")
Back to Journals » Journal of Pain Research » Volume 13
Design of Phase 3 Studies Evaluating Vixotrigine for Treatment of Trigeminal Neuralgia
Authors Kotecha M, Cheshire WP , Finnigan H, Giblin K, Naik H, Palmer J, Tate S, Zakrzewska JM
Received 11 February 2020
Accepted for publication 14 May 2020
Published 1 July 2020 Volume 2020:13 Pages 1601—1609
DOI https://doi.org/10.2147/JPR.S247182
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Michael Schatman
Mona Kotecha,1 William P Cheshire,2 Helen Finnigan,3 Kathryn Giblin,1 Himanshu Naik,1 Joanne Palmer,4 Simon Tate,4 Joanna M Zakrzewska5
1Biogen, Cambridge, MA, USA; 2Department of Neurology, Mayo Clinic Florida, Jacksonville, FL, USA; 3Biogen, Maidenhead, UK; 4Convergence Pharmaceuticals, a Biogen Company, Cambridge, UK; 5Facial Pain Unit and Pain Management Centre, University College London Hospitals NHS Foundation Trust/University College London, London, UK
Correspondence: Mona Kotecha Email [email protected]
Purpose: Vixotrigine (BIIB074) is a voltage- and use-dependent sodium channel blocker. These studies will evaluate the efficacy and safety of vixotrigine in treating pain experienced by patients with trigeminal neuralgia (TN) using enriched enrollment randomized withdrawal trial designs.
Patients and Methods: Two double-blind randomized withdrawal studies are planned to evaluate the efficacy and safety of vixotrigine compared with placebo in participants with TN (NCT03070132 and NCT03637387). Participant criteria include ≥ 18 years old who have classical, purely paroxysmal TN diagnosed ≥ 3 months prior to study entry, who experience ≥ 3 paroxysms of pain/day. The two studies will include a screening period, 7-day run-in period, a 4- or 6-week single-dose-blind dose-optimization period (Study 1) or 4-week open-label period (Study 2), and 14-week double-blind period. Participants will receive vixotrigine 150 mg orally three times daily in the dose-optimization and open-label periods. The primary endpoint of both studies is the proportion of participants classified as responders at Week 12 of the double-blind period. Secondary endpoints include safety measures, quality of life, and evaluation of vixotrigine population pharmacokinetics.
Conclusion: There is a need for an effective, well-tolerated, noninvasive treatment for the neuropathic pain associated with TN. The proposed studies will evaluate the efficacy and safety of vixotrigine in treating pain experienced by patients with TN.
Keywords: facial pain, neuropathic pain, voltage-gated sodium channels, enriched enrollment randomized withdrawal, EERW, Penn Facial Pain Scale-Revised, PENN-FPS-R
Introduction
Trigeminal neuralgia (TN) is a chronic pain condition characterized by recurrent episodes of brief, usually unilateral, very severe pain in the distribution of one or more branches of the trigeminal nerve.1 TN is a relatively rare disease, with an estimated prevalence of 1.08 per 10,000 in the European Union and <200,000 cases in the USA, and is more common in women.2 Population-based prevalence studies are limited in the USA, but a medical record–based study identified an age- and sex-adjusted incidence of 4.7 per 100,000 person-years.3 Disease onset is typically after the age of 40 years and increases with age.2 A single episode or “paroxysm” usually lasts from less than a second and up to a few minutes, followed by a refractory period of several minutes.1,2,4 Symptomatic paroxysms typically occur in “bouts” lasting weeks to months, with periods of remission of up to 6 months. It has been assumed that the natural history of TN shows that paroxysms typically increase in severity, duration, and frequency, and periods of remission become shorter; however, this does not occur in all patients.2 When patients with TN are in a severe “bout,” their quality of life is significantly affected, as they are unable to work or carry out usual activities of daily living.5 A hallmark of TN is that although patients may report spontaneous pain, paroxysms are almost always triggered by seemingly innocuous stimuli.6
TN can be classified into types; the diagnostic criteria and terminology for the different types of TN has changed slightly over the existing editions of the International Classification of Headache Disorders. In the current third edition, classical TN is associated with vascular compression of the root of the trigeminal nerve near the pons of the brainstem, leading to morphological changes of the nerve root.1,6,7 Secondary TN is caused by a lesion other than vascular compression, eg, multiple sclerosis.1,2,6 TN may also be classified as idiopathic in cases where no neurovascular compression or other lesions are noted.
Current first-line therapies for pain control in patients with TN include the anticonvulsant drugs carbamazepine and oxcarbazepine; carbamazepine is the only drug approved by the United States Food and Drug Administration for the treatment of TN.2,7,8 Both are voltage-gated sodium channel (Nav) blockers that raise the threshold of excitability, thus decreasing the increased frequency of neuronal firings thought to underlie TN pain.2 Although these drugs have proven efficacy in decreasing TN pain, their use is limited by the need for dose titration, monitoring for hyponatremia and decreased hematologic cell counts, the eventual development of tachyphylaxis in most patients, and, for many, poor tolerability, including the potential for life-threatening side effects such as Stevens-Johnson syndrome, cognitive impairment, and drug interactions.9 In a prospective observational survey of 161 participants with TN receiving either carbamazepine or oxcarbazepine, 31.3% reported tiredness and 22.7% reported memory problems.10
Second-line drugs for the treatment of TN pain include lamotrigine, gabapentin, and baclofen; however, these medications are not approved for TN treatment and limited evidence is available to support their use.7,11 If pharmacotherapy is ineffective or poorly tolerated, surgical options including microvascular decompression, radiofrequency thermocoagulation or glycerol rhizotomy, balloon compression, and stereotactic radiosurgery are available.7,11 Of the surgical options available, microvascular decompression is the only nonablative procedure available.7,11 Although ~90% of patients experience pain relief soon after the procedure, this percentage declines to 73% at 5 years, at which point patients need to restart their medications.7,12 There is an unmet need for an effective, well-tolerated, pharmacologic treatment for the pain associated with TN.
Vixotrigine (BIIB074) is a voltage- and use-dependent sodium channel blocker.13 Results from previous clinical trials suggest that vixotrigine, up to 150 mg 3 times daily (TID), may be a safe and effective treatment for TN pain.13 Vixotrigine demonstrates good tolerability and can be administered without lengthy titration based on Phase I studies.13 In a randomized Phase IIa proof-of-concept study in participants with TN, those treated with vixotrigine displayed a significant reduction in the average number of TN paroxysms experienced compared with those receiving placebo.13 The average number of paroxysms was 45% lower (P=0.028), the average daily pain score was 50% lower (P=0.0009), and the average severity of paroxysms was 26% lower (P=0.085), after adjusting for placebo.13 Significant improvement in neuropathic pain was also observed in a Phase II study of participants with painful lumbosacral radiculopathy (PLSR), although a follow-up study did not confirm the results.14 The difference in the changes in average daily pain intensity numerical rating scale (PI-NRS) for neuropathic pain from baseline to Week 3 was –0.43 (P=0.0265) for vixotrigine (350 mg twice daily) compared with placebo.15 Vixotrigine was well tolerated, with similar incidences of adverse events (AEs) experienced by both treatment groups and few reports of cognitive impairment. The most common AEs in the Phase IIa study were headache and dizziness.13 The current studies will evaluate the efficacy and safety of vixotrigine up to 250 mg 3 times daily in treating pain experienced by patients with TN using enriched enrollment randomized withdrawal trial designs, which are an option in those conditions for which the pain severity precludes the use of long-term placebo.16
Patients and Methods
Study Design
Two double-blind randomized withdrawal studies are planned to evaluate the efficacy and safety of vixotrigine compared with placebo in participants with TN (NCT03070132, Study 1, and NCT03637387, Study 2). The 2 studies will include a screening period, 7-day run-in period, 4- or 6-week single-dose-blind dose-optimization period (Study 1) or 4-week open-label period (Study 2), and 14-week double-blind period (Figure 1). Participants will be given an electronic diary to record daily and weekly efficacy assessments and will receive training on its use during the screening period.
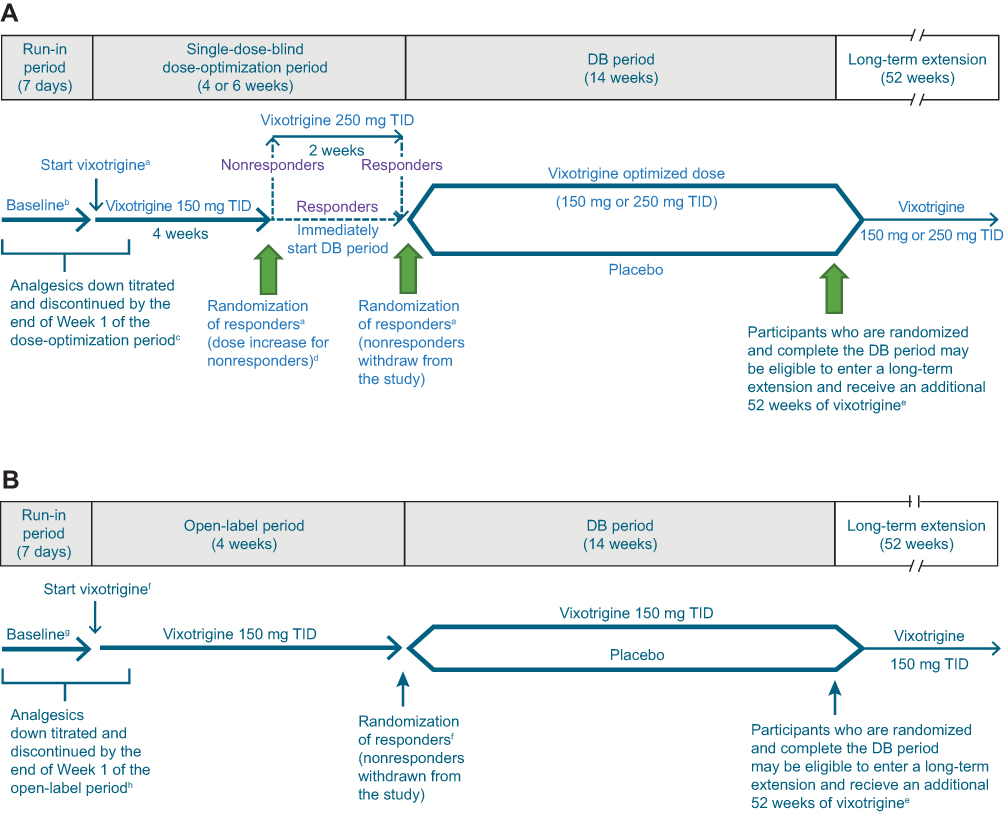 |
Figure 1 Study design for (A) Study 1 and (B) Study 2. Notes: Responders are defined as participants with ≥30% reduction in mean pain score from run-in period baseline to the last week of the dose-optimization/open-label period. aParticipants must meet all eligibility criteria on Day 1 to enter the dose-optimization period and all randomization criteria at Week 4 (Day 29) or Week 6 (Day 43) to be randomized to DB treatment. bFor efficacy endpoints based on daily participant diary (pain score, worst pain score, and number of paroxysms), baseline will be defined as the means of the diary data recorded over the 7 days preceding the first dose of study treatment in the dose-optimization period. For other efficacy endpoints, baseline will be the last measurement before the first dose of study treatment. cParticipants taking >1 TN medication at study entry will be required to gradually titrate down and discontinue their medications so that they are receiving no more than 1 TN medication at the start of the dose-optimization period. The remaining TN medication should be at a low enough dose at the start of the dose-optimization period so that it can be safely stopped by the end of Week 1. Participants taking carbamazepine or oxcarbazepine will be required to reduce their dose by the start of the dose-optimization period and will take their last dose by Day 7, prior to the start of Week 2 of the dose-optimization period. dThe increase in dose at the end of Week 4 for nonresponders will occur only if participants have recorded their pain score in the electronic diary on ≥5 of the last 7 days of the dose-optimization period; participants who are noncompliant with the electronic diary will be withdrawn from the study. eIncludes participants who discontinue DB study treatment for reasons other than adverse events but remain in the study and complete the DB period through Week 14. Participants who discontinue DB study treatment and withdraw from the study and participants who exceed dosing limits for acetaminophen/paracetamol, pregabalin, or immediate-release oxycodone during the DB period will not be eligible for the long-term extension. fParticipants must meet all eligibility criteria on Day 1 to enter the open-label period and all randomization criteria at Week 4 (Day 29) to be randomized to DB treatment. gFor efficacy endpoints based on daily participant diary (pain score, worst pain score, and number of paroxysms), baseline will be defined as the means of the diary data recorded over the 7 days preceding the first dose of study treatment in the open-label period. For other efficacy endpoints, baseline will be the last measurement before the first dose of study treatment. hParticipants taking >1 TN medication at study entry will be required to gradually titrate down and discontinue their medications so that they are receiving no more than 1 TN medication at the start of the open-label period. The remaining TN medication should be at a low enough dose at the start of the open-label period so that it can be safely stopped by the end of Week 1. Participants taking carbamazepine or oxcarbazepine will be required to reduce their dose by the start of the open-label period and will take their last dose by Day 7, prior to the start of Week 2 of the open-label period. Abbreviations: DB, double-blind; TID, 3 times daily; TN, trigeminal neuralgia. |
During the run-in period for both studies, gradual discontinuation of TN medications will be initiated so that by Week 2 of the dose-optimization or open-label period, only vixotrigine is in continuous use. In the enrichment period, Study 1 and Study 2 have slightly different methodologies, as described below.
During the dose-optimization period in Study 1, participants will initially receive vixotrigine 150 mg orally TID for 4 weeks. Participants will be told that there are 2 doses of vixotrigine used in the study but not which dose they are receiving at any time during the dose-optimization period. At the end of Week 4, participants who are classified as responders (≥30% reduction in mean pain score from the run-in period baseline to the last week of the dose-optimization period) will be randomized to receive either vixotrigine 150 mg TID or placebo and will start the double-blind period. Those who do not meet responder criteria will receive vixotrigine 250 mg TID for an additional 2 weeks. At the end of Week 6, participants who now meet the responder criteria will be randomized to receive either vixotrigine 250 mg TID or placebo during the subsequent double-blind period of 14 weeks, whereas those who do not meet the responder criteria will be withdrawn from the study.
In the open-label period in Study 2, participants will receive vixotrigine 150 mg TID for 4 weeks. At the end of Week 4, participants who meet the responder criteria (≥30% reduction in mean pain score from the run-in period baseline to the last week of the open-label period) will be randomized to receive either vixotrigine 150 mg TID or placebo during the subsequent double-blind period of 14 weeks, whereas those who do not meet the responder criteria will be withdrawn from the study.
In the double-blind period, participants who respond to vixotrigine during the open-label or dose-optimization period will be randomized 1:1 to receive either vixotrigine at the dose to which they responded (Study 1, 150 mg or 250 mg TID; Study 2, 150 mg TID) or placebo. The 150 mg TID dose regimen was selected based on results from the Phase II study in participants with TN, in which favorable effects on the primary and secondary endpoints were observed following treatment with vixotrigine 150 mg TID compared with placebo.13 A total of 11 of 44 participants who completed the open-label period had some reduction in pain severity or number of paroxysms but failed to meet the responder criteria for randomization into the double-blind period.13 The results suggested that a higher dose of vixotrigine (250 mg TID) may provide benefit to participants who do not have an adequate response to vixotrigine 150 mg TID.13 A further study in PLSR using vixotrigine 350 mg twice daily (700 mg/day) for up to 3 weeks (a dose regimen that provides similar exposure to the proposed dose of 250 mg TID) showed that this dosage was generally well tolerated, and showed a statistically significant reduction in neuropathic pain scores compared with placebo.14
Randomization will be stratified by whether participants failed 1 previous pharmacologic treatment for TN or failed >1 previous treatment, including ≥1 pharmacologic treatment (both studies); by the dose received at the end of the dose-optimization period (Study 1 only); and by region (Study 2 only; USA, Japan, and the rest of the world). In both studies, participants who complete the double-blind period may be eligible for a long-term extension (LTE) study in which they would receive up to an additional 52 weeks of vixotrigine treatment. Participants who prematurely discontinue the double-blind study treatment for reasons other than AEs but remain in the study may be eligible for the LTE.
The studies will be performed in accordance with the International Council for Harmonisation Guideline on Good Clinical Practice, European Union Clinical Trial Directive, United States Code of Federal Regulations, and the Declaration of Helsinki and in compliance with all federal and local guidance. Ethics committee approval at each site will be obtained.
Participants
Both studies will include participants aged ≥18 years, who have classical, purely paroxysmal TN (based on the International Headache Society International Classification of Headache Disorders beta criteria 13.1.1.118) diagnosed ≥3 months prior to study entry, who experience ≥3 attacks of pain/day with a mean pain score of ≥4 to ≤8 on an 11-point numerical rating scale during the run-in period, and who have failed ≥1 prior standard-of-care pharmacologic TN treatment. A sufficient number of participants (~200–250 for Study 1; ~270–320 for Study 2) will be recruited to ensure that 88 participants are randomized into the double-blind period per study. 88 participants gives 90% power at the 5% significance level, assuming the underlying difference in response rates is 35%. Of participants in the double-blind period, it is estimated that ~60 will enter the LTE period in each study.
Endpoints
The primary endpoint of both studies is the proportion of participants classified as responders at Week 12 of the double-blind period, where a responder is a participant who has ≥30% reduction in weekly mean pain score at Week 12 of the double-blind period from baseline, has not discontinued randomized study treatment before the end of Week 12 of the double-blind period, and has not taken prohibited pain medication before the end of Week 12 of the double-blind period (Figure 2). The 3 secondary endpoints relate to the proportion of participants who meet the definition of responder based on 1) a Patient Global Impression of Change (PGIC) response of “much improved” or “very much improved”; 2) ≥50% reduction in mean number of paroxysms from baseline; and 3) ≥50% reduction in mean pain score from baseline (Figure 2). For all responder-based endpoints, nonresponders will be classified as participants who did not meet all the criteria that define a responder. Other secondary endpoints will include the incidence of AEs and serious AEs (SAEs), quality of life, and evaluation of vixotrigine population pharmacokinetics (area under the concentration-time curve [AUC] and maximum concentration at steady state [Cmax]). Additional endpoints include proportion of responders based on the following criteria (Figure 2): 1) ≥30% reduction in mean worst pain score from baseline and 2) ≥30% reduction in mean number of paroxysms from baseline; change from baseline in mean pain score, mean worst pain score, and mean number of paroxysms at double-blind Week 12; change from baseline in mean pain score, mean worst pain score, and mean number of paroxysms by week during the dose-optimization/open-label and double-blind periods; average amount (dose in mg) of 1) acetaminophen, 2) pregabalin, and 3) immediate-release oxycodone used for TN pain per week while receiving randomized treatment; change from baseline to double-blind Week 12 in the Penn Facial Pain Scale-Revised (PENN-FPS-R) score, EuroQoL 5-Dimensions 5-Level version (EQ-5D-5L) score, and Work Productivity and Activity Impairment (WPAI): Neuropathic Pain v2.0 score; and change from baseline in PENN-FPS-R score by week and change from baseline in EQ-5D-5L and WPAI: Neuropathic Pain v2.0 scores by visit, during the dose-optimization/open-label and double-blind periods.
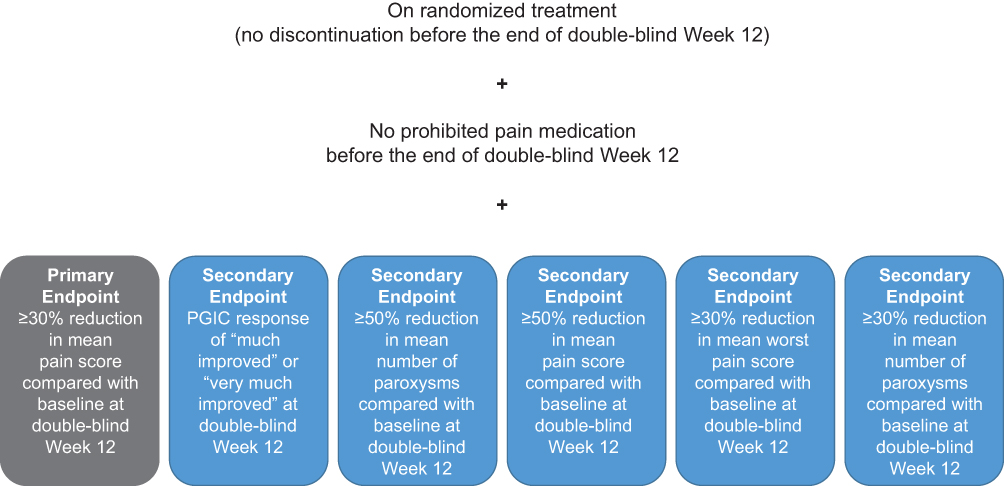 |
Figure 2 Double-blind Week 12 primary, secondary, and other responder endpoints. Abbreviation: PGIC, Patient Global Impression of Change. |
The primary endpoint of the LTE is the incidence of AEs and SAEs. Secondary LTE endpoints relate to the maintenance of vixotrigine effects and include the proportion of participants with a response based on the following criteria: 1) ≥30% reduction in mean pain score and 2) ≥50% reduction in mean number of paroxysms from baseline (recorded during the run-in period) during 4-week periods through Week 52 of the LTE; change from baseline in mean pain score, mean worst pain score, and mean number of paroxysms during 4-week periods through Week 52 of the LTE; proportion of participants with a PGIC response of “much improved” or “very much improved” at each LTE visit; and change from baseline in PENN-FPS-R, EQ-5D-5L, or WPAI: Neuropathic Pain v2.0 scores at each LTE visit.
Assessments
Clinical efficacy, safety, and laboratory assessments are the same for both studies. Clinical efficacy assessments include pain score, worst pain score, and number of paroxysms over the last 24 hrs (recorded daily in the evening through the LTE [if participating]) using an 11-point numerical rating scale in which participants rate pain from no pain (0) to worst pain imaginable (10); PGIC (relative to Day 1) and PENN-FPS-R19 score (recorded weekly through the LTE [if participating]); EQ-5D-5L and WPAI: Neuropathic Pain v2.0 scores (recorded at Day 1 and Week 4 of the dose-optimization/open-label periods and [for nonresponders receiving 150 mg TID in Study 1 only] Week 6 visits of the dose-optimization period; Week 4, 8, 12, and 14 visits of the double-blind period; upon premature treatment discontinuation; and at Day 1 through Week 52 of the LTE [if participating]); and amount of acetaminophen, pregabalin, immediate-release oxycodone, and immediate-release oxycodone/acetaminophen combination used for TN pain (recorded daily).
Clinical safety assessments will be conducted at screening and every subsequent visit in the full protocol and include physical examinations; 12-lead electrocardiograms; vital signs including temperature, heart rate, systolic and diastolic blood pressure, and respiratory rate while sitting; Columbia Suicide Severity Rating Scale (C-SSRS); and concomitant therapy and procedure review and recording of AEs and SAEs (monitoring and recording from the point of signing informed consent through end of LTE [if participating]).
Laboratory safety assessments will be conducted at screening and at subsequent visits, and will include pregnancy tests, standard hematology and chemistry parameters, and urinalyses.
Statistical Analyses
Detailed plans for statistical analysis will be specified in the statistical analysis plan. Two statistical analyses are planned for each study: 1) analyses related to the double-blind period will be performed when all participants have completed the double-blind period (first database lock) and 2) all analyses related to the LTE will be performed after all participants have completed the LTE (second database lock). Efficacy data, demographics, and baseline disease characteristics will be summarized by summary statistics. For continuous variables, the number of participants with data, mean, standard deviation, median, minimum, and maximum will generally be presented. For categorical variables, the number of participants with data and the percentage of those with data in each category will be presented.
For the primary endpoint, the proportion of responders at Week 12 of the double-blind period (based on ≥30% reduction from baseline in mean pain score) will be presented by treatment group (vixotrigine vs placebo). Logistic regression with the following independent variables will be used to assess the effect of vixotrigine vs placebo on response rates: treatment group, stratification factors, mean pain score at baseline, and mean pain score in the last week of the dose-optimization/open-label period. Odds ratios with 95% confidence intervals and P-values for treatment effect will be calculated. If there are few (<5) responders or nonresponders in either or both treatment groups, Fisher’s exact test will be used to compare the treatment groups.
For the 3 secondary endpoints, the proportions of responders at Week 12 of the double-blind period based on 1) a PGIC response of “much improved” or “very much improved”; 2) ≥50% reduction from baseline in mean number of paroxysms; or 3) ≥50% reduction from baseline in mean pain score), will be presented by treatment group (vixotrigine vs placebo). Logistic regression with the following independent variables will be used to assess the effect of vixotrigine vs placebo on response rates: treatment group; stratification factors; PGIC score at the end of the dose-optimization/open-label period (endpoint 1 above only); mean number of paroxysms at baseline and at the last week of the dose-optimization/open-label period (endpoint 2 above only); and mean pain score at baseline and at the last week of the dose-optimization/open-label period (endpoint 3 above only). If there are <5 responders or nonresponders in either or both treatment groups, Fisher’s exact test will be used to compare treatment groups.
For the responder endpoints at Week 12 of the double-blind period based on pain scores and paroxysms, participants who complete 12 weeks of double-blind treatment and do not take prohibited medications will have any missing Week 12 mean values imputed using a missing-at-random approach; intermittent missing weekly mean values will also be imputed using a missing-at-random approach. For the responder endpoints at Week 12 of the double-blind period based on PGIC, participants who have completed 12 weeks of double-blind treatment and not taken prohibited medications will be considered nonresponders if the Week 12 result is missing.
To control the overall type I error rate caused by multiple endpoints (the primary and 3 secondary endpoints), a sequential (closed) testing procedure will be used in which the endpoints are rank prioritized in the following order: 1) response based on ≥30% reduction from baseline in mean pain score (primary endpoint); 2) response based on PGIC score; 3) response based on ≥50% reduction from baseline in mean number of paroxysms; and 4) response based on ≥50% reduction from baseline in mean pain score. If treatment group comparisons do not reach statistical significance for any endpoint, all lower ranked endpoints will not be considered statistically significant. No multiple comparison adjustments will be made for other endpoints.
Only treatment-emergent AEs will be summarized; their incidence will be summarized by severity and relationship to study treatment. Treatment emergent is defined for the dose-optimization (Study 1) and open-label (Study 2) periods as having an onset/worsening date that is on or after the first dose of study treatment but before the first dose of double-blind treatment (if applicable), and for the double-blind period (both studies) it is defined as having an onset/worsening date that is on or after the first dose of randomized treatment. AEs of abuse potential will be reported, including timing relative to first dose, duration, severity, seriousness, and relatedness. These AEs will be summarized by treatment group and by severity, seriousness, and relatedness if incidences allow, and other subgroup analyses may be conducted. Shift tables will be used to summarize select laboratory data as specified in the statistical analysis plan. Potentially clinically significant abnormalities in laboratory values will be summarized.
Discussion
There is an unmet need for an effective, well-tolerated, noninvasive treatment for the neuropathic pain associated with TN. The current proposed studies will evaluate the efficacy and safety of vixotrigine in treating pain experienced by patients with TN. Current drugs are reasonably effective, but tolerability is a major problem that vixotrigine aims to address. Unlike other neuropathic pain conditions, TN is episodic and has its own natural remission periods, which make it challenging to evaluate. Patients remain ambivalent as to what is more important, reduction in intensity or reduction in frequency, hence the need for a composite value.20,21
There are multiple challenges from a previously published Phase II design17 that are addressed by this Phase III design. Participants with a mean pain score >8 on the 11-point PI-NRS during the run-in were excluded, given the potential ethical considerations of subjecting participants with extreme pain to a clinical trial with limitations on concomitant medications, and the potential of randomization to placebo. In addition, based on the Phase II study, potential participants with the highest levels of pain were considered to be higher risk of dropout due to desire for surgical intervention.17 A responder endpoint was chosen because it allows all participants to be included in the analysis, and ensures consistent handling of participants that have discontinued study treatment, taken prohibited medications, or have missing data. A 30% reduction in mean pain score is widely agreed to represent a clinically important improvement in chronic pain,22 and has been used in other chronic pain studies.23 Moreover, 28% reduction in average pain intensity has been determined to be the minimal clinically important difference for facial pain specifically.24 In addition, classification of nonresponders is based on the European Medicines Agency Guideline on the Clinical Development of Medicinal Products Intended for the Treatment of Pain.25 Demonstration of efficacy over 12 weeks is standard for support of a chronic pain indication26 but it was felt that participant awareness and anxiety about the end of the study may potentially confound or otherwise bias endpoint assessments; therefore, the study continues for 14 weeks despite assessing efficacy at 12 weeks.
The rationale for dose selection is based on the previous published study.13 In the Phase II study, vixotrigine 150 mg TID was well tolerated and yielded favorable effects on primary and secondary endpoints.13 A total of 11 of 44 participants who completed the open-label period had some reduction in pain severity or number of paroxysms but failed to meet the responder criteria for randomization into the double-blind period.13 A higher dose of vixotrigine (250 mg TID) may provide additional benefit to those not demonstrating an adequate response to 150 mg TID.13
Abbreviations
AE, adverse event; AUC, area under the concentration-time curve; Cmax, maximum concentration at steady state; C-SSRS, Columbia Suicide Severity Rating Scale; EQ-5D-5L, EuroQoL 5-Dimensions 5-Level version; LTE, long-term extension; Nav, voltage-gated sodium channel; PENN-FPS-R, Penn Facial Pain Scale-Revised; PGIC, Patients’ Global Impression of Change; PI-NRS, pain intensity numerical rating scale; SAE, serious adverse event; TID, 3 times daily; TN, trigeminal neuralgia; WPAI, Work Productivity and Activity Impairment.
Ethics Approval and Informed Consent
These studies will be performed in alignment with the ethical principles outlined in the Declaration of Helsinki. Informed consent will be obtained from all participants in accordance with local practice and regulations.
Acknowledgments
The authors would like to thank Deborah J. Steiner, MD, Amy Chappell, MD, and Valerie Morisset, PhD, for their contribution to the development of this paper. Biogen provided funding for medical writing support in the development of this paper; Yien Liu, PhD (Excel Scientific Solutions, Fairfield, CT) wrote the first draft of the manuscript based on input from the authors, and Miranda Dixon (Excel Scientific Solutions, Horsham, UK) copyedited and styled the manuscript per journal requirements. Biogen reviewed and provided feedback on the manuscript. The authors had full editorial control of the manuscript, and provided their final approval of all content.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
MK, HF, and HN are employees of and hold stock/stock options in Biogen. KG was an employee of Biogen at the time design was in development, holds stock/stock options in Biogen. KG is currently employed by Sarepta Therapeutics. WPC has received consulting fees from Biogen and reports personal fees from Biogen during the conduct of the study. JP and ST were employees of Convergence, subsequently Biogen when they acquired Convergence, and held stock/stock options in Biogen and Convergence. JMZ is supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre, has received consulting fees from Biogen, and reports grants from Rosetrees Trust outside the submitted work. The authors report no other possible conflicts of interest in this work.
References
1. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 2018;38(1):1–211. doi:10.1177/0333102417738202.
2. Feller L, Khammissa RAG, Fourie J, Bouckaert M, Lemmer J. Postherpetic neuralgia and trigeminal neuralgia. Pain Res Treat. 2017;2017:1681765. doi:10.1155/2017/1681765
3. Katusic S, Beard CM, Bergstralh E, Kurland LT. Incidence and clinical features of trigeminal neuralgia, Rochester, Minnesota, 1945-1984. Ann Neurol. 1990;27(1):89–95. doi:10.1002/ana.410270114
4. Di Stefano G, La Cesa S, Truini A, Cruccu G. Natural history and outcome of 200 outpatients with classical trigeminal neuralgia treated with carbamazepine or oxcarbazepine in a tertiary centre for neuropathic pain. J Headache Pain. 2014;15:34. doi:10.1186/1129-2377-15-34
5. Zakrzewska JM, Wu J, Mon-Williams M, Phillips N, Pavitt SH. Evaluating the impact of trigeminal neuralgia. Pain. 2017;158(6):1166–1174. doi:10.1097/j.pain.0000000000000853
6. Cruccu G, Finnerup NB, Jensen TS, et al. Trigeminal neuralgia: new classification and diagnostic grading for practice and research. Neurology. 2016;87(2):220–228. doi:10.1212/WNL.0000000000002840
7. Bendtsen L, Zakrzewska JM, Abbott J, et al. European Academy of Neurology guideline on trigeminal neuralgia. Eur J Neurol. 2019;26(6):831–849. doi:10.1111/ene.13950
8. Zakrzewska JM. Multi-dimensionality of chronic pain of the oral cavity and face. J Headache Pain. 2013;14:37. doi:10.1186/1129-2377-14-37
9. Tentolouris‐Piperas V, Lee G, Reading J, O’Keeffe A, Zakrzewska J, Cregg R. Adverse effects of anti‐epileptics in trigeminal neuralgiform pain. Acta Neurol Scand. 2018;137(6):566–574. doi:10.1111/ane.12901
10. Besi E, Boniface DR, Cregg R, Zakrzewska JM. Comparison of tolerability and adverse symptoms in oxcarbazepine and carbamazepine in the treatment of trigeminal neuralgia and neuralgiform headaches using the Liverpool Adverse Events Profile (AEP). J Headache Pain. 2015;16:563. doi:10.1186/s10194-015-0563-z
11. Zakrzewska JM, Linskey ME. Trigeminal neuralgia. BMJ. 2014;348(feb17 9):g474. doi:10.1136/bmj.g474
12. Gronseth G, Cruccu G, Alksne J, et al. Practice parameter: the diagnostic evaluation and treatment of trigeminal neuralgia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the European Federation of Neurological Societies. Neurology. 2008;71(15):1183–1190. doi:10.1212/01.wnl.0000326598.83183.04
13. Zakrzewska JM, Palmer J, Morisset V; for the Study Investigators, et al. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 2017;16(4):291–300. doi:10.1016/S1474-4422(17)30005-4.
14. Forrestal F, Naik H, Shinobu L, et al. Assessment of the efficacy and safety of vixotrigine in participants with lumbosacral radiculopathy: results of a randomized, placebo-controlled trial [PS02]. Presented at: 7th International Congress on Neuropathic Pain; May 9–11, 2019; London, UK.
15. Versavel M. Efficacy and safety of the novel sodium channel blocker CNV1014802 in trigeminal neuralgia and lumbosacral radiculopathy. J Pain Relief. 2015;4(3):36. doi:10.4172/2167-0846.S1.002
16. Moore RA, Wiffen PJ, Eccleston C, et al. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. Pain. 2015;156(8):1382–1395. doi:10.1097/j.pain.0000000000000088
17. Zakrzewska JM, Palmer J, Bendtsen L, et al. Challenges recruiting to a proof of concept pharmaceutical trial in a rare disease: the trigeminal neuralgia experience. Trials. 2018;19(1):704. doi:10.1186/s13063-018-3045-1
18. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629–808. doi:10.1177/0333102413485658.
19. Symonds T, Randall JA, Hoffman DL, Zakrzewska JM, Gehringer W, Lee JYK. Measuring the impact of trigeminal neuralgia pain: the Penn Facial Pain Scale-Revised. J Pain Res. 2018;11:1067–1073. doi:10.2147/JPR.S152958
20. Lee JYK. Measurement of trigeminal neuralgia pain: Penn Facial Pain Scale. Neurosurg Clin N Am. 2016;27(3):327–336. doi:10.1016/j.nec.2016.02.003
21. Kumar S, Rastogi S, Kumar S, Mahendra P, Bansal M, Chandra L. Pain in trigeminal neuralgia: neurophysiology and measurement: a comprehensive review. J Med Life. 2013;6(4):383–388.
22. Farrar JT, Young JP
23. Crofford LJ, Mease PJ, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain. 2008;136(3):419–431. doi:10.1016/j.pain.2008.02.027
24. Sandhu SK, Halpern CH, Vakhshori V, Mirsaeedi-Farahani K, Farrar JT, Lee JY. Brief Pain Inventory–Facial minimum clinically important difference. J Neurosurg. 2015;122(1):180–190. doi:10.3171/2014.8.JNS132547
25. European Medicines Agency. Guideline on the Clinical Development of Medicinal Products Intended for the Treatment of Pain; 2016. London, UK. Available from. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-development-medicinal-products-intended-treatment-pain-first-version_en.pdf.
26. Dworkin RH, Turk DC, Peirce-Sandner S, et al. Research design considerations for confirmatory chronic pain clinical trials: IMMPACT recommendations. Pain. 2010;149(2):177–193. doi:10.1016/j.pain.2010.02.018
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.