecules form centrosymmetric dimers via N-H…N, C-H…N, C-H…π (Cg1) hydrogen bonds and weak π…π stacking interactions along the crystallographic b-axis wherein these dimers are linked with weak C-H…F (involving H20, F2) hydrogen bond and F…F (involving F1, F3) interactions (Figure 3B). Hence, it is noteworthy to mention that interactions involving fluorine atom are one of the important contributors to the overall packing.54–56 The list of all the intermolecular interactions is given in Table 4.
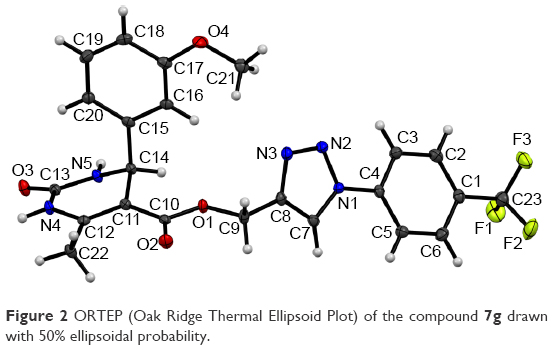 | Figure 2 ORTEP (Oak Ridge Thermal Ellipsoid Plot) of the compound 7g drawn with 50% ellipsoidal probability. |
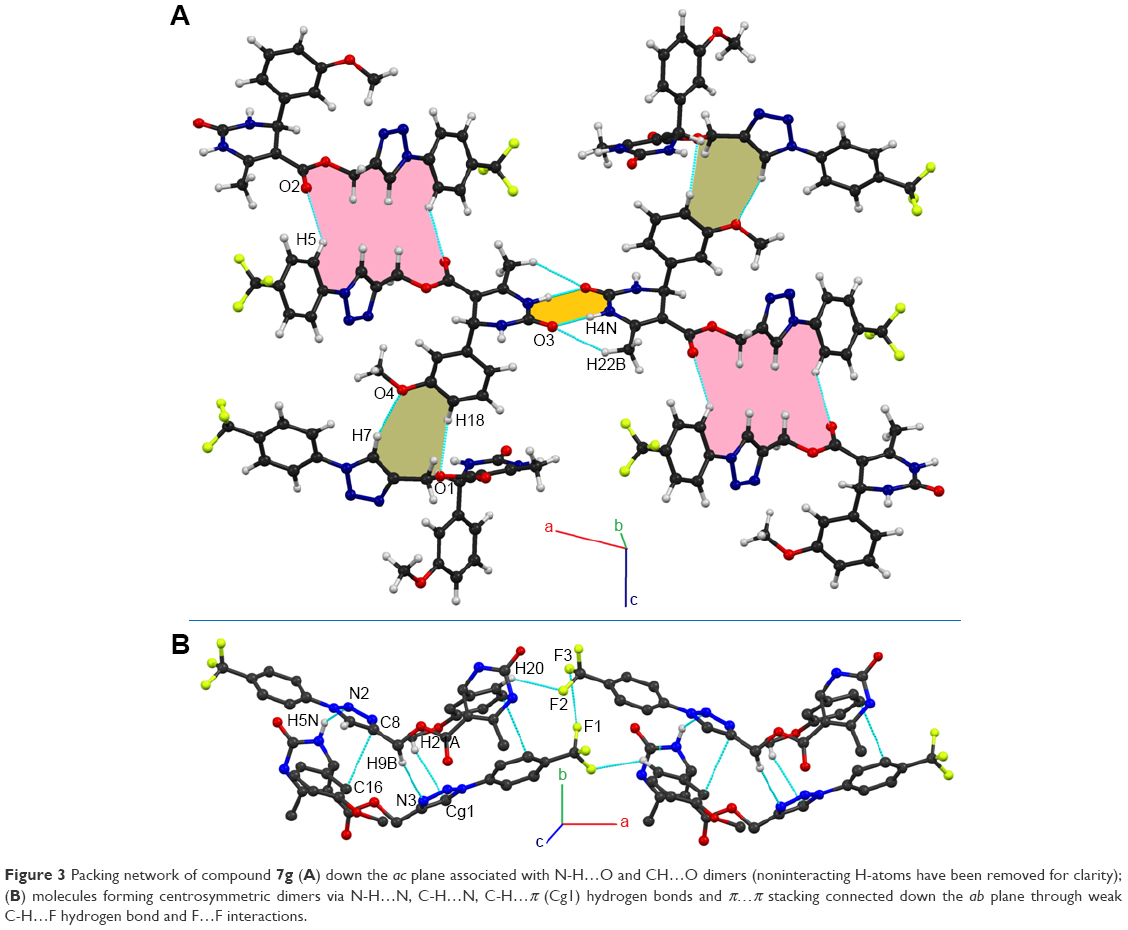 | Figure 3 Packing network of compound 7g (A) down the ac plane associated with N-H…O and CH…O dimers (noninteracting H-atoms have been removed for clarity); (B) molecules forming centrosymmetric dimers via N-H…N, C-H…N, C-H…π (Cg1) hydrogen bonds and π…π stacking connected down the ab plane through weak C-H…F hydrogen bond and F…F interactions. |
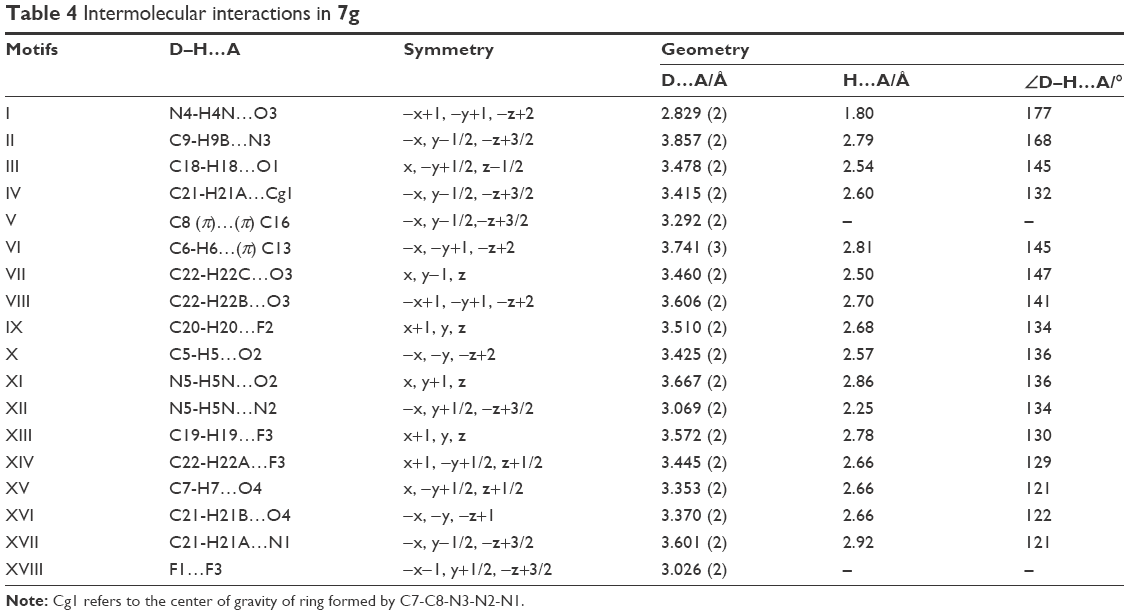 | Table 4 Intermolecular interactions in 7g |
Safely studies
Test compounds 7a-l were evaluated for safely studies by an MTT assay and it was found that up to 500 μg/mL no toxicity on PBM cell lines was observed.
Antitubercular activity
Anti-TB activity of the test compounds 7a-l was evaluated against H37Rv and MDR-MTB by resazurin microplate assay plate method and the results are tabulated in Table 3. Compound 7d with trifluoro methyl group at fourth position of two-phenyl ring appeared as a promising agent against H37Rv and MDR-MTB at 10 and 15 μg/mL, respectively. However, compound 7f with methoxy group on phenyl ring of pyrimidine nucleus and trifluoromethyl group on phenyl ring of triazole ring exhibited activity at 10 μg/mL against H37Rv and no activity against MDR-MTB. Test compounds 7b, 7g, and 7l exhibited similar activity against H37Rv and MDR-MTB in spite of varying functional groups on phenyl rings that are on pyrimidine and triazole nucleus.
Conclusion
We have established an operationally simple and straightforward one-pot synthesis for the synthesis of 1,2,3-triazole hybrid with DHPMs analogs via click chemistry. The purity of the compound was over 99% and yield of the compounds was excellent. Compound 7d emerged as a promising compound from the series for anti-TB activity. Crystallographic studies for the compound 7g revealed that the interplay of strong (such as N-H…O, N-H…N, C-H…O) and weak interactions (eg, C-H…F, C-H…π, F…F) stabilizes the overall crystal packing in the solid state.
Acknowledgments
The authors are grateful to Deanship of Scientific Research (150199), King Faisal University, Kingdom of Saudi Arabia, and IISER Bhopal for providing access to their facility and for their encouragement. GBDR thanks IISER Bhopal for postdoctoral fellowship. DC thanks IISER Bhopal for research facilities and infrastructure.
Disclosure
The authors report no conflicts of interest in this work.
References
Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286(5441):971–974. | ||
Hurst EW, Hull R. Two new synthetic substances active against viruses of the psittacosis-lymphogranuloma-trachoma group. J Med Pharm Chem. 1961;3(2):215–229. | ||
Zhu X, Zhao G, Zhou X, et al. 2,4-Diaryl-4,6,7,8-tetrahydroquinazolin-5(1H)-one derivatives as anti-HBV agents targeting at capsid assembly. Bioorg Med Chem Lett. 2010;20(1):299–301. | ||
Karnail SA, Brian NS, Steven EU, et al. Dihydropyrimidine calcium channel blockers. 3, 3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-pyrimidinecarboxylic acid esters as orally effective antihypertensive agents. J Med Chem. 1991;34(2):806–811. | ||
Jauk B, Pernat T, Kappe CO. Design and synthesis of a conformationally rigid mimic of the dihydropyrimidine calcium channel modulator SQ 32,926. Molecules. 2000;5:227–239. | ||
Venugopala KN, Nayak SK, Pillay M, Prasanna R, Coovadia YM, Odhav B. Synthesis and antitubercular activity of 2-(substituted phenyl/benzyl-amino)-6-(4-chlorophenyl)-5-(methoxycarbonyl)-4-methyl-3,6-dihydropyrimidin-1-ium chlorides. Chem Biol Drug Des. 2013;81(2):219–227. | ||
Wael AES, Ibrahim FN, Adel AH, Abdel R. C-furyl glycosides, II: synthesis and antimicrobial evaluation of C-furyl glycosides bearing pyrazolines, isoxazolines, and 5,6-dihydropyrimidine-2(1H)-thiones. Monatsh Chem. 2009;140:365–370. | ||
Shah TB, Gupte A, Patel MR, Chaudhari VS, Patel H, Patel VC. Synthesis and in vitro study of biological activity of heterocyclic N-mannich bases of 3,4-dihydropyrimidine-2(1H)-thiones. Ind J Chem. 2010;49B(05):578–586. | ||
Sushilkumar SB, Devanand BS. Synthesis and anti-inflammatory activity of some 2-amino-6-(4-substituted aryl)-4-(4-substituted phenyl)-1,6-dihydropyrimidine-5-yl-acetic acid derivatives. Acta Pharm. 2003;53:223–229. | ||
Nofal ZM, Fahmy HH, Zarea ES, El-Eraky W. Synthesis of new pyrimidine derivatives with evaluation of their anti-inflammatory and analgesic activities. Acta Pol Pharm. 2011;68(4):507–517. | ||
Rajanarendar E, Reddy MN, Murthy KR, et al. Synthesis, antimicrobial, and mosquito larvicidal activity of 1-aryl-4-methyl-3,6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2,3,6,10b-tetrahydro-1H-pyrimido[5,4-c]quinolin-5-ones. Bioorg Med Chem Lett. 2010;20(20):6052–6055. | ||
Venugopala KN, Gleiser RM, Chalannavar RK, Odhav B. Antimosquito properties of 2-substituted phenyl/benzylamino-6-(4-chlorophenyl)-5-methoxycarbonyl-4-methyl-3,6-dihydropyrimidinium chlorides against Anopheles arabiensis. Med Chem. 2014;10(2):211–219. | ||
Buckle DR, Outred DJ, Rockell CJ, Smith H, Spicer BA. Studies on v-triazoles. 7. Antiallergic 9-oxo-1H,9H-benzopyrano[2,3-d]-v-triazoles. J Med Chem. 1983;26(2):251–254. | ||
Głowacka IE, Balzarini J, Wróblewski AE. The synthesis, antiviral, cytostatic and cytotoxic evaluation of a new series of acyclonucleotide analogues with a 1,2,3-triazole linker. Eur J Med Chem. 2013;70:703–722. | ||
Dabiri M, Salehi P, Bahramnejad M, Koohshari M, Aliahmadi A. One-pot synthesis of (1,2,3-triazolyl)methyl 3,4-dihydro-2-oxo-1H-pyrimidine-5-carboxylates as potentially active antimicrobial agents. Helv Chim Acta. 2014;97(3):375–383. | ||
Patpi SR, Pulipati L, Yogeeswari P, et al. Design, snthesis, and structure–activity correlations of novel Dibenzo[b,d]furan, Dibenzo[b,d]thiophene, and N-Methylcarbazole clubbed 1,2,3-triazoles as potent inhibitors of mycobacterium tuberculosis. J Med Chem. 2012;55(8):3911–3922. | ||
Giffin MJ, Heaslet H, Brik A, et al. A copper(I)-catalyzed 1,2,3-triazole azide-alkyne click compound is a potent inhibitor of a multidrug-resistant HIV-1 protease variant. J Med Chem. 2008;51(20):6263–6270. | ||
Zou Y, Zhao Q, Liao J, et al. New triazole derivatives as antifungal agents: synthesis via click reaction, in vitro evaluation and molecular docking studies. Bioorg Med Chem Lett. 2012;22(8):2959–2962. | ||
Billing JF, Nilsson UJ. C2-symmetric macrocyclic carbohydrate/amino acid hybrids through copper(I)-catalyzed formation of 1,2,3-triazoles. J Org Chem. 2005;70(12):4847–4850. | ||
Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J Am Chem Soc. 2003;125(16):4686–4687. | ||
Collman JP, Devaraj NK, Chidsey CED. “Clicking” functionality onto electrode surfaces. Langmuir. 2004;20(4):1051–1053. | ||
Löber S, Rodriguez-Loaiza P, Gmeiner P. Click linker: efficient and high-yielding synthesis of a new family of SPOS resins by 1,3-dipolar cycloaddition. Org Lett. 2003;5(10):1753–1755. | ||
Genin MJ, Allwine DA, Anderson DJ, et al. Substituent effects on the antibacterial activity of nitrogen–carbon-linked (azolylphenyl)oxazolidinones with expanded activity against the fastidious gram-negative organisms haemophilus influenzae and moraxella catarrhalis. J Med Chem. 2000;43(5):953–970. | ||
Thibault RJ, Takizawa K, Lowenheilm P, et al. A versatile new monomer family: functionalized 4-vinyl-1,2,3-triazoles via click chemistry. J Am Chem Soc. 2006;128(37):12084–12085. | ||
Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40(11):2004–2021. | ||
Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discov Today. 2003;8(24):1128–1137. | ||
Claes P, Cappoen D, Uythethofken C, et al. 2,4-Dialkyl-8,9,10,11-tetrahydrobenzo[g]pyrimido[4,5-c]isoquinoline-1,3,7,12(2H,4H)-tetraones as new leads against Mycobacterium tuberculosis. Eur J Med Chem. 2014;77:409–421. | ||
Cappoen D, Claes P, Jacobs J, et al. 1,2,3,4,8,9,10,11-Octahydrobenzo[j]phenanthridine-7,12-diones as new leads against Mycobacterium tuberculosis. J Med Chem. 2014;57(7):2895–2907. | ||
Claes P, Cappoen D, Mbala BM, et al. Synthesis and antimycobacterial activity of analogues of the bioactive natural products sampangine and cleistopholine. E J Med Chem. 2013;67:98–110. | ||
Smits R, Cadicamo CD, Burger K, Koksch B. Synthetic strategies to alpha-trifluoromethyl and alpha-difluoromethyl substituted alpha-amino acids. Chem Soc Rev. 2008;37(8):1727–1739. | ||
Boechat N, Ferreira VF, Ferreira SB, et al. Novel 1,2,3-triazole derivatives for use against Mycobacterium tuberculosis H37Rv (ATCC 27294) strain. J Med Chem. 2011;54(17):5988–5999. | ||
Leeson P. Drug discovery: chemical beauty contest. Nature. 2012;481(7382):455–456. | ||
van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol. 2011;731:237–245. | ||
Venugopala KN, Krishnappa M, Nayak SK, et al. Synthesis and antimosquito properties of 2,6-substituted benzo[d]thiazole and 2,4-substituted benzo[d]thiazole analogues against Anopheles arabiensis. Eur J Med Chem. 2013;65:295–303. | ||
Venugopala KN, Nayak SK, Gleiser RM, Sanchez-Borzone ME, Garcia DA, Odhav B. Synthesis, polymorphism, and insecticidal activity of methyl 4-(4-chlorophenyl)-8-iodo-2-methyl-6-oxo-1,6-dihydro-4H-pyrimido[2,1-b]quinazoline-3-carboxylate against Anopheles arabiensis mosquito. Chem Biol Drug Des. Epub 2016 Feb 3. | ||
Nayak SK, Venugopala KN, Chopra D, Row TNG. Insights into conformational and packing features in a series of aryl substituted ethyl-6-methyl-4-phenyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates. CrystEngComm. 2011;13(2):591–605. | ||
Panini P, Venugopala KN, Odhav B, Chopra D. Polymorphism in two biologically active dihydropyrimidinium hydrochloride derivatives: quantitative inputs towards the energetics associated with crystal packing. Acta Crystallogr Sect B. 2014;70(4):681–696. | ||
Panini P, Venugopala KN, Odhav B, Chopra D. Quantitative analysis of intermolecular interactions in 7-hydroxy-4-methyl-2H-chromen-2-one and its hydrate. Proc Natl Acad Sci India Sect A Phy Sci. 2014;84(2):281–295. | ||
Puripat M, Ramozzi R, Hatanaka M, Parasuk W, Parasuk V, Morokuma K. The biginelli reaction is a urea-catalyzed organocatalytic multicomponent reaction. J Org Chem. 2015;80(14):6959–6967. | ||
Apex2, Version 2 User Manual, M86-E01078, Bruker analytical X-ray systems Madison, WI; 2006. | ||
Siemens, SMART System, Siemens Analytical X-ray Instruments Inc. Madison, MI; 1995. | ||
Burla MC, Caliandro R, Carrozzini B, et al. Crystal structure determination and refinement via SIR2014. J Appl Cryst. 2015;48(1):306–309. | ||
Sheldrick G. A short history of SHELX. Acta Cryst Sect A. 2008;64(1):112–122. | ||
Farrugia L. WinGX suite for small-molecule single-crystal crystallography. J Appl Cryst. 1999;32(4):837–838. | ||
Sheldrick GM. SADABS. Madison, WI: Bruker AXS, Inc.; 2007. | ||
Macrae CF, Bruno IJ, Chisholm JA, et al. Mercury CSD 2.0 – new features for the visualization and investigation of crystal structures. J Appl Cryst. 2008;41(2):466–470. | ||
Nardelli M. PARST95 – an update to PARST: a system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses. J Appl Cryst. 1995;28(5):659. | ||
Spek A. Single-crystal structure validation with the program PLATON. J Appl Cryst. 2003;36(1):7–13. | ||
Mossman T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. | ||
Martin A, Morcillo N, Lemus D, et al. Multicenter study of MTT and resazurin assays for testing susceptibility to first-line anti-tuberculosis drugs. Int J Tuberc Lung Dis. 2005;9(8):901–906. | ||
Venugopala KN, Albericio F, Coovadia YM, et al. Total synthesis of a depsidomycin analogue by convergent solid-phase peptide synthesis and macrolactonization strategy for antitubercular activity. J Pep Sci. 2011;17(10):683–689. | ||
Rao GBD, Anjaneyulu B, Kaushik MP. Greener and expeditious one-pot synthesis of dihydropyrimidinone derivatives using non-commercial [small beta]-ketoesters via the Biginelli reaction. RSC Adv. 2014;4(82):43321–43325. | ||
Kutonova KV, Trusova ME, Postnikov PS, Filimonov VD, Parello J. A simple and effective synthesis of aryl azides via arenediazonium tosylates. Synthesis. 2013;45(19):2706–2710. | ||
Chopra D. Is organic fluorine really “not” polarizable? Cryst Growth Des. 2012;12(2):541–546. | ||
Panini P, Chopra D. In hydrogen bonded supramolecular structures. In: Li Z, Wu L, editors. Lecture Notes in Chemistry Berlin, Heidelberg: Springer-Verlag; 2015;87:37–67. | ||
Dey D, Thomas SP, Spackman MA, Chopra D. ‘Quasi-isostructural polymorphism’ in molecular crystals: inputs from interaction hierarchy and energy frameworks. Chem Comm. 2016;52(10):2141–2144. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.