")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Design, Synthesis and Biological Evaluation of New 3,4-Dihydro-2(1H)-Quinolinone-Dithiocarbamate Derivatives as Multifunctional Agents for the Treatment of Alzheimer’s Disease
Authors Guo J, Xu A, Cheng M, Wan Y, Wang R, Fang Y, Jin Y, Xie SS, Liu J
Received 6 January 2022
Accepted for publication 10 May 2022
Published 18 May 2022 Volume 2022:16 Pages 1495—1514
DOI https://doi.org/10.2147/DDDT.S354879
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Jie Guo,1,* Airen Xu,2,* Maojun Cheng,1 Yang Wan,1 Rikang Wang,1 Yuanying Fang,1 Yi Jin,1 Sai-Sai Xie,1 Jing Liu3
1National Pharmaceutical Engineering Center for Solid Preparation in Chinese Herbal Medicine, Jiangxi University of Chinese Medicine, Nanchang, Jiangxi, People’s Republic of China; 2Clinical Pharmacology Research Center, The Second Hospital of Yinzhou, Ningbo, Zhejiang, People’s Republic of China; 3School of Pharmacy, Jiangxi University of Chinese Medicine, Nanchang, Jiangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Sai-Sai Xie, National Pharmaceutical Engineering Center for Solid Preparation in Chinese Herbal Medicine, Jiangxi University of Chinese Medicine, No. 56, Yangming Road, Donghu District, Nanchang City, Jiangxi Province, 330006, People’s Republic of China, Email [email protected] Jing Liu, School of Pharmacy, Jiangxi University of Chinese Medicine, No. 56, Yangming Road, Donghu District, Nanchang City, Jiangxi Province, 330006, People’s Republic of China, Email [email protected]
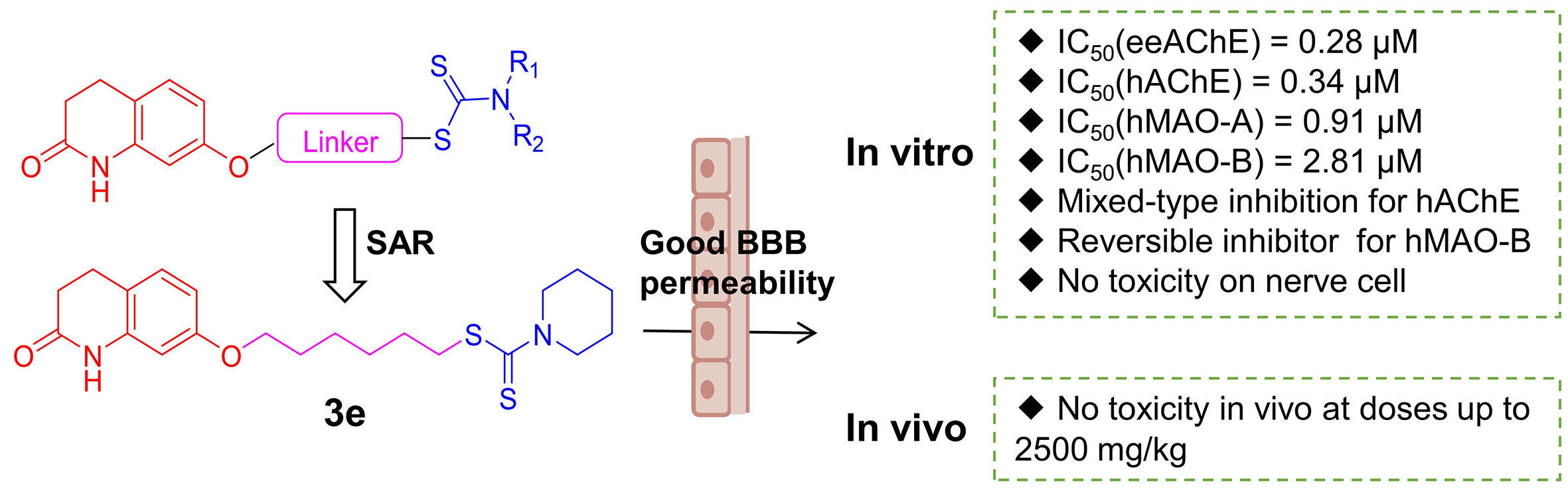
Background: Alzheimer’s disease (AD) belongs to neurodegenerative disease, and the increasing number of AD patients has placed a heavy burden on society, which needs to be addressed urgently. ChEs/MAOs dual-target inhibitor has potential to treat AD according to reports.
Purpose: To obtain effective multi-targeted agents for the treatment of AD, a novel series of hybrid compounds were designed and synthesized by fusing the pharmacophoric features of 3,4-dihydro-2 (1H)-quinolinone and dithiocarbamate.
Methods: All compounds were evaluated for their inhibitory abilities of ChEs and MAOs. Then, further biological activities of the most promising candidate 3e were determined, including the ability to cross the blood-brain barrier (BBB), kinetics and molecular model analysis, cytotoxicity in vitro and acute toxicity studies in vivo.
Results: Most compounds showed potent and clear inhibition to AChE and MAOs. Among them, compound 3e was considered to be the most effective and balanced inhibitor to both AChE and MAOs (IC50=0.28 μM to eeAChE; IC50=0.34 μM to hAChE; IC50=2.81 μM to hMAO-B; IC50=0.91 μM to hMAO-A). In addition, 3e showed mixed inhibition of hAChE and competitive inhibition of hMAO-B in the enzyme kinetic studies. Further studies indicated that 3e could penetrate the BBB and showed no toxicity on PC12 cells and HT-22 cells when the concentration of 3e was lower than 12.5 μM. More importantly, 3e lacked acute toxicity in mice even at high dose (2500 mg/kg, P.O.).
Conclusion: This work indicated that compound 3e with a six-carbon atom linker and a piperidine moiety at terminal position was a promising candidate and was worthy of further study.
Keywords: Alzheimer’s disease, cholinesterase, monoamine oxidase, 3,4-dihydro-2(1H)-quinolinone, dithiocarbamate
Graphical Abstract:
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disease that is characterized by memory loss, decreased language ability, and cognitive impairment.1 According to the World Alzheimer Report, nearly 47 million people worldwide are now suffering from AD, and this number of AD patients is expected to triple by 2050.2 Therefore, AD has become a serious public health problem, which places a heavy burden on the public health system in the world and needs to be solved urgently.3
For many years, a lot of drug researchers and institutions have been committed to the development of new drugs, but most of them end in failure.4 Due to the insidious onset and unclarified etiopathogenesis of AD, many factors and targets are related to the occurrence and development of AD, such as acetylcholine (ACh) deficiency, Aβ aggregation, metabolic homeostasis disruption of bio-metals, oxidative stress, tau protein hyperphosphorylation, etc.5,6 At present, the clinical treatment of AD mainly focuses on the defects of acetylcholine (ACh).7 There are three AChE inhibitors of the four clinical drugs (Donepezil, Galantamine, Rivastigmine and Memantine) approved by the US Food and Drug Administration (FDA) for the treatment of AD.8 Many related studies have shown that decline of ACh level in brain of AD patients can lead to memory deficits and cognitive impairment, so reducing ACh metabolism may alleviate these terrible symptoms.9,10 In the central nervous system (CNS), there are two types of cholinesterases (ChEs), including acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). In contrast to BuChE, the AChE accounts for about 80% of the hydrolysis of ACh in the normal brains. Thus, AChE has received considerable attention over the past few decades.11 The crystallographic structure of AChE shows that the enzyme has a deep and narrow gorge which includes two binding active sites: a catalytic active site (CAS) at the bottom of the gorge and a peripheral anion site (PAS) at the entrance of the gorge. A good AChE inhibitor should have a core ring system that binds to PAS and a basic center that interacts with CAS, as well as a linking structure that can connect the two parts.12–15
Apart from AChE, monoamine oxidase (MAOs) is also an effective therapeutic target for AD. MAOs is an enzyme containing flavin adenine dinucleotide (FAD), which is responsible for the oxidative deamination of various biogenic and xenobiotic amines.16 MAOs is widely distributed in the human body, especially in tissue cells such as liver, brain and kidney. MAOs exists as two distinct enzymatic isoforms, namely MAO-A and MAO-B.17 MAO-A focuses on the decomposition and deamination of polar aromatic amines, such as serotonin (5-HT), norepinephrine and epinephrine, while MAO-B prefers non-polar amines. Selective MAO-A inhibitors can be used to treat depression. Meanwhile, selective MAO-B inhibitors are usually used to treat Parkinson’s disease.18 According to research, it has been found that the level of MAO-B has been increased in the brains of AD patients, which increases the metabolism of amines and generates a large amount of hydrogen peroxide to induce oxidative stress that damages the nerve cells, so inhibiting the activity of MAO-B may also play an important role in the treatment of AD.19 In addition, the researchers find that AD patients usually experience depressive symptoms during the onset of the disease. Based on this, it is believed that elevated MAO-A level is also a risk factor for the development of AD.20 Therefore, dual inhibition of MAO-A and MAO-B, rather than MAO-B alone, may be more valuable for the treatment of AD.
Due to the complex pathogenesis of AD, its occurrence and development involve multiple targets and molecular signaling pathways.21 More and more studies have proved that the “one molecule, one target” approach cannot achieve satisfactory therapeutic effects, although these single-target drugs can temporarily delay the progression of patients’ cognitive decline. Currently, two approaches are commonly used for multi-target therapeutics. The first approach is to combine drugs with one target or a single active ingredient, respectively, while the second approach is multi-target directed ligands (MTDLs).22 The former approach can provide better dosing flexibility and lower treatment costs by directly adjusting the ratio of the mixed drugs. However, drug combinations often suffer from more side effects, such as dose-limiting toxicities, drug–drug interactions and poor patient compliance. MTDLs strategy means that one drug can act on multiple targets at the same time and participate in the regulation of different mechanisms.23 Compared with a mixture, MTDLs increase therapeutic efficacy through low-dose synergy and reduce adverse effects, resulting in a larger therapeutic window.
Our group has made great efforts to find potential MTDLs targeting AChE and MAOs. As shown in Figure 1, in the development of MAOs inhibitors for the treatment of neurodegenerative diseases, 3,4-dihydro-2(1H)-quinolinone derivatives are obvious MAOs inhibitors, among which C-7 substituted compound 49 possesses the best inhibitory on MAO-B with IC50 values of 0.0029 μM according to reports.24 The 3,4-dihydro-2(1H)-quinolinone core (marked in the red color as shown in Figure 1) is the main structure of 49, which can occupy the catalytic site of MAO-B to achieve the effect of inhibiting MAO-B. Moreover, the 3,4-dihydro-2(1H)-quinolinone core can also act on the PAS site of AChE. On the other hand, the structure of dithiocarbamate (marked in the blue color as shown in Figure 1) is a multifunctional pharmacophore with a wide range of biological activities, such as antioxidant, antiviral, antibacterial and antitumor, etc. However, it is rarely reported to have activities related to neurodegenerative diseases. Our group found that the structure of dithiocarbamate can interact with CAS site of AChE to achieve the purpose of inhibiting AChE.25 Therefore, in this work, we design a new series of 3,4-dihydro-2(1H)-quinolinone-dithiocarbamate derivatives for the treatment of AD. The designed compounds were synthesized and evaluated for their biological activities including the inhibitory abilities of ChEs and MAOs, the ability to cross the blood-brain barrier (BBB), kinetics and molecular model analysis, cytotoxicity in vitro and acute toxicity studies in vivo.
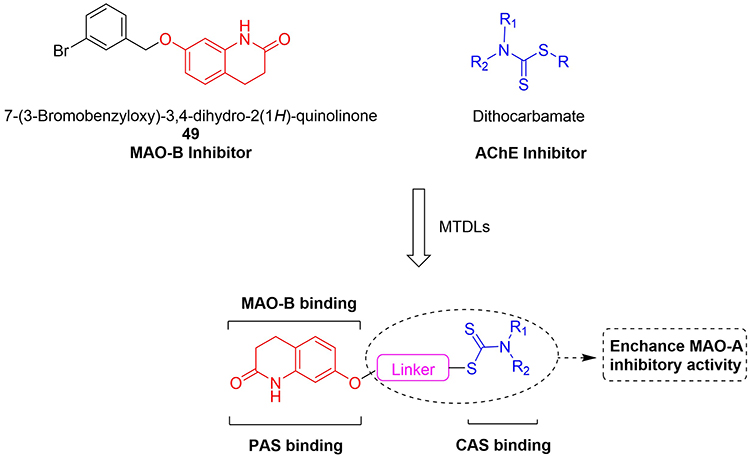 |
Figure 1 Design strategy of 3,4-dihydro-2(1H)-quinolinone-dithiocarbamate derivatives. |
Design Strategy
Like our previous design, we attempted to connect the 3,4-dihydro-2(1H)-quinolinone core with dithiocarbamate moiety through different lengths of the carbon chain linker to design novel multifunctional compounds for AD treatment (Figure 1). 3,4-dihydro-2(1H)-quinolinone core was chosen to inhibit the MAO-B and interact with the PAS of AChE. The dithiocarbamate moiety was utilized to bind to the CAS of AChE. At the same time, the change of the alkylamine side chain might increase the inhibitory activities of MAO-A.26 In the initial step, compounds with different linker lengths were first synthesized to obtain the optimal linker length for balanced inhibition of AChE and MAO-B. Once the optimal linker length was determined, various secondary amine groups were introduced into the dithiocarbamate moiety for SAR studies.
Materials and Methods
Chemistry
All chemical reagents and solvents used in the experiment were purchased from commercial suppliers and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) on silica gel GF254 (Qingdao Haiyang Chemical Plant, Qingdao, China), and then visualized with a UV lamp (254 nm). Column chromatography was performed using silica gel (100–200 mesh; Qingdao Haiyang Chemical Plant, Qingdao, China). Melting points were measured on an XT-4 micromelting point apparatus and uncorrected. 1H NMR spectra (600 MHz) and 13C NMR spectra (151 MHz) were recorded on a Bruker ACF-600 spectrometer at 25°C using the TMS as internal standard and CDCl3 or DMSO-d6 as the solvent. The purity of these compounds was determined by analytical high performance liquid chromatography (HPLC) performed on a Waters ACQUITY Arc HPLC system equipped with a 2998 PDA detector (Column: Agilent XDB-C18, 5 mm particle size, 4.6 mm×250 mm; mobile phase: A=CH3OH, B=H2O (0.1% acetic acid); isocratic elution, A = 80%, B = 20%; flow rate = 1 mL/min; λ=254 nm; 10μL injection). High-resolution mass spectra were conducted on an AB Sciex Triple TOF 5600 spectrometer (HR-ESI-MS) and the error analysis of the HRMS results was summarized in Table S1 (Table S1 in the Supporting Information).
General Procedures for the Preparation of Compounds 2a-i
Compound 1 (0.2 g, 1.23 mmol) and anhydrous K2CO3 (2eq, 0.34 g, 2.45 mmol) were suspended in acetone (20 mL). The suitable α, ω-dibromoalkanes (30eq, 6.91 g, 36.77 mmol) were added, and the mixture was stirred under reflux for 6 h. After cooling, the reaction mixture was filtered, and the filtrate was evaporated under reduced pressure. The obtained residue was purified by silica gel chromatography with PE/EA (1:1 to 3:1) as eluent to give compounds 2a-i as white solid.
7-(2-bromoethoxy)-3,4-dihydroquinolin-2(1H)-one (2a). Yield 65%; white solid; mp 143.7–145.9°C; 1H NMR (600 MHz, CDCl3) δ 8.34 (s, 1H), 7.08 (d, J=8.3 Hz, 1H), 6.56 (dd, J=8.3, 2.4 Hz, 1H), 6.39 (d, J=2.4 Hz, 1H), 4.29 (t, J=6.2 Hz, 2H), 3.64 (t, J=6.2 Hz, 2H), 2.93 (t, 2H), 2.65 (t, 2H) (Figure S1 in the Supporting Information).
7-(3-bromopropoxy)-3,4-dihydroquinolin-2(1H)-one (2b). Yield 82%; white solid; mp 110.8–113.8°C; 1H NMR (600 MHz, CDCl3) δ 8.28 (s, 1H), 7.08 (d, J=8.3 Hz, 1H), 6.56 (dd, J=8.3, 2.4 Hz, 1H), 6.37 (d, J=2.4 Hz, 1H), 4.10 (t, J=5.8 Hz, 2H), 3.62 (t, J=6.4 Hz, 2H), 2.92 (t, 2H), 2.65 (t, 2H), 2.37–2.25 (m, J=6.1 Hz, 2H) (Figure S2 in the Supporting Information).
7-(4-bromobutoxy)-3,4-dihydroquinolin-2(1H)-one (2c). Yield 85%; white solid; mp 107.2–110.1°C; 1H NMR (600 MHz, CDCl3) δ 8.20 (s, 1H), 7.07 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.35 (d, J=2.4 Hz, 1H), 3.99 (t, J=6.1 Hz, 2H), 3.51 (t, J=6.6 Hz, 2H), 2.92 (t, 2H), 2.64 (t, 2H), 2.15–2.02 (m, 2H), 2.03–1.90 (m, 2H) (Figure S3 in the Supporting Information).
7-((5-bromopentyl) oxy)-3,4-dihydroquinolin-2(1H)-one (2d). Yield 71%; white solid; mp 122.4–124.1°C; 1H NMR (600 MHz, CDCl3) δ 8.26 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.35 (d, J=2.0 Hz, 1H), 3.96 (t, J=6.3 Hz, 2H), 3.46 (t, J=6.8 Hz, 2H), 2.92 (t, 2H), 2.64 (t, 2H), 1.99–1.90 (m, 2H), 1.88–1.77 (m, 2H), 1.66–1.61 (m, 2H) (Figure S4 in the Supporting Information).
7-((6-bromohexyl) oxy)-3,4-dihydroquinolin-2(1H)-one (2e). Yield 89%; white solid; mp 83.5–86.2°C; 1H NMR (600 MHz, CDCl3) δ 8.17 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.35 (d, J=2.3 Hz, 1H), 3.95 (t, J=6.4 Hz, 2H), 3.45 (t, J=6.8 Hz, 2H), 2.92 (t, 2H), 2.69–2.59 (m, 2H), 1.96–1.87 (m, 2H), 1.85–1.76 (m, 2H), 1.59–1.46 (m, 4H) (Figure S5 in the Supporting Information).
7-((7-bromoheptyl) oxy)-3,4-dihydroquinolin-2(1H)-one (2f). Yield 88%; white solid; mp 83.9–84.9°C; 1H NMR (600 MHz, CDCl3) δ 8.19 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.35 (d, J=2.3 Hz, 1H), 3.94 (t, J=6.4 Hz, 2H), 3.44 (t, J=6.8 Hz, 2H), 2.92 (t, J=7.5 Hz, 2H), 2.66–2.62 (m, 2H), 1.95–1.85 (m, 2H), 1.84–1.74 (m, 2H), 1.49 (m, J=15.0, 7.3 Hz, 4H), 1.42–1.36 (m, 2H) (Figure S6 in the Supporting Information).
7-((8-bromooctyl) oxy)-3,4-dihydroquinolin-2(1H)-one (2g). Yield 75%; white solid; mp 82.2–84.3°C; 1H NMR (600 MHz, CDCl3) δ 8.10 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.34 (d, J=2.4 Hz, 1H), 3.94 (t, J=6.5 Hz, 2H), 3.43 (t, J=6.8 Hz, 2H), 2.92 (t, 2H), 2.64 (t, 2H), 1.92–1.83 (m, 2H), 1.83–1.74 (m, 2H), 1.52–1.42 (m, 4H), 1.43–1.31 (m, 4H) (Figure S7 in the Supporting Information).
7-((9-bromononyl) oxy)-3,4-dihydroquinolin-2(1H)-one (2h). Yield 78%; white solid; mp 83.1–84.6°C; 1H NMR (600 MHz, CDCl3) δ 8.02 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.34 (d, J=2.3 Hz, 1H), 3.94 (t, J=6.5 Hz, 2H), 3.43 (t, J=6.8 Hz, 2H), 2.92 (t, 2H), 2.64 (t, 2H), 1.92–1.84 (m, 2H), 1.82–1.74 (m, 2H), 1.50–1.42 (m, 4H), 1.41–1.33 (m, 6H) (Figure S8 in the Supporting Information).
7-((10-bromodecyl) oxy)-3,4-dihydroquinolin-2(1H)-one (2i). Yield 71%; white solid; mp 84.7–86.1°C; 1H NMR (600 MHz, CDCl3) δ 7.90 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.33 (d, J=2.3 Hz, 1H), 3.94 (t, J=6.5 Hz, 2H), 3.43 (t, J=6.9 Hz, 2H), 2.92 (t, 2H), 2.64 (t, 2H), 1.92–1.84 (m, 2H), 1.83–1.72 (m, 2H), 1.50–1.40 (m, 4H), 1.35 (dd, J=19.2, 8.2 Hz, 8H) (Figure S9 in the Supporting Information).
General Procedure for the Preparation of Compounds 3a-i and 3e1-e11
To a solution of appropriate secondary amine (2.2eq, 0.14 g, 1.63 mmol) and TEA (2eq, 0.15 g, 1.48 mmol) in DMF (2 mL) was added CS2 (2.4eq, 0.14 g 1.78 mmol) dropwise. After stirring for 5 min, another solution containing intermediate compounds 2a-i (0.2g, 0.74 mmol) in DMF (8 mL) was added, and the mixture was stirred at 25°C for 12 h. After completion, the mixture was diluted with 20 mL of water, and extracted with ethyl acetate. The combined organic phase was washed with water (20 mL×3), dried over anhydrous Na2SO4, concentrated under vacuum, and purified by silica gel chromatography with PE/EA (1:2 to 4:1) as eluent to give the desired compounds 3a-i and 3e1-e11.
2-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) ethyl piperidine-1-carbodithioate (3a). Yield 85%; white solid; mp 170.5–172.8°C; 1H NMR (600 MHz, CDCl3) δ 7.73 (s, 1H), 7.07 (d, J=8.3 Hz, 1H), 6.60 (dd, J=8.3, 2.5 Hz, 1H), 6.41 (d, J=2.4 Hz, 1H), 4.25 (br s, 2H), 4.24 (d, J=6.3 Hz, 2H), 3.93 (br s, 2H), 3.76 (t, J=6.4 Hz, 2H), 2.90 (t, J=7.2 Hz, 2H), 2.64 (t, J=6.6 Hz, 2H), 1.92–1.51 (m, 6H) (Figure S10 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 194.72, 171.45, 158.13, 138.07, 128.75, 116.14, 108.88, 102.37, 66.78, 53.25, 51.49, 35.78, 31.08 (2C), 24.61 (2C), 24.27 (Figure S11 in the Supporting Information). HRMS: calcd for [M+H]+ 351.1196, found 351.1196 (Figure S12 in the Supporting Information). HPLC purity 99.28% (Figure S13 in the Supporting Information).
3-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) propyl piperidine-1-carbodithioate (3b). Yield 87%; white solid; mp 127.7–128.6°C; 1H NMR (600 MHz, CDCl3) δ 7.83 (s, 1H), 7.08 (d, J=7.8 Hz, 1H), 6.56 (dd, J=8.3, 2.4 Hz, 1H), 6.34 (d, J=2.4 Hz, 1H), 4.32 (br s, 2H), 4.06 (t, J=6.1 Hz, 2H), 3.91 (br s, 2H), 3.51 (t, J=7.1 Hz, 2H), 3.05–2.81 (m, 2H), 2.73–2.57 (m, 2H), 2.32–2.11 (m, 2H), 1.79–1.65 (m, 6H) (Figure S14 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 195.38, 171.56, 158.48, 138.08, 128.73, 115.90, 108.72, 102.20, 66.65, 52.99, 51.29, 33.46, 31.10, 28.68 (2C), 24.61 (2C), 24.32 (Figure S15 in the Supporting Information). HRMS: calcd for [M+H]+ 365.1352, found 365.1348 (Figure S16 in the Supporting Information). HPLC purity 99.69% (Figure S17 in the Supporting Information).
4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) butyl piperidine-1-carbodithioate (3c). Yield 90%; white solid; mp 115.5–116.1°C; 1H NMR (600 MHz, CDCl3) δ 8.00 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.34 (d, J=2.4 Hz, 1H), 4.32 (br s, 2H), 4.10–3.86 (m, 2H), 3.92 (br s, 2H), 3.47–3.30 (m, 2H), 2.97–2.83 (m, 2H), 2.64 (t, J=7.8 Hz, 2H), 1.93–1.91 (m, 4H), 1.81–1.47 (m, 6H) (Figure S18 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 195.69, 171.67, 158.60, 138.08, 128.69, 115.77, 108.69, 102.23, 67.59, 52.93, 51.33, 36.75, 31.11, 28.46, 25.53 (2C), 24.60 (2C), 24.34 (Figure S19 in the Supporting Information). HRMS: calcd for [M+H]+ 379.1509, found 379.1497 (Figure S20 in the Supporting Information). HPLC purity 99.65% (Figure S21 in the Supporting Information).
5-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) pentyl piperidine-1-carbodithioate (3d). Yield 91%; white solid; mp 109.7–110.4°C; 1H NMR (600 MHz, CDCl3) δ 7.74 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.33 (d, J=2.4 Hz, 1H), 4.32 (br s, 2H), 3.96 (t, J=6.4 Hz, 2H), 3.92 (br s, 2H), 3.36 (t, J=7.2 Hz, 2H), 2.91 (t, J=6.0 Hz, 2H), 2.64 (t, J=6.6 Hz, 2H), 2.13–1.02 (m, 12H) (Figure S22 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 195.87, 171.49, 158.69, 138.02, 128.72, 115.72, 108.72, 102.16, 67.93, 52.81, 51.21, 36.98, 31.12, 28.77, 28.57, 25.47 (2C), 24.60 (2C), 24.35 (Figure S23 in the Supporting Information). HRMS: calcd for [M+H]+ 393.1665, found 393.1659 (Figure S24 in the Supporting Information). HPLC purity 99.79% (Figure S25 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl piperidine-1-carbodithioate (3e). Yield 89%; white solid; mp 110.7–113.2°C; 1H NMR (600 MHz, CDCl3) δ 8.00 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (dd, J=8.3, 2.4 Hz, 1H), 6.34 (d, J=2.4 Hz, 1H), 4.32 (br s, 2H), 3.94 (t, J=6.5 Hz, 2H), 3.91 (br s, 2H), 3.33 (t, J=7.8 Hz, 2H), 2.91 (t, J=7.8 Hz, 2H), 2.64 (t, J=7.8 Hz, 2H), 1.89–1.61 (m, 14H) (Figure S26 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 195.98, 171.67, 158.71, 138.08, 128.67, 115.65, 108.73, 102.16, 68.04, 52.73, 51.18, 37.09, 31.13, 29.03, 28.70, 28.68, 25.60 (2C), 24.60 (2C), 24.35 (Figure S27 in the Supporting Information). HRMS: calcd for [M+H]+ 407.1822, found 407.1821 (Figure S28 in the Supporting Information). HPLC purity 99.98% (Figure S29 in the Supporting Information).
7-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) heptyl piperidine-1-carbodithioate (3f). Yield 87%; white solid; mp 116.6–118.7°C; 1H NMR (600 MHz, CDCl3) δ 8.07 (s, 1H), 7.06 (d, J=8.1 Hz, 1H), 6.54 (d, J=8.2 Hz, 1H), 6.35 (s, 1H), 4.31 (s, 2H), 3.94 (t, J=5.9 Hz, 2H), 3.92 (s, 2H), 3.32 (t, J=7.2 Hz, 2H), 2.91 (t, J=7.2 Hz, 2H), 2.63 (t, J=7.2 Hz, 2H), 1.81–1.76 (m, 2H), 1.71 (d, J=15.6 Hz, 8H), 1.45 (dd, J=29.2, 6.5 Hz, 6H) (Figure S30 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.05, 171.79, 158.73, 138.09, 128.63, 115.61, 108.71, 102.22, 68.14, 52.75, 51.19, 37.20, 31.12, 29.12, 28.89, 28.82, 28.65, 25.99, 25.89, 25.46, 24.60, 24.35 (Figure S31 in the Supporting Information). HRMS: calcd for [M+H]+ 421.1978, found 421.1980 (Figure S32 in the Supporting Information). HPLC purity 98.54% (Figure S33 in the Supporting Information).
8-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) octyl piperidine-1-carbodithioate (3g). Yield 70%; white solid; mp 86.9–89.1°C; 1H NMR (600 MHz, CDCl3) δ 8.12 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (d, J=8.3 Hz, 1H), 6.35 (s, 1H), 4.31 (s, 2H), 3.94 (d, J=6.4 Hz, 2H), 3.92 (s, 2H), 3.31 (t, J=7.4 Hz, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 1.82–1.76 (m, J=14.1, 6.9 Hz, 2H), 1.75–1.69 (m, 8H), 1.49–1.36 (m, 8H) (Figure S34 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.07, 171.75, 158.74, 138.06, 128.64, 115.60, 108.71, 102.18, 68.16, 52.78, 51.22, 37.25, 31.12, 29.23, 29.17, 29.09, 28.94, 28.68, 26.04, 25.95, 25.43, 24.59, 24.35 (Figure S35 in the Supporting Information). HRMS: calcd for [M+H]+ 435.2135, found 435.2140 (Figure S36 in the Supporting Information). HPLC purity 96.07% (Figure S37 in the Supporting Information).
9-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) nonyl piperidine-1-carbodithioate (3h). Yield 74%; white solid; mp 100.6–102.7°C; 1H NMR (600 MHz, CDCl3) δ 7.96 (s, 1H), 7.06 (d, J=8.1 Hz, 1H), 6.54 (d, J=8.2 Hz, 1H), 6.34 (s, 1H), 4.31 (s, 2H), 3.94 (d, J=6.0 Hz, 2H), 3.90 (s, 2H), 3.31 (t, J=7.3 Hz, 2H), 2.92 (s, J=8.7 Hz, 2H), 2.63 (t, J=7.2 Hz, 2H), 1.81–1.75 (m, 2H), 1.70 (d, J=6.0 Hz, 8H), 1.44 (s, J=56.0 Hz, 10H) (Figure S38 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.12, 171.72, 158.76, 138.07, 128.63, 115.60, 108.69, 102.20, 68.18, 52.73, 51.19, 37.29, 31.12, 29.36, 29.25, 29.19, 29.09, 29.00, 28.69, 26.09, 25.96, 25.46, 24.60, 24.35 (Figure S39 in the Supporting Information). HRMS: calcd for [M+H]+ 449.2291, found 449.2292 (Figure S40 in the Supporting Information). HPLC purity 99.48% (Figure S41 in the Supporting Information).
10-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) decyl piperidine-1-carbodithioate (3i). Yield 80%; white solid; mp 80.4–83.8°C; 1H NMR (600 MHz, CDCl3) δ 7.96 (s, 1H), 7.06 (d, J=8.2 Hz, 1H), 6.54 (d, J=8.2 Hz, 1H), 6.34 (s, 1H), 4.31 (s, 2H), 3.94 (d, J=6.1 Hz, 2H), 3.89 (s, 2H), 3.31 (t, J=7.2 Hz, 2H), 2.91 (t, J=7.3 Hz, 2H), 2.63 (t, J=7.2 Hz, 2H), 1.81–1.76 (m, 2H), 1.75–1.70 (m, J=6.5 Hz, 8H), 1.47–1.32 (m, 12H) (Figure S42 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.14, 171.70, 158.77, 138.06, 128.64, 115.59, 108.71, 102.17, 68.20, 52.72, 51.23, 37.31, 31.12, 29.46, 29.39, 29.31, 29.21, 29.16, 29.03, 28.69, 26.10, 26.00, 25.45, 24.60, 24.35 (Figure S43 in the Supporting Information). HRMS: calcd for [M+H]+ 463.2448, found 463.2450 (Figure S44 in the Supporting Information). HPLC purity 99% (Figure S45 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl dimethyl-carbamodithioate (3e1). Yield 85%; white solid; mp 115.9–116.9°C; 1H NMR (600 MHz, CDCl3) δ 8.22 (s, 1H), 7.05 (d, J=8.3 Hz, 1H), 6.53 (d, J=8.2 Hz, 1H), 6.36 (s, 1H), 3.94 (t, J=6.4 Hz, 2H), 3.57 (s, 3H), 3.38 (s, 3H), 3.31 (t, J=7.4 Hz, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 1.83–1.77 (m, 2H), 1.75 (dd, J=14.2, 7.2 Hz, 2H), 1.55–1.48 (m, 4H) (Figure S46 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 197.56, 171.80, 158.69, 138.09, 128.64, 115.63, 108.75, 102.18, 68.02, 45.29, 41.47, 37.51, 31.11, 29.03, 28.66, 28.61, 25.59, 24.59 (Figure S47 in the Supporting Information). HRMS: calcd for [M+H]+ 367.1509, found 367.1506 (Figure S48 in the Supporting Information). HPLC purity 99.16% (Figure S49 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexylethyl (methyl)- carbamodithioate (3e2). Yield 77%; white solid; mp 90.1–92.8°C; 1H NMR (600 MHz, CDCl3) δ 8.13 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.54 (d, J=8.2 Hz, 1H), 6.35 (s, 1H), 4.14 (dd, J=13.8, 6.8 Hz, 1H), 3.94 (t, J=6.4 Hz, 2H), 3.83 (dd, J=13.8, 6.8 Hz, 1H), 3.51 (s, 2H), 3.32 (s, J=7.1 Hz, 3H), 2.91 (t, J=7.5 Hz, 2H), 2.64 (t, J=7.5 Hz, 2H), 1.82–1.77 (m, 2H), 1.77–1.72 (m, 2H), 1.52–1.26 (m, 7H) (Figure S50 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.50, 171.75, 158.69, 138.07, 128.65, 115.64, 108.73, 102.17, 68.03, 51.82, 49.31, 42.88, 38.85, 31.12, 29.04, 25.60, 24.59, 12.15, 11.33 (Figure S51 in the Supporting Information). HRMS: calcd for [M+H]+ 381.1665, found 381.1666 (Figure S52 in the Supporting Information). HPLC purity 99.63% (Figure S53 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl diethyl-carbamodithioate (3e3). Yield 72%; white solid; mp 85.9–87.1°C; 1H NMR (600 MHz, CDCl3) δ 8.25 (s, 1H), 7.05 (d, J=8.3 Hz, 1H), 6.53 (d, J=8.2 Hz, 1H), 6.36 (s, 1H), 4.05 (dd, J=13.7, 6.8 Hz, 2H), 3.94 (t, J=6.4 Hz, 2H), 3.76 (dd, J=13.8, 6.8 Hz, 2H), 3.31 (t, J=7.5 Hz, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 1.83–1.78 (m, 2H), 1.77–1.73 (m, 2H), 1.51–1.27 (m, 10H) (Figure S54 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 195.92, 171.84, 158.70, 138.08, 128.64, 115.63, 108.74, 102.19, 68.04, 49.38, 46.66, 37.06, 31.12, 29.06, 28.73, 28.61, 25.62, 24.59, 12.43, 11.64 (Figure S55 in the Supporting Information). HRMS: calcd for [M+H]+ 395.1822, found 395.1810 (Figure S56 in the Supporting Information). HPLC purity 99.92% (Figure S57 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl pyrrolidine-1-carbodithioate (3e4). Yield 88%; white solid; mp 120.7–124.1°C; 1H NMR (600 MHz, CDCl3) δ 8.29 (s, 1H), 7.05 (d, J=8.2 Hz, 1H), 6.53 (d, J=8.2 Hz, 1H), 6.36 (s, 1H), 3.96 (d, J=7.1 Hz, 2H), 3.93 (s, J=6.7 Hz, 2H), 3.66 (t, J=6.9 Hz, 2H), 3.32 (t, J=7.4 Hz, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 2.11–2.06 (m, 2H), 2.02–1.96 (m, 2H), 1.82–1.76 (m, 4H), 1.51 (s, 4H) (Figure S58 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 193.09, 171.85, 158.68, 138.09, 128.63, 115.62, 108.77, 102.18, 68.03, 54.93, 50.61, 36.34, 31.11, 29.02, 28.84, 28.62, 26.04, 25.58, 24.59, 24.31 (Figure S59 in the Supporting Information). HRMS: calcd for [M+H]+ 393.1665, found 393.1655 (Figure S60 in the Supporting Information). HPLC purity 99.18% (Figure S61 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl 2-methyl piperidine-1-carbodithioate (3e5). Yield 91%; white solid; mp 76.8–78.6°C; 1H NMR (600 MHz, CDCl3) δ 8.24 (s, 1H), 7.05 (d, J=8.3 Hz, 1H), 6.53 (d, J=8.2 Hz, 1H), 6.36 (s, 1H), 5.75 (d, J=209.6 Hz, 1H), 4.78 (d, J=287.2 Hz, 1H), 3.94 (t, J=6.4 Hz, 2H), 3.33 (t, J=7.4 Hz, 2H), 3.15 (s, 1H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 1.80 (d, J=6.6 Hz, 2H), 1.75 (d, J=2.6 Hz, 2H), 1.72–1.50 (m, 9H), 1.34–1.24 (m, 4H) (Figure S62 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.33, 171.83, 158.70, 138.08, 128.64, 115.63, 108.74, 102.19, 68.04, 53.89, 53.86, 36.91, 31.11, 30.17, 30.06, 29.71, 29.05, 28.75, 28.59, 25.62, 24.59, 18.69 (Figure S63 in the Supporting Information). HRMS: calcd for [M+H]+ 421.1978, found 421.1980 (Figure S64 in the Supporting Information). HPLC purity 99.56% (Figure S65 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl 4-methylpiperidine-1-carbodithioate (3e6). Yield 95%; white solid; mp 78.2–79.7°C; 1H NMR (600 MHz, CDCl3) δ 8.25 (s, 1H), 7.05 (d, J=8.3 Hz, 1H), 6.53 (d, J=8.3 Hz, 1H), 6.36 (s, 1H), 5.56 (d, J=3.5 Hz, 1H), 4.63 (d, J=3.4 Hz, 1H), 3.94 (t, J=6.4 Hz, 2H), 3.32 (d, J=2.2 Hz, 2H), 3.13 (d, J=47.1 Hz, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 1.80 (s, 2H), 1.74 (s, 2H), 1.73–0.94 (m, J=241.8, 75.6 Hz, 12H) (Figure S66 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.05, 171.84, 158.69, 138.09, 128.64, 115.63, 108.75, 102.19, 68.03, 52.05, 50.40, 37.14, 33.84, 31.11, 30.99, 29.71, 29.04, 28.70, 28.67, 25.61, 24.59, 21.31 (Figure S67 in the Supporting Information). HRMS: calcd for [M+H]+ 421.1978, found 421.1976 (Figure S68 in the Supporting Information). HPLC purity 100% (Figure S69 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl 2,6-dimethylpiperidine-1-carbodithioate (3e7). Yield 94%; white solid; mp 110.0–113.1°C; 1H NMR (600 MHz, CDCl3) δ 8.13 (s, 1H), 7.05 (d, J=8.3 Hz, 1H), 6.53 (d, J=8.3 Hz, 1H), 6.35 (s, 1H), 5.61 (s, 1H), 4.69 (s, 1H), 3.94 (t, J=6.4 Hz, 2H), 3.31 (s, 2H), 3.13 (d, J=43.4 Hz, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.69–2.54 (m, 4H), 1.95–1.73 (m, 6H), 1.64–1.45 (m, 10H) (Figure S70 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.45, 171.72, 158.68, 138.08, 128.65, 115.64, 108.73, 102.15, 68.02, 62.11, 50.95, 50.24, 49.24, 37.28, 31.12, 29.71, 29.03, 28.68, 28.62, 26.17, 25.59, 24.59, 24.58 (Figure S71 in the Supporting Information). HRMS: calcd for [M+H]+ 435.2135, found 435.2131 (Figure S72 in the Supporting Information). HPLC purity 99.53% (Figure S73 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl 4-hydroxypiperidine-1-carbodithioate (3e8). Yield 86%; white solid; mp 129.4–131.1°C; 1H NMR (600 MHz, CDCl3) δ 8.23 (s, 1H), 7.05 (d, J=8.3 Hz, 1H), 6.54 (d, J=8.3 Hz, 1H), 6.36 (s, 1H), 4.67 (s, 1H), 4.17 (d, J=49.1 Hz, 2H), 4.09 (t, J=6.9 Hz, 1H), 3.94 (t, J=6.4 Hz, 2H), 3.78 (s, 1H), 3.32 (s, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 1.98 (s, 2H), 1.83–1.73 (m, J=14.2, 6.7 Hz, 6H), 1.51 (s, 5H) (Figure S74 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 196.60, 171.89, 158.68, 138.04, 128.66, 115.64, 108.80, 102.19, 68.00, 66.26, 48.37, 46.76, 37.30, 33.62, 31.10, 29.71, 28.99, 28.60, 28.58, 25.57, 24.56 (Figure S75 in the Supporting Information). HRMS: calcd for [M+H]+ 423.1771, found 423.1771 (Figure S76 in the Supporting Information). HPLC purity 99.38% (Figure S77 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl [1,4’-bipiperidine]-1’-carbodithioate (3e9). Yield 78%; white solid; mp 143.6–144.7°C; 1H NMR (600 MHz, CDCl3) δ 8.20 (s, 1H), 7.05 (d, J=8.3 Hz, 1H), 6.54 (d, J=10.0 Hz, 1H), 6.36 (s, 1H), 5.98–5.71 (m, J=9.8, 3.3 Hz, 1H), 5.02–4.86 (m, 1H), 3.94 (t, J=6.4 Hz, 2H), 3.44–3.21 (m, 2H), 2.91 (t, J=7.4 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 1.97–1.84 (m, 1H), 1.85–1.74 (m, 6H), 1.75–1.18 (m, 18H) (Figure S78 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 197.01, 171.81, 158.70, 138.08, 128.64, 115.63, 108.74, 102.19, 68.05, 53.47, 53.41, 52.78, 52.76, 36.93, 31.12, 30.42, 30.17, 29.71, 29.06, 28.80, 28.44, 25.63, 24.59, 19.77, 18.73, 13.99 (Figure S79 in the Supporting Information). HRMS: calcd for [M+H]+ 490.2557, found 490.2542 (Figure S80 in the Supporting Information). HPLC purity 99.87% (Figure S81 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl morpholine-4-carbodithioate (3e10). Yield 89%; white solid; mp 141.7–142.9°C; 1H NMR (600 MHz, CDCl3) δ 8.28 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.53 (d, J=8.3 Hz, 1H), 6.36 (s, 1H), 4.37 (s, 2H), 4.08–3.85 (m, 4H), 3.78 (s, 4H), 3.35 (t, J=7.4 Hz, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 1.82–1.73 (m, 4H), 1.58–1.46 (m, 4H) (Figure S82 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 197.98, 171.85, 158.68, 138.11, 128.64, 115.65, 108.73, 102.17, 67.99, 66.29, 66.16, 51.16, 50.27, 36.92, 31.11, 29.03, 28.67, 28.57, 25.59, 24.58 (Figure S83 in the Supporting Information). HRMS: calcd for [M+H]+ 409.1614, found 409.1609 (Figure S84 in the Supporting Information). HPLC purity 99.93% (Figure S85 in the Supporting Information).
6-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) oxy) hexyl 4-methylpiperazine-1-carbodithioate (3e11). Yield 91%; white solid; mp 115.4–117.1°C; 1H NMR (600 MHz, CDCl3) δ 8.09 (s, 1H), 7.06 (d, J=8.3 Hz, 1H), 6.53 (d, J=8.2 Hz, 1H), 6.34 (s, 1H), 4.39 (s, 2H), 4.09–3.85 (m, J=17.2, 10.8 Hz, 4H), 3.33 (t, J=7.4 Hz, 2H), 2.91 (t, J=7.5 Hz, 2H), 2.63 (t, J=7.5 Hz, 2H), 2.51 (s, 4H), 2.35 (s, 3H), 1.79–1.75 (m, 4H), 1.57–1.48 (m, 4H) (Figure S86 in the Supporting Information). 13C NMR (151 MHz, CDCl3) δ 197.38, 171.71, 158.68, 138.07, 128.66, 115.66, 108.72, 102.15, 68.02, 54.46, 54.41, 50.94, 49.57, 45.64, 37.06, 31.11, 29.03, 28.68, 28.60, 25.59, 24.59 (Figure S87 in the Supporting Information). HRMS: calcd for [M+H]+ 422.1935, found 422.1916 (Figure S88 in the Supporting Information). HPLC purity 99.71% (Figure S89 in the Supporting Information).
Biological Evaluation
In vitro Inhibition of ChEs
The in vitro inhibitory activities of designed compounds against ChEs were determined according to classic Ellman’s method. Acetylcholinesterase (AChE) from electric eel (eeAChE, C3389) and human erythrocytes (hAChE, C1682), butylcholinesterase (BuChE) from equine serum (eqBuChE, C1057) and human serum (hBuChE, B4186), acetylthiocholine iodide (ATCI, A5751), S-butylthiocholine iodide (BTCI, 20820) and 5,5'-dithiobis-(2-nitrobenzoic acid) (Ellman’s reagent, DTNB, D8130) were obtained from Sigma-Aldrich (St. Louis, MO, USA), and their detailed procedures could be configured according to the product instructions (for example, 0.22 U/mL eeAChE: 1 mL of Tris-HCl buffer solution was added to the purchased product to form 500 U/mL eeAChE solution. Then, the 500 U/mL eeAChE solution was divided and diluted to 0.22 U/mL).
The specific experimental operations were as follows: Firstly, the test compounds were dissolved in DMSO, and then diluted with Tris-HCl buffer solution (50 mM, pH = 8.0, 0.1 M NaCl, 0.02 M MgCl2·H2O) (DMSO <0.01%) to yield corresponding concentrations. Then, the experimental protocol was performed on 96-well plate. For each test well, 160 μL of DTNB (1.5 mM) and 50 μL of AChE (0.22 U/mL eeAChE or 0.05 U/mL hAChE) or 50 μL of BuChE (0.12 U/mL eqBuChE or 0.024 U/mL hBuChE) were incubated with 10 μL of different concentrations of test compounds at 37°C for 5 min. After this, 30 μL of substrate (15 mM, ATCI or BTCI) was added quickly and the absorbance was measured at a wavelength of 405 nm at different time intervals (0, 60, 120, and 180 s). The inhibition activity of the target compound was reported with IC50 value that was calculated as concentration of the compound that produced 50% enzyme activity inhibition. These results were expressed as mean±SD of three independent experiments. Data analysis was performed using Graph Pad Prism 4.03 software (San Diego, CA, USA).
In vitro Inhibition of MAOs
The inhibitory activities of the compounds 3a-i and 3e1-e11 towards MAOs were assayed by the Amplex Red fluorescence method. The recombinant human MAOs (hMAO-A, M7316 and hMAO-B, M7441) and p-tyramine (T90344) were obtained from Sigma-Aldrich (St. Louis, MO, USA). hMAO-A and hMAO-B and Amplex Red assay kit were used to determine the production of H2O2 from substrate p-tyramine. The test compounds were dissolved in DMSO and then diluted to different concentrations with PBS buffer solution (DMSO <0.01%). 80 μL of hMAO-A or hMAO-B and 20 μL of different concentrations of compounds were added to a 96-well black microtiter plate, and then incubated for 15 min at 37°C in the dark. After this period, a substrate mixture was added quickly and the results were tested by a multi-detection microplate fluorescence reader with excitation/emission wavelengths of 544/590 nm. Data were shown as mean±SD of three independent experiments.
In vitro Blood-Brain Barrier Permeation Assay
The permeability of all synthetic compounds on the blood-brain barrier (BBB) was detected by parallel artificial membrane penetration assay (PAMPA). The materials of experiment included porcine brain lipid (PBL, Avanti Polar Lipids, 141101P), dodecane (Sigma-Aldrich, D221104), donor 96-well filter microplate (PVDF membrane, pore size 0.45 μm, Millipore, MAIPN4550), acceptor indented 96-well microplate (Millipore, MATRNPS50) and the 96-well UV plate (Corning Incorporated, 3635). Compound 3e was configured with DMSO to 5 mg/mL, and then diluted to 50 μg/mL with buffer solution (PBS:EtOH = 70:30). The PBL was dissolved in dodecane, and 5 μL was added dropwise on the acceptor microplate. And then, 300 μL of diluted solution was added to each test donor well and 200 μL of buffer solution (PBS:EtOH = 70:30) was added to the corresponding acceptor wells. The donor microplate was carefully put on the acceptor microplate to form a sandwich, which maintained undisturbedly for 18 h at room temperature. After incubation, the donor microplate was carefully removed, and the concentration of test compound in acceptor wells was determined by a UV plate reader (SpectraMax Plus 384, Molecular Devices, Sunnyvale, CA, USA). The calculation formula of Pe is as follows: Pe={-VdVa/[(Vd+Va)At]}ln(1-drugacceptor /drugequilibrium), where Vd is the volume of donor well, Va is volume in acceptor well, A is the filter area, t is the permeation time, drugacceptor is the absorbance obtained in the acceptor well and drugequilibrium is the theoretical equilibrium absorbance. Each compound was analyzed at five wavelengths in four wells at least three independent runs, and the results were expressed as mean±SD.
Kinetic Study of Inhibition on hAChE
In order to determine the inhibitory mechanism of the potential compound, compound 3e was selected for kinetic studies of hAChE inhibition using Ellman’s method. The specific operation was similar to that mentioned above in enzyme inhibition assay. Four varied concentrations of compound 3e (0, 0.25, 0.5, 1 μM) were selected for the assay and evaluated against six concentrations of substrates (0.05, 0.075, 0.1, 0.15, 0.2, 0.5 mM for ATCI). Lineweaver-Burk reciprocal plots were established by reciprocal plots of 1/velocity against 1/[substrate]. The effect of concentrations of compound 3e on the activity for the catalysis of hAChE at 37°C was also studied. Assay conditions were the same as described in protocol except for the final concentrations of enzyme (0–0.2 U/mL). Concentrations of compound 3e (0, 0.25, 0.5, 1 μM) were used for the determination of reversible as well as irreversible binding of inhibitors at enzyme, respectively. All measurements were performed in triplicate and data analysis was performed with Graph Pad Prism 4.03 software (San Diego, CA, USA).
Kinetic Study of Inhibition on hMAO-B
The inhibitory mechanism of hMAO-B by compound 3e was obtained by construction of the Lineweaver-Burk reciprocal plots. Compound 3e at four different concentrations (0, 1.5, 3, 6 μM) was applied for kinetic study. The establishment of a curve was based on the initial catalytic rate of hMAO-B in the absence and presence of the corresponding concentration of compound 3e and six different concentrations (0.05, 0.1, 0.5, 1.0, 1.5, and 3.0 mM) of p-tyramine. The results were analyzed by Graph Pad Prism 4.03 software (San Diego, CA, USA).
Molecular Docking and Molecular Dynamics Simulations
Molecular Operating Environment (MOE) software (Montreal, Canada, version 2015.10) was used for molecular modeling calculations and docking studies. The crystal structures of the hAChE (PDB ID: 4EY7) and hMAO-B (PDB ID: 2V61) enzymes were obtained from the Protein Data Bank (PDB). Firstly, all water molecules in downloaded PDB files were removed and several charges were added. Then, compound 3e was constructed using the builder module, and energy minimized using the Merck Molecular force field (MMFF94x, RMSD gradient: 0.05 kal mol−1 Å−1). Next, 3e was docked into the active site of the protein by the “Triangle Matcher” method. All postures produced by the docking were analyzed, and the highest-score posture with the lowest binding energy of each compound was selected to further study the interaction with the corresponding enzyme.
After docking, the best pose of 3e-hAChE and 3e-hMAO-B complexes were imported into MOE software for molecular dynamics (MD) simulations. The Amber 10 EHT force field was chosen for energy minimization combined with Reaction Field electrostatics and configured the partial charges in all atoms through Hydrogen and lone pairs. In addition, we kept all crystal parameters at their default settings, and the Nosé-Poincaré-Andersen (NPA) algorithm of motion was used for MD simulations with a multi-stage protocol, which included heating the system for 100 ps (0 to 300 K), then stabilizing for 300 ps (T = 300), and finally cooling to 0 K. Figure S90 was obtained by plotting atomic potential energy vs time.
Cell Culture and Toxicity Assay
All cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cytotoxicity of compound 3e on PC12 cells (rat adrenal pheochromocytoma cells) and HT-22 cells (Mouse hippocampal neuron cell) was determined by CCK-8 assay. All cells were cultured in high-glucose DEME containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 U/mL streptomycin in a humidified atmosphere of 5% CO2 at 37°C. PC12 cells or HT-22 cells (1×104 cells/well) were grown in 96-well plates for 24 h. Then, the cells were treated with different concentrations of compound 3e (3.125–100 μM) for 1 day. The cell viability was determined by the CCK-8 assay according to the manufacturer’s instructions. All experimental data were presented as the mean±SD of three independent experiments.
Acute Toxicity Test
All Kunming (KM) mice (half male and half female, 18–22g) were obtained from Hunan SJA Laboratory Animal Co., Ltd (Hunan, People’s Republic of China). They were maintained under a 12 h light/dark cycle and allowed free access to tap water and standard laboratory chow. The room was maintained at temperature of 23±2°C with a relative humidity of 55±5%. All procedures were carried out according to the Animal Management Rules of the Chinese Ministry of Health (Revised 2017). The animal study complied with ARRIVE guidelines and was approved by the Ethical Committee at Jiangxi University of Chinese Medicine (No. JZLLSC2021-0310).
A total of 40 KM mice were randomly divided into corresponding groups. Before experiment, mice were fasted overnight and allowed free access to water. Three doses of compound 3e (625, 1250, and 2500 mg/kg) were uniformly dispersed in 0.5% carboxymethyl cellulose sodium (CMC-Na) salt solution and were given by oral administration.27 The mice were observed continuously for the first 4 h including tremor, spasm, death and other neurotoxic reactions after administration of the compound. The mice were continuously observed for 14 days. All animals were sacrificed on the 14th day and the liver, spleen and other organs of the mice were taken to observe the pathological changes of the tissues and organs.
Results and Discussion
Chemistry
The specific synthesis process of the target compounds 3a-i and 3e1-e11 was described in Scheme 1. Firstly, 3,4-dihydro-2(1H)-quinolinone and the corresponding α, ω-dibromoalkanes underwent a substitution reaction in acetone. To make the reaction proceed smoothly, K2CO3 was added as an acid binding agent and catalyst. Then, the intermediate compounds 2a-i were obtained. Finally, according to reported method,28 compounds 2a-i were reacted with CS2, a suitable secondary amine and TEA in DMF to obtain the target compounds 3a-i and 3e1-e11.
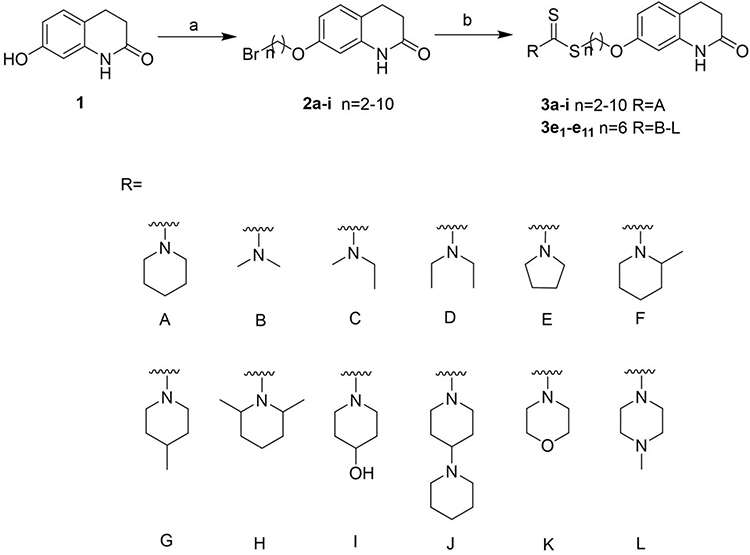 |
Scheme 1 Synthesis of compounds 3a-i and 3e1-e11. Reagents and conditions: (a) α, ω-dibromoalkanes, K2CO3, acetone, reflux, 6 h, yield: 65%-90%; (b) appropriate secondary amines, CS2, TEA, DMF, r.t., 12 h, yield: 70%–95%. |
Biological Evaluation
In vitro Biological Activity Evaluation
Firstly, Ellman’s method was used to determine the inhibitory activities of compounds 3a-i and 3e1-e11 on electric eel acetylcholinesterase (eeAChE) and equine serum butyrylcholinesterase (eqBuchE) (Table 1), and then compounds 3b-f, 3e2, 3e5 and 3e9 with good and selective inhibition on eeAChE were selected to determine their inhibitory activities towards human cholinesterase (hChEs) (Table 2).29 The inhibitory activities of target compounds 3a-i and 3e1-e11 against human monoamine oxidase (hMAOs) were determined by Amplex Red fluorescence method.30,31 Donepezil and iproniazid were used as positive controls to reflect the inhibitory effects of all the resulting compounds. As shown in Table 1, most of the target compounds show effective inhibitory activities towards eeAChE with IC50 values ranging from micromolar to submicromolar. Among these compounds, compound 3e held the best inhibitory activity on eeAChE with IC50 value of 0.28 μM. For hMAOs, the target compounds also had moderate to good inhibitory activities. Compound 3e4 was the most effective inhibitor of hMAOs in these series with IC50 value of 0.76 μM on hMAO-B and IC50 value of 0.78 μM on hMAO-A, which was higher than those of iproniazid (IC50=6.52 μM to hMAO-B; IC50=7.48 μM to hMAO-A). More meaningfully, almost all compounds could selectively inhibit eeAChE, and possessed almost no inhibitory effects on eqBuChE, which might be more conducive to the treatment of AD.32,33 Meanwhile, this series of compounds retained inhibitory activities on both hMAO-B and hMAO-A, so these compounds showed certain potential for the treatment of AD.
 |
Table 1 Inhibition of ChEs and hMAOs by Compounds 3a-i and 3e1-e11 |
 |
Table 2 Inhibition of Human ChEs by Selected Compounds |
Our previous studies demonstrated that the length of the linker was related to the eeAChE and hMAOs inhibitory activities of compounds.34 Therefore, compounds 3a-i with different linker lengths (n = 2–10) were prepared to study the inhibitory activities. It can be seen from Table 1 that the inhibitory activities of these compounds on eeAChE are varied significantly with changing the alkyl chain length. When the linker length was increased from two- to six-carbon atoms, the inhibitory activities of the compounds on eeAChE were gradually improved (3a: IC50=44.21 µM; 3e: IC50=0.28 µM). However, the activity was decreased when the linker length was extended to seven-carbon atoms (3f: IC50=4.53 µM). Simultaneously, the same trend was appeared in the determination of hMAOs inhibitory activities of target compounds. Taken together, compound 3e with six-carbon atoms showed better activity than other compounds, which indicated that linker length with six-carbon atoms was the best.
With compound 3e as the lead compound, compounds 3e1-e11 were obtained by changing the terminal amine group to discuss the structure–activity relationship (Table 1).35,36 It was found that the change of the terminal amine group had a greater impact on the eeAChE and hMAOs activities of the compounds. When the terminal group was a ring-opened alkylamine, the inhibitory activities of compounds on eeAChE had a certain degree of decrease. However, the inhibitory activities on hMAOs existed no obvious change compared with compound 3e. Compound 3e2 (IC50=14.43 µM to eeAChE; IC50=2.08 µM to hMAO-B; IC50=2.59 µM to hMAO-A) bearing a methylethylamine group exhibited more potent activity than its congeners 3e1 (IC50=62.33 µM to eeAChE; IC50=5.17 µM to hMAO-B; IC50=1.44 µM to hMAO-A) and 3e3 (IC50=24.05 µM to eeAChE; IC50=2.8 µM to hMAO-B; IC50=3.26 µM to hMAO-A).
Compounds 3e6, 3e8, 3e9 were obtained by modification of piperidine at position 4 with different substituents. Compared with compound 3e, the introduction of the 4-substituent resulted in a sharp reduction in the inhibitory activities of eeAChE. Surprisingly, compound 3e6 (IC50=0.99 µM to hMAO-B; IC50=0.90 µM to hMAO-A) bearing 4-methylpiperidine held better hMAOs inhibitory activities than compound 3e. When the 4-position methyl group was moved to the 2-position or 2,6-dimethyl, this trend was reversed compared with 3e6. The contraction of the piperidine ring to pyrrole ring made the inhibitory activity on eeAChE of compound 3e4 (38.65% inhibition rate to eeAChE; IC50=0.76 µM to hMAO-B; IC50=0.78 µM to hMAO-A) almost lost. To our surprise, its inhibitory activities on hMAOs were significantly improved. Compounds e10 (IC50=57.9 µM to eeAChE; IC50=41.42 µM to hMAO-B; IC50=1.76 µM to hMAO-A) and e11 (IC50=91.98 µM to eeAChE; 33.53% inhibition rate to hMAO-B; IC50=13.00 µM to hMAO-A) possessing a morpholinyl moiety and a 4-methylpiperazine moiety, respectively, displayed weak inhibitory activities on eeAChE and hMAO-B. However, they were provided with good selective inhibitory activities on hMAO-A. This implied that the presence of heteroatoms at the terminal amine group would increase the inhibitory activities on hMAO-A.
To evaluate the inhibitory activities of the designed compounds on ChEs more reasonably, eight compounds 3b-f, 3e2, 3e5 and 3e9 with good inhibitory activities on eeAChE were selected for further determination of the inhibitory activities on human ChEs. The results in Table 2 show that the compounds 3b-f, 3e2, 3e5 and 3e9 had almost no inhibitory activities on hBuChE, while they still maintained the high inhibitory activities and good selectivity on hAChE with IC50 values fluctuating from micromolar to submicromolar. Among them, compound 3e, the best inhibitor on eeAChE, also grasped the most effective inhibitory activity on hAChE with IC50 value of 0.34 µM.
In vitro Blood-Brain Barrier (BBB) Permeation Assay
Blood-brain barrier (BBB) permeability is the primary requirement for successful CNS drugs.37 Therefore, to determine whether the selected compound 3e could penetrate blood-brain barrier, a parallel artificial membrane penetration assay (PAMPA-BBB) was performed.38,39 Firstly, the permeability of 10 commercial drugs was used to verify the method in Table 3. A plot of experiment data versus the bibliographic values gave a good linear correlation: Pe (exp.)=0.9286×Pe (bibl.)-0.0268 (R2=0.9602) (Figure 2). From this equation and taking into account the limits established by Di et al.40 For BBB permeability, we classified compounds as follows:
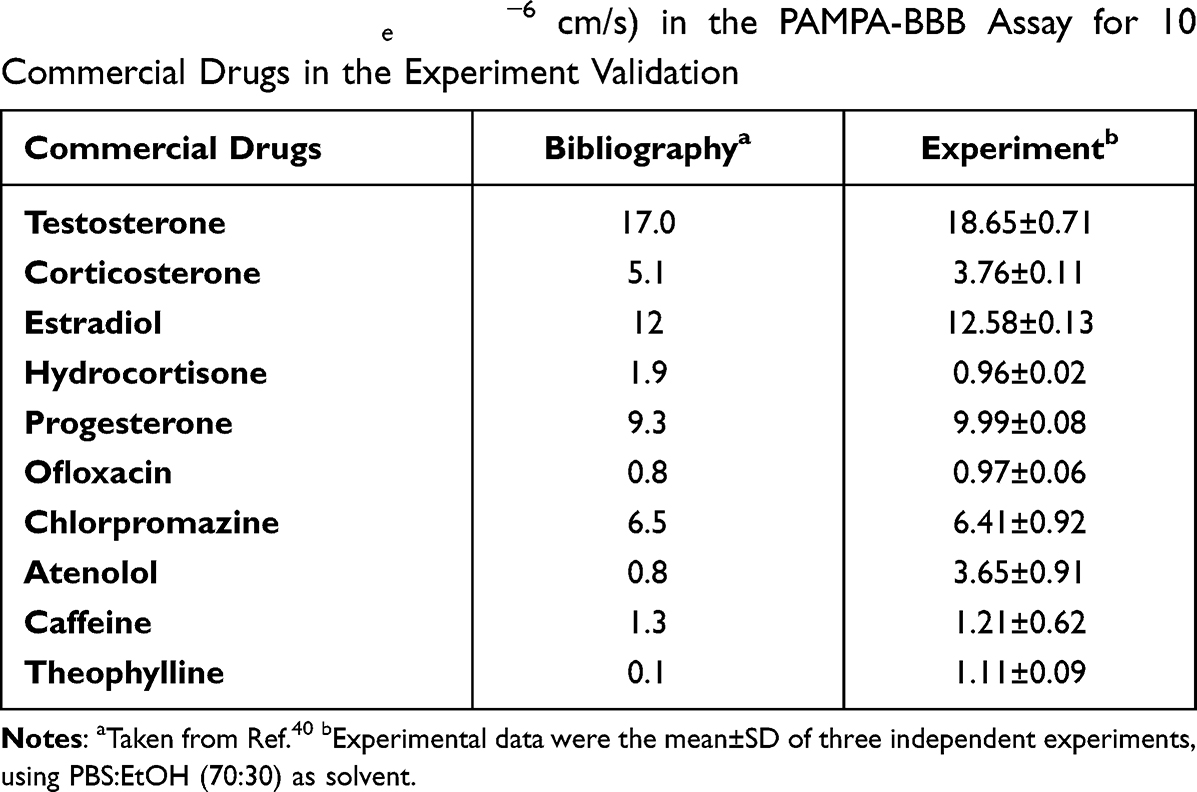 |
Table 3 Permeability Pe (× 10−6 cm/s) in the PAMPA-BBB Assay for 10 Commercial Drugs in the Experiment Validation |
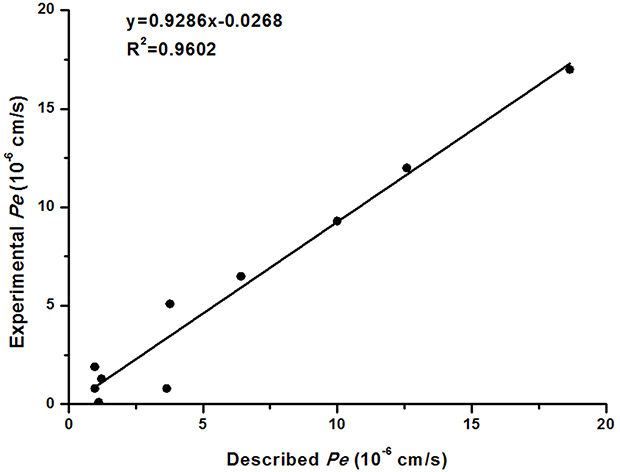 |
Figure 2 Lineal correlation between experimental and reported permeability of commercial drugs using the PAMPA-BBB assay. Pe (exp.)=0.9286×Pe (bibl.)-0.0268 (R2=0.9602). |
(a) “CNS+” (high BBB permeation predicted): Pe (10−6 cm/s)>3.69
(b) “CNS-” (low BBB permeation predicted): Pe (10−6 cm/s)<1.83
(c) “CNS±” (uncertain BBB permeation): 1.83<Pe (10−6 cm/s)<3.69
According to the measured permeability, the Pe value of compound 3e showed 13.74, which was higher than 3.69 and indicated that the selected compound 3e could penetrate the BBB and exert biological effects in the CNS.
Kinetic Study of Inhibition on hAChE and hMAO-B
In order to explore the inhibition mechanism of the synthesized compounds on hAChE and hMAO-B, the most representative compound 3e was selected for enzyme kinetic study. Firstly, we figured out the enzyme kinetic study of compound 3e on hAChE. As can be seen in Figure 3A, the plots of the remaining enzyme activity versus the concentration of enzyme (0, 0.025, 0.05, 0.1, 0.2 U/mL) in the presence of different concentrations of compound 3e for the catalysis of hAChE give a series of straight lines. All the lines intersected at the same point and the increasing inhibitor concentration resulted in a decrease in the slope of the lines, which indicated that compound 3e was reversible hAChE inhibitor. The Lineweaver-Burk double reciprocal plots are shown in Figure 3B. The slopes and intercepts increased with the concentration of compound 3e increasing, which indicated that compound 3e was mixed-type inhibitor. The above results explained that compound 3e could interact with PAS and CAS of hAChE, belonging to dual-site binding reversible inhibitor.
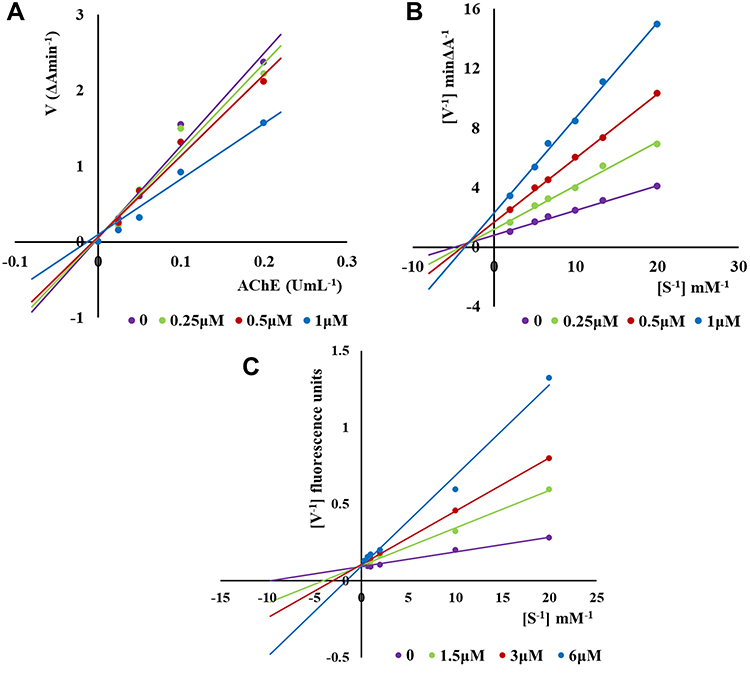 |
Figure 3 Kinetic study on the inhibition mechanism of hAChE and hMAO-B by compound 3e: (A) Lineweaver-Burk plots of hAChE inhibition kinetics of compound 3e; (B) The Lineweaver-Burk secondary plots of compound 3e on hAChE; (C) and on hMAO-B. |
Then, the interaction mechanism between compound 3e and hMAO-B was investigated. As shown in Figure 3C, the Lineweaver-Burk double reciprocal plots were established by the rates of the catalytic oxidation of hMAO-B for p-tyramine (0.05–3.0 mM) as substrate at different concentrations (0, 1.5, 3 and 6 μM) of compound 3e. All lines were crossed on the Y-axis, which pointed out that compound 3e was a competitive inhibitor on hMAO-B.
Molecular Docking and Molecular Dynamics Simulations
To further clarify the binding mode of compound 3e with the amino acid residues of hAChE and hMAO-B, molecular docking studies were carried out using Molecular Operating Environment (MOE) software based on the X-ray crystal structures of hAChE (PDB code: 4EY7) and hMAO-B (PDB code: 2V61).
The docking results of 3e-hAChE showed that compound 3e occupied enzymatic catalytic site (CAS), the mid-gorge site and peripheral anionic site (PAS) of hAChE completely (Figure 4A and 4C). Among them, the benzene ring of 3,4-dihydro-2(1H)-quinolinone core of compound 3e could bind to the Typ286 of PAS site via a π-π stacking interaction. In addition, the nitrogen atom and oxygen atom of 3,4-dihydro-2(1H)-quinolinone core were found to bind to ser293 via two hydrogen bonds which made the connection between compound 3e and the PAS site stronger. The flexible alkyl chain passed through the mid-gorge extending into the CAS site. In mid-gorge, the long dimethyl chain interacted with the Tyr341 via “arene-H”. The dithiocarbamate moiety of 3e was oriented towards the CAS of hAChE, exhibiting a hydrogen bond with the residues Gly121. The above results indicated that compound 3e occupied the entire active sites of AChE enzyme and was a dual-site binding inhibitor.
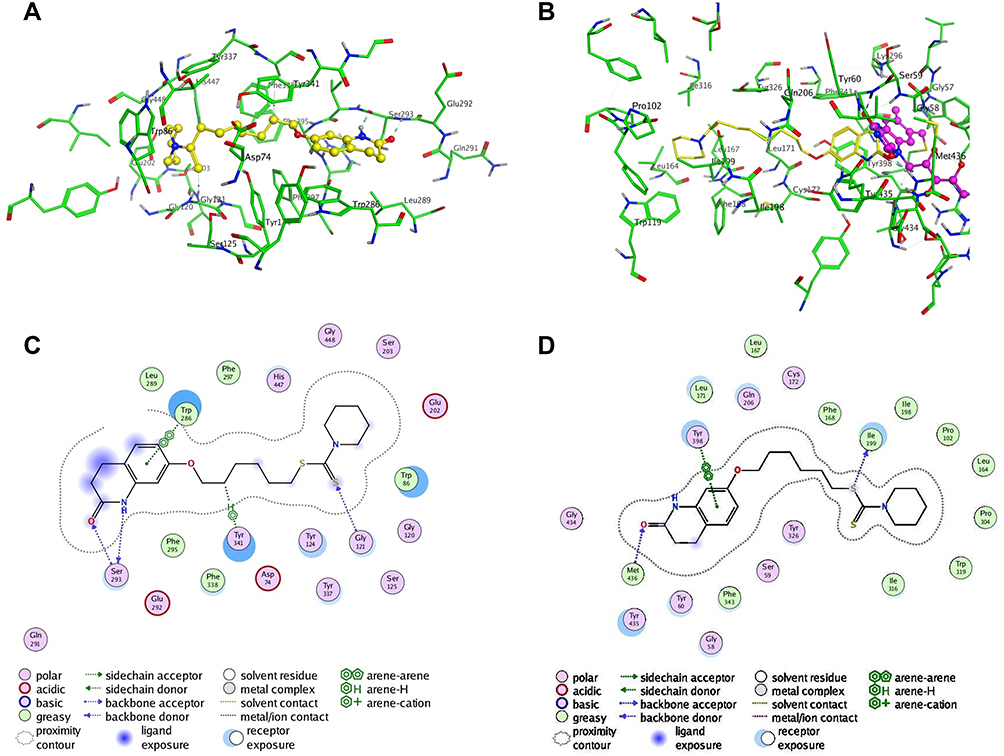 |
Figure 4 Molecule docking results: (A and C) were 3D and 2D docking models of compound 3e with hAChE; (B and D) were 3D and 2D docking models of compound 3e with hMAO-B. Atom colors: yellow-carbon atoms of 3e, green-carbon atoms of residues of hAChE, blue-nitrogen atoms, red-oxygen atoms, light yellow-sulfur atoms. The figure was generated with MOE. |
In 3e-hMAO-B complex (Figure 4B and 4D), compound 3e passed through the substrate cavity and the entrance cavity of hMAO-B. The benzene ring of the 3,4-dihydro-2(1H)-quinolinone moiety of compound 3e established a π-π stacking interaction with Tyr398 in the substrate cavity which was faced to the flavin adenine dinucleotide (FAD) cofactor. Besides, the carbonyl oxygen of this moiety generated a hydrogen bond with Met436. The piperidinyldithiocarbamate moiety interacted with Phe168, Ile198, Pro102, Leu164, Pro104, Ile316 through van der waals and hydrophobic interactions in entrance cavity. In addition, the oxygen atom in the side chain was observed to bind to Ile199 through hydrogen bond interaction.
To explore the dynamic stability of the 3e-hAChE complex and 3e-hMAO-B complex, the time-dependent potential energy of the complexes was calculated during molecular dynamics (MD) simulations. It was found that 3e-hAChE complex achieved equilibrium around 100 ps. Meanwhile, 3e-hMAO-B complex achieved the equilibrium around 100 ps (Figure S90 in the Supporting Information), which demonstrated that they were stable during the MD simulations.
In summary, the results of molecular docking and MD simulations of compound 3e with hAChE and hMAO-B were consistent with kinetic study, which explained the rationality of the compounds molecular design.
In vitro Cytotoxicity Evaluation
To determine the potential toxicity of compound 3e, nerve cells (PC12 and HT-22) were used to evaluate its cytotoxicity, and donepezil was taken as a reference compound. After treating the cells with compound 3e or donepezil at different concentrations (3.125, 6.25, 12.5, 25, 50 and 100 μM) for 24 h, the viability of the cells was determined by the CCK-8 method.41 As shown in Figure 5, when the concentration of compound 3e is less than 12.5 μM, it has almost no toxicity to nerve cells (PC12 and HT-22).
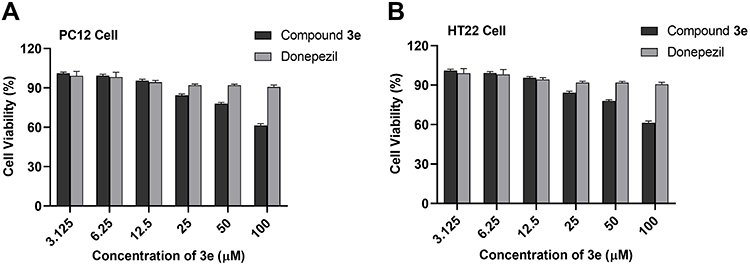 |
Figure 5 (A) Cytotoxicity of compound 3e and donepezil on PC12 cells. (B) on HT-22 cells. PC12 and HT-22 cells were incubated with different concentrations of compound 3e or donepezil (3.125–100 µM) for 24 h. The results were shown as cell viability after treated with compound 3e or donepezil vs untreated control cells. Date were expressed as mean±SD from three independent experiments. |
Acute Toxicity Assay
The toxicity of new chemicals is a major obstacle on the road to drug discovery and development.42 Therefore, compound 3e was selected for acute toxicity assay in vivo to further test its safety. After delivering compound 3e to Kunming (KM) mice with different dosages (625, 1250 and 2500 mg/kg) by oral administration, no dead mice were found. As shown in Figure 6, there are no obvious abnormal fluctuations in the weight of the mice in the two weeks after the administration. Furthermore, all mice were sacrificed on the 14th day after drug administration. Hematoxylin-eosin (HE) staining showed that compared to control mice, no significant histopathological changes in the vital organs (heart, liver, lung, kidney and brain) of mice at the maximum tested dosage 2500 mg/kg were discovered (Figure 7).43 Overall, compound 3e did not demonstrate acute toxicity and exhibited safety in vivo.
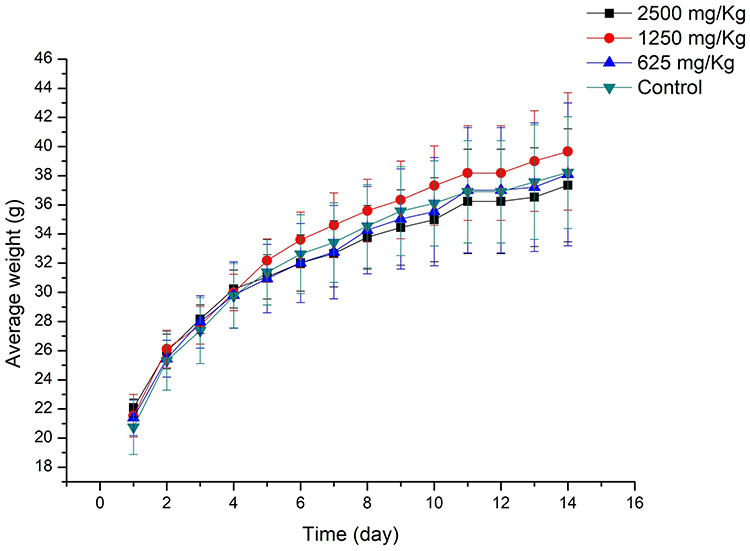 |
Figure 6 The change on body weight of mice after oral administration of different concentrations of compound 3e. Data were expressed as the average weight±SD of mice. |
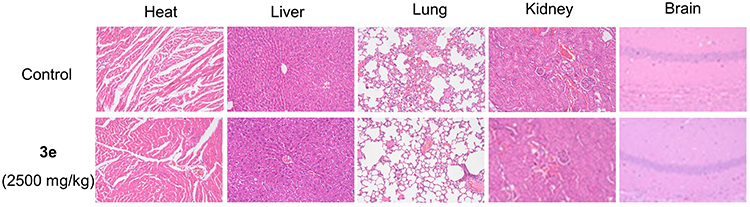 |
Figure 7 Histological analysis of heart, liver, lung, kidney, and brain for the acute toxicity studies of compound 3e at dosage of 2500 mg/kg in mice; 200 mm indicated the scale bar of images (HE staining). Representative images of HE-stained heart, liver, lung, kidney and brain for each group were shown. |
Conclusion
In order to find effective drugs for the treatment of Alzheimer’s disease, a series of novel inhibitors 3a-i and 3e1-e11 by hybridization of 3,4-dihydro-2(1H)-quinolinone and dithiocarbamate were designed, synthesized and evaluated. All the target compounds were investigated for their abilities to inhibit the ChEs and MAOs. The results indicated that compound 3e with a six-carbon atom linker and a piperidine moiety at terminal position displayed the balanced activities to inhibit AChE and MAOs. Kinetic and molecular modelling studies showed that compound 3e was a dual-site binding inhibitor that could simultaneously bind PAS and CAS of AChE and a competitive inhibitor for MAO-B. Moreover, it showed good ability to penetrate the BBB and no significant toxicity on PC12 cells and HT-22 cells when the concentration of 3e was lower than 12.5 µM. In vivo, compound 3e did not display any acute toxicity in mice at doses up to 2500 mg/kg (P.O.). Taken together, these results proposed that compound 3e was a promising candidate and was worthy of further study.
Supporting Information
The 1H NMR charts of intermediates 2a-i, 1H NMR, 13C NMR, HRMS, HPLC purity charts and HRMS error analysis data of the target compounds 3a-i, 3e1-e11 were available as Supporting Information. The figure of molecular dynamics simulation was also available as Supporting Information.
Ethical Approval
The animal study complied with ARRIVE guidelines and was approved by the research ethics committee of Jiangxi University of Chinese Medicine (No. JZLLSC2021-0310). All procedures were carried out according to the Animal Management Rules of the Chinese Ministry of Health (Revised 2017).
Author Contributions
All authors made a significant contribution to this article, whether that is in the experimental protocol design, execution, data acquisition, analysis, and interpretation, or in all these areas; participated in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have reached an agreement on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Science Foundation of China [No. 81760622, 21807052]; Support Program for Outstanding Youth Talents in Jiangxi province [Grant No. 20192BCB23018]; the Program of Natural Science Foundation of Jiangxi province of China [Grant No. 20192ACBL21034, 20202BAB216042]; the 1050 Youth Talent Project of Jiangxi University of Traditional Chinese Medicine [Grant No. 1141900616]; Nanchang Double-Hundred Talents Project [Grant No.20200729]; Foundation of Jiangxi Educational Committee [Grant No. GJJ190673].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dunn B, Stein P, Temple R, Cavazzoni P. An appropriate use of accelerated approval - aducanumab for Alzheimer’s disease. N Engl J Med. 2021;385:856–857.
2. Zhang DF, Xu M, Bi R, et al. Genetic analyses of Alzheimer’s disease in China: achievements and perspectives. ACS Chem Neurosci. 2019;10:890–901. doi:10.1021/acschemneuro.8b00435
3. Rochoy M, Rivas V, Chazard E, et al. Factors associated with Alzheimer’s disease: an overview of reviews. J Prev Alzheimers Dis. 2019;6:121–134. doi:10.14283/jpad.2019.7
4. Ju Y, Tam KY. Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen Res. 2022;17:543–549. doi:10.4103/1673-5374.320970
5. Mishra P, Kumar A, Panda G. Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer’s disease (1998–2018). Bioorg Med Chem. 2019;27:895–930. doi:10.1016/j.bmc.2019.01.025
6. Villavicencio Tejo F, Quintanilla RA. Contribution of the Nrf2 pathway on oxidative damage and mitochondrial failure in Parkinson and Alzheimer’s disease. Antioxidants. 2021;10:1069–1100. doi:10.3390/antiox10071069
7. Bohnen NI, Grothe MJ, Ray NJ, et al. Recent advances in cholinergic imaging and cognitive decline-revisiting the cholinergic hypothesis of dementia. Curr Geriatr Rep. 2018;7:1–11. doi:10.1007/s13670-018-0234-4
8. Kabir MT, Uddin MS, Mamun AA, et al. Abdel-Daim, combination drug therapy for the management of Alzheimer’s disease. Int J Mol Sci. 2020;21:3272–3295. doi:10.3390/ijms21093272
9. Perry EK, Tomlinson BE, Blessed G, et al. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J. 1978;2:1457–1459. doi:10.1136/bmj.2.6150.1457
10. Kucukoglu K, Gul H, Taslimi P, et al. Investigation of inhibitory properties of some hydrazone compounds on hCA I, hCA II and AChE enzymes. Bioorg Chem. 2019;86:316–321. doi:10.1016/j.bioorg.2019.02.008
11. Zhang Z, Guo J, Cheng M, et al. Design, synthesis, and biological evaluation of novel xanthone-alkylbenzylamine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem. 2021;213:113154. doi:10.1016/j.ejmech.2021.113154
12. Fu J, Bao F, Gu M, et al. Design, synthesis and evaluation of quinolinone derivatives containing dithiocarbamate moiety as multifunctional AChE inhibitors for the treatment of Alzheimer’s disease. J Enzyme Inhib Med Chem. 2020;35:118–128. doi:10.1080/14756366.2019.1687460
13. Han X, He G. Toward a rational design to regulate beta-Amyloid fibrillation for Alzheimer’s disease treatment. ACS Chem Neurosci. 2018;9:198–210. doi:10.1021/acschemneuro.7b00477
14. Shidore M, Machhi J, Shingala K, et al. Benzylpiperidine-linked diarylthiazoles as potential anti-Alzheimer’s agents: synthesis and biological evaluation. J Med Chem. 2016;59:5823–5846. doi:10.1021/acs.jmedchem.6b00426
15. Gao HW, Jiang YY, Zhan JY, et al. Pharmacophore-based drug design of AChE and BChE dual inhibitors as potential anti-Alzheimer’s disease agents. Bioorg Chem. 2021;114:105149. doi:10.1016/j.bioorg.2021.105149
16. Tripathi AC, Upadhyay S, Paliwal S, et al. Privileged scaffolds as MAO inhibitors: retrospect and prospects. Eur J Med Chem. 2018;145:445–497. doi:10.1016/j.ejmech.2018.01.003
17. Kumar B, Sheetal AK, Mantha AK, et al. Synthesis, biological evaluation and molecular modeling studies of phenyl-/benzhydrylpiperazine derivatives as potential MAO inhibitors. Bioorg Chem. 2018;77:252–262. doi:10.1016/j.bioorg.2018.01.020
18. Pathania A, Kumar R, Sandhir R. Hydroxytyrosol as anti-parkinsonian molecule: assessment using in-silico and MPTP-induced Parkinson’s disease model. Biomed Pharmacother. 2021;139:111525. doi:10.1016/j.biopha.2021.111525
19. Xie SS, Liu J, Tang C, et al. Design, synthesis and biological evaluation of rasagiline-clorgyline hybrids as novel dual inhibitors of monoamine oxidase-B and amyloid-beta aggregation against Alzheimer’s disease. Eur J Med Chem. 2020;202:112475. doi:10.1016/j.ejmech.2020.112475
20. Bolea I, Juarez-Jimenez J, de Los Rios C, et al. Samadi, Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl) methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J Med Chem. 2011;54:8251–8270. doi:10.1021/jm200853t
21. Jalal K, Khanb K, Haleema DJ, et al. In silico study to identify new monoamine oxidase type a (MAO-A) selective inhibitors from natural source by virtual screening and molecular dynamics simulation. J Mol Struct. 2022;1254:132244. doi:10.1016/j.molstruc.2021.132244
22. Rossi M, Freschi M, de Camargo Nascente L, et al. Sustainable drug discovery of multi-target-directed ligands for Alzheimer’s disease. J Med Chem. 2021;64:4972–4990. doi:10.1021/acs.jmedchem.1c00048
23. Cavalli A, Bolognesi ML, Minarini A, et al. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem. 2008;51:347–372. doi:10.1021/jm7009364
24. Wang J, Cai P, Yang XL, et al. Novel cinnamamide-dibenzylamine hybrids: potent neurogenic agents with antioxidant, cholinergic, and neuroprotective properties as innovative drugs for Alzheimer’s disease. Eur J Med Chem. 2017;139:68–83. doi:10.1016/j.ejmech.2017.07.077
25. Carradori S, Silvestri R. New frontiers in selective human MAO-B inhibitors. J Med Chem. 2015;58:6717–6732. doi:10.1021/jm501690r
26. Jiang N, Ding J, Liu J, et al. Novel chromanone-dithiocarbamate hybrids as multifunctional AChE inhibitors with beta-amyloid anti-aggregation properties for the treatment of Alzheimer’s disease. Bioorg Chem. 2019;89:103027. doi:10.1016/j.bioorg.2019.103027
27. Sang Z, Pan W, Wang K, et al. Design, syntHesis and biological evaluation of 3,4-diHydro-2(1 H)-quinoline- O -alkylamine derivatives as new multipotent cHolinesterase/monoamine oxidase inHibitors for tHe treatment of AlzHeimer’s disease. Bioorg Med Chem. 2017;25:3006–3017. doi:10.1016/j.bmc.2017.03.070
28. Jiang N, Huang Q, Liu J, et al. Design, synthesis and biological evaluation of new coumarin-dithiocarbamate hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem. 2018;146:287–298. doi:10.1016/j.ejmech.2018.01.055
29. Ellman G, Courtney K, Andres V, et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95.
30. Holt A, Sharman DF, Baker GB, et al. A continuous spectrophotometric assay for monoamine oxidase and related enzymes in tissue homogenates. Anal Biochem. 1997;244:384–392. doi:10.1006/abio.1996.9911
31. Yip LY, Aw CC, Lee SH, et al. The liver-gut microbiota axis modulates hepatotoxicity of tacrine in the rat. Hepatology. 2018;67:282–295. doi:10.1002/hep.29327
32. Knez D, Colettis N, Iacovino LG, et al. Stereoselective activity of 1-propargyl-4-styrylpiperidine-like analogues that can discriminate between monoamine oxidase isoforms A and B. J Med Chem. 2020;63:1361–1387. doi:10.1021/acs.jmedchem.9b01886
33. Greig NH, Utsuki T, Yu Q, et al. A new therapeutic target in Alzheimer’s disease treatment: attention to butyrylcholinesterase. Curr Med Res Opin. 2001;17:159–165. doi:10.1185/03007990152673800
34. He Q, Liu J, Lan JS, et al. Coumarin-dithiocarbamate hybrids as novel multitarget AChE and MAO-B inhibitors against Alzheimer’s disease: design, synthesis and biological evaluation. Bioorg Chem. 2018;81:512–528. doi:10.1016/j.bioorg.2018.09.010
35. Prashanth MK, Revanasiddappa HD, Lokanatha Rai KM, et al. Synthesis, characterization, antidepressant and antioxidant activity of novel piperamides bearing piperidine and piperazine analogues. Bioorg Med Chem Lett. 2012;22:7065–7070. doi:10.1016/j.bmcl.2012.09.089
36. Fu J, Cheng K, Zhang ZM, et al. Synthesis, structure and structure-activity relationship analysis of caffeic acid amides as potential antimicrobials. Eur J Med Chem. 2010;45:2638–2643. doi:10.1016/j.ejmech.2010.01.066
37. Wang Z, Wang Y, Li W, et al. Design, synthesis, and evaluation of multitarget-directed selenium-containing clioquinol derivatives for the treatment of Alzheimer’s disease. ACS Chem Neurosci. 2014;5:952–962. doi:10.1021/cn500119g
38. Bicker J, Alves G, Fortuna A, et al. A new PAMPA model using an in-house brain lipid extract for screening the blood-brain barrier permeability of drug candidates. Int J Pharm. 2016;501:102–111. doi:10.1016/j.ijpharm.2016.01.074
39. Clemons K, Kretsch A, Verbeck G. Parallel artificial membrane permeability assay for blood-brain permeability determination of illicit drugs and synthetic analogues. Sci Justice. 2014;54:351–355. doi:10.1016/j.scijus.2014.06.004
40. Di L, Kerns EH, Fan K, et al. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem. 2003;38:223–232. doi:10.1016/S0223-5234(03)00012-6
41. Zhang Y, Huang N, Lu H, et al. Icariin protects against sodium azide-induced neurotoxicity by activating the PI3K/Akt/GSK-3beta signaling pathway. Peer J. 2020;8:e8955. doi:10.7717/peerj.8955
42. Wang K, Shi J, Zhou Y, et al. Design, synthesis and evaluation of cinnamic acid hybrids as multi-target-directed agents for the treatment of Alzheimer’s disease. Bioorg Chem. 2021;112:104879. doi:10.1016/j.bioorg.2021.104879
43. Zhong T, Feng M, Su M, et al. Qihuzha granule attenuated LPS-induced acute spleen injury in mice via Src/MAPK/Stat3 signal pathway. J Ethnopharmacol. 2021;281:114458. doi:10.1016/j.jep.2021.114458
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.