")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Design of an epitope-based peptide vaccine against spike protein of human coronavirus: an in silico approach
Authors Oany A, Emran AA, Jyoti TP
Received 16 May 2014
Accepted for publication 11 June 2014
Published 21 August 2014 Volume 2014:8 Pages 1139—1149
DOI https://doi.org/10.2147/DDDT.S67861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Arafat Rahman Oany,1 Abdullah-Al Emran,1,2 Tahmina Pervin Jyoti3
1Department of Biotechnology and Genetic Engineering, Life Science Faculty, Mawlana Bhashani Science and Technology University, Tangail, Bangladesh; 2Translational Research Institute, University of Queensland, Brisbane, Australia; 3Biotechnology and Genetic Engineering Discipline, Life Science School, Khulna University, Khulna, Bangladesh
Abstract: Human coronavirus (HCoV), a member of Coronaviridae family, is the causative agent of upper respiratory tract infections and “atypical pneumonia”. Despite severe epidemic outbreaks on several occasions and lack of antiviral drug, not much progress has been made with regard to an epitope-based vaccine designed for HCoV. In this study, a computational approach was adopted to identify a multiepitope vaccine candidate against this virus that could be suitable to trigger a significant immune response. Sequences of the spike proteins were collected from a protein database and analyzed with an in silico tool, to identify the most immunogenic protein. Both T cell immunity and B cell immunity were checked for the peptides to ensure that they had the capacity to induce both humoral and cell-mediated immunity. The peptide sequence from 88–94 amino acids and the sequence KSSTGFVYF were found as the most potential B cell and T cell epitopes, respectively. Furthermore, conservancy analysis was also done using in silico tools and showed a conservancy of 64.29% for all epitopes. The peptide sequence could interact with as many as 16 human leukocyte antigens (HLAs) and showed high cumulative population coverage, ranging from 75.68% to 90.73%. The epitope was further tested for binding against the HLA molecules, using in silico docking techniques, to verify the binding cleft epitope interaction. The allergenicity of the epitopes was also evaluated. This computational study of design of an epitope-based peptide vaccine against HCoVs allows us to determine novel peptide antigen targets in spike proteins on intuitive grounds, albeit the preliminary results thereof require validation by in vitro and in vivo experiments.
Keywords: vaccinomics, HLA, atypical pneumonia, allergenicity, docking
Introduction
Human coronavirus (HCoV) belongs to the Coronaviridae family (alphacoronavirus 1) and comprises a large group of enveloped, positive-sense, single-stranded polyadenylated RNA virus.1,2 It consists of the largest known viral RNA genomes, ranging from 27.6 to 31.6 kb. Usually, coronaviruses are classified into three groups (group I to III), based on their serological cross-reactivity.3 Their classification is also supported by evolutionary analysis.1 The group I viruses are animal pathogens, including porcine epidemic diarrhea virus and feline infectious peritonitis virus. The group II viruses are responsible for domestic animal pathogenic infections, and the final group III viruses are responsible for avian species infection.4 However both the group I and group II viruses are considered HCoV. The protein molecules that usually contribute the structure of all coronaviruses are the spike (S), envelope (E), membrane (M) and nucleocapsid (N). HCoV is usually the causative agent of upper respiratory tract infections and also the causative agent of “atypical pneumonia”, which was first identified in the People’s Republic of China.5 As nowadays, an environmental resistance is shown by these viruses,6 it is urgent to develop an effective prevention for HCoV. Currently, there is no available treatment or vaccine to cure HCoV infections. Due to the ever rising spread of this viral infection, the development of vaccines or antiviral drugs against HCoVs infections is crucial.
A novel approach integrating immunogenetics and immunogenomics with bioinformatics for the development of vaccines is known as vaccinomics.7 This approach has been used to address the development of new vaccines. The present conventional approach for vaccine development relies on antigen expression, in sufficient amount, from in vitro culture models; however, many antigens, while expressed sufficiently, may not be good candidates for vaccine. With these conventional approaches, it has not been possible to control different types of outbreaks of viral pathogens, such as recent avian and swine influenza strains, due to their time-consuming development process. Hence, the rapid in silico informatics-based approach has gained much popularity with the recent advancement in the sequencing of many pathogen genomes and protein sequence databases.8 The “vaccinomics” approach has already proven to be essential for combating diseases such as multiple sclerosis,9 malaria,10 and tumors.11 However, these methods of vaccine development usually work through the identification of human leukocyte antigens (HLA) ligands and T cell epitopes,12 which specify the selection of the potent vaccine candidates associated with the transporter of antigen presentation (TAP) molecules.13–16 Allergenicity assessment is one of the vital steps in the development of a peptide vaccine because when we provide the vaccine into the human body, it is detected as a foreign substance. As a result, inflammation occurs, demonstrating an allergic reaction. For the prediction of a B-cell epitope, hydrophilicity is an important criterion which is usually in the beta turns region. These assessments strengthen the possibility of the vaccine candidates. Therefore, our present study was undertaken to design an epitope-based peptide vaccine against HCoVs (229E, NL63, HKU1, EMC, and OC43) using the vaccinomics approach, with the wet lab researcher expected to validate our prediction.
Materials and methods
The flow chart summarizing the protocols for the complete epitope prediction is depicted in Figure 1.
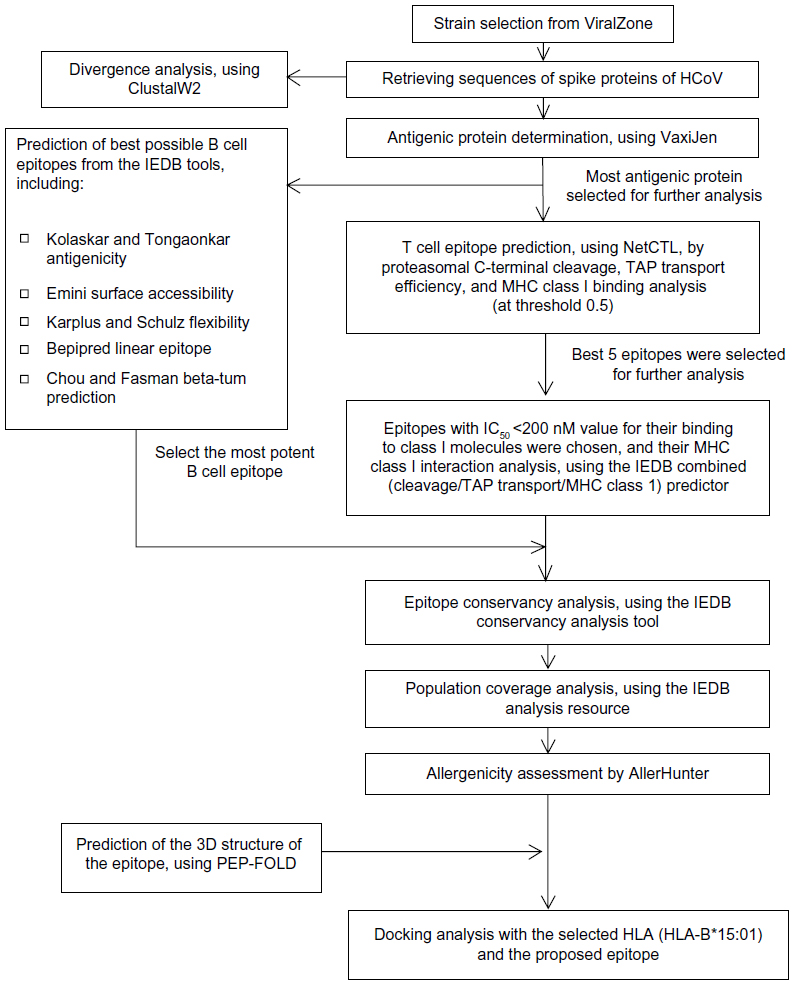 | Figure 1 Flow chart summarizing the protocols for the complete epitope prediction. |
Viral strain selection
ViralZone, a database of the ExPASy Bioinformatics Resource Portal was used for the selection of HCoVs and their associated information, including their genus, family, host, transmission, disease, genome, and proteome.
Protein sequence retrieval
The outer membrane protein (spike protein) sequences of HCoV were retrieved from the UniProtKB database.17 Then all the sequences were stored as a FASTA format for further analysis.
Evolution analysis
For the analysis of the evolutionary divergence in the membrane proteins of HCoV, a phylogenetic tree was constructed, using the ClustalW2 multiple sequence alignment tool.18
Antigenic protein identification
VaxiJen v2.0,19 a server for the prediction of protective antigens and subunit vaccines, was used for the determination of the most potent antigenic protein. Here, we used the default parameter of this server for the determination of the antigenic protein.
T Cell epitope identification
The NetCTL 1.2 server was used for the identification of the T cell epitope.20 The prediction method integrated peptide major histocompatibility complex class I (MHC-I) binding; proteasomal C terminal cleavage, and TAP transport efficiency. The epitope prediction was restricted to 12 MHC-I supertypes. MHC-I binding and proteasomal cleavage were performed through artificial neural networks, and the weight matrix was used for TAP transport efficiency. The parameter we used for this analysis was set at threshold 0.5 to maintain sensitivity and specificity of 0.89 and 0.94, respectively. This allowed us to recruit more epitopes for further analysis. A combined algorithm of MHC-I binding, TAP transport efficiency, and proteasomal cleavage efficiency were selected to predict overall scores.
A tool from the Immune Epitope Database21 was used to predict the MHC-I binding. The stabilized matrix base method (SMM)22 was used to calculate the half-maximal inhibitory concentration (IC50) values of peptide binding to MHC-I molecules from different prediction methods. For the binding analysis, all the alleles were selected, and the length was set at 9.0 before prediction was done. For the selected epitopes, a web-based tool was used to predict proteasomal cleavage, TAP transport, and MHC-I.23 This tool combines predictors of proteasomal processing, TAP transport, and MHC-I binding to produce an overall score for each peptide’s intrinsic potential as a T cell epitope. SMM was also implemented for this prediction.
Epitope conservancy analysis
For the analysis of the epitope conservancy, the web-based tool from IEDB24 analysis resource was used.
Prediction of population coverage
Population coverage for each individual epitope was selected by the IEDB population coverage calculation tool analysis resource. Here we used the allelic frequency of the interacting HLA alleles for the prediction of the population coverage for the corresponding epitope.
Allergenicity assessment
The web-based AllerHunter server25 was used to predict the allergenicity of our proposed epitope for vaccine development. This server predicts allergenicity through a combinational prediction, by using both integration of the Food and Agriculture Organization (FAO)/World Health Organization (WHO) allergenicity evaluation scheme and support vector machines (SVM)-pairwise sequence similarity. AllerHunter predicts allergens as well as nonallergens with high specificity. This makes AllerHunter is a very useful program for allergen cross-reactivity prediction.26,27
Molecular docking analysis and HLA allele interaction
Design of the three-dimensional (3D) epitope structure
For the docking analysis, the KSSTGFVYF epitope was subjected to PEP-FOLD web-based server28 for 3D structure conversion, in order to analyze the interactions with different HLAs. This server modeled five 3D structures of the proposed epitope, and the best one was selected for the docking analysis.
Docking analysis
To ensure the binding between HLA molecules and our predicted epitope, a docking study was performed using Molegro Virtual Docker, version 6.0 (CLC bio, Aarhus, Denmark).29 The HLA-B*15:01 was selected for docking on the basis of the available Protein Data Bank (PDB) structure deposited in the database, which interacted with our proposed epitope. The Protein Data Bank structure 1XR8, of Epstein – Barr virus EBNA-3 complexed with human UbcH6 peptide, was retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) protein database30 and simplified to HLA-B*15:01. Finally the docking was established at a grid of X: 24.81, Y: 29.16, and Z: 40.59.
Identification of the B cell epitope
Prediction of potentially immunogenic epitopes in a given protein sequence may significantly reduce wet lab effort needed to discover the epitopes required for the design of vaccines and for immunodiagnostics. The aim of the prediction of the B cell epitope was to find the potential antigen that would interact with B lymphocytes and initiate an immunoresponse.31 Tools from IEDB were used to identify the B cell antigenicity, including the Kolaskar and Tongaonkar antigenicity scale,32 Emini surface accessibility prediction,33 Karplus and Schulz flexibility prediction,34 and Bepipred linear epitope prediction analysis.35 The Chou and Fasman beta turn prediction tool36 was used because the antigenic parts of a protein belong to the beta turn regions.37
Results
Divergence analysis of the retrieved sequences
A total of 56 outer membrane protein (spike protein) sequences from the different variants belonging to five types (229E, NL63, HKU1, EMC, and OC43) of HCoVs were retrieved from the UniProtKB database. Then, the sequences were subjected to multiple sequence alignments in order to construct a phylogenetic tree (Figure S1). The phylogram showed evolutionary divergence among the different strains of HCoV.
Antigenic protein prediction
The VaxiJen server assessed all of the retrieved protein sequences in order to find the most potent antigenic protein. UniprotKB id: B2KKT9 was selected as the most potent antigenic protein, with a highest total prediction score of 0.6016. Then, the protein was used for further analysis.
T cell epitope identification
In a preselected environment, the NetCTL server predicted the potent T cell epitopes from the selected protein sequence. Based on the high combinatorial score, the five best epitopes (Table 1) were selected for further analysis.
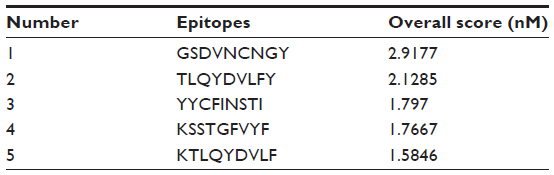 | Table 1 The selected epitopes, on the basis of their overall score predicted by the NetCTL server |
MHC-I binding prediction, which was run through SMM, predicted a wide range of MHC-I allele interactions with the five T cell epitopes. The MHC-I alleles for which the epitopes showed higher affinity (IC50 <200 nM) were selected for further analysis (Table 2).
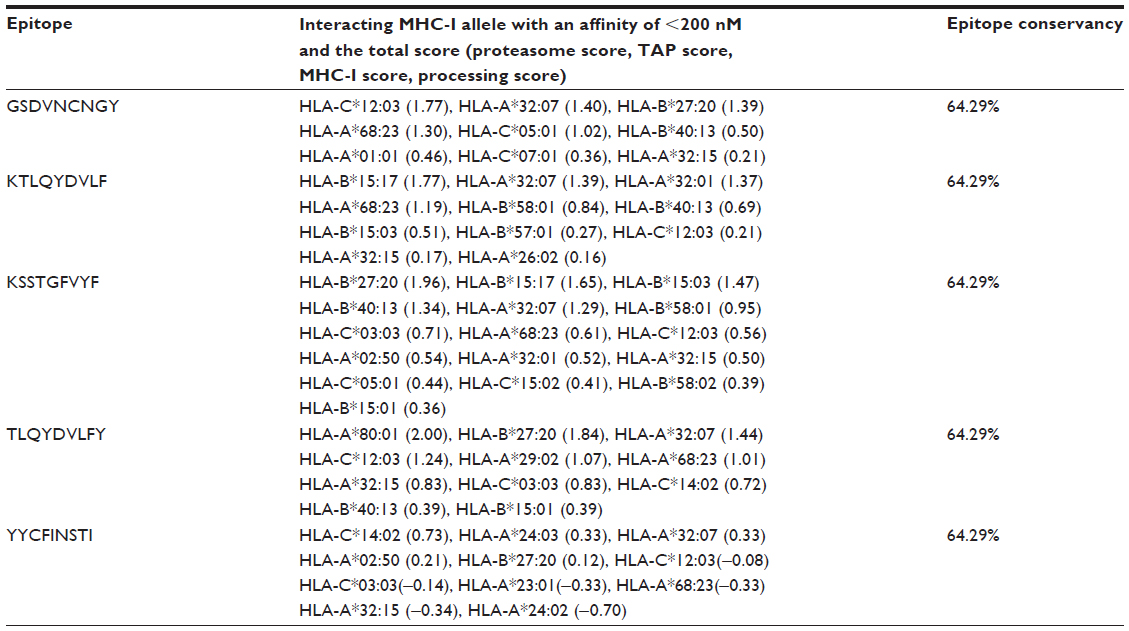 | Table 2 The five potential T cell epitopes, along with their interacting MHC-I alleles and total processing score, and epitope conservancy result |
MHC-I processing (proteasomal cleavage/TAP transport/MHC-I combined predictor) predicted an overall score for each peptide’s intrinsic potential to be a T cell epitope from the protein sequence. Proteasome complex, which cleaved the peptide bonds, thus converted the proteins into peptides. The peptide molecules from proteasome cleavage associated with class-I MHC molecules, and the peptide-MHC molecule then were transported to the cell membrane where they were presented to T helper cells. Here, higher overall score for each peptide denotes higher processing capabilities (Table 2).
Among the five T cell epitopes, a 9 mer epitope, KSSTGFVYF, was found to interact with most MHC-I alleles, including HLA-B*27:20; HLA-B*15:17; HLA-B*15:03; HLA-B*40:13; HLA-A*32:07; HLA-B*58:01; HLA-C*03:03; HLA-A*68:23; HLA-C*12:03; HLA-A*02:50; HLA-A*32:01; HLA-A*32:15; HLA-C*05:01; HLA-C*15:02; HLA-B*58:02; HLA-B*15:01 with higher affinity (Table 2).
Epitope conservancy and population coverage analysis
The IEDB conservancy analysis tool analyzed the conservancy of the predicted epitopes, which are shown in Table 2. The population coverage of the predicted epitopes is depicted in Figure 2.
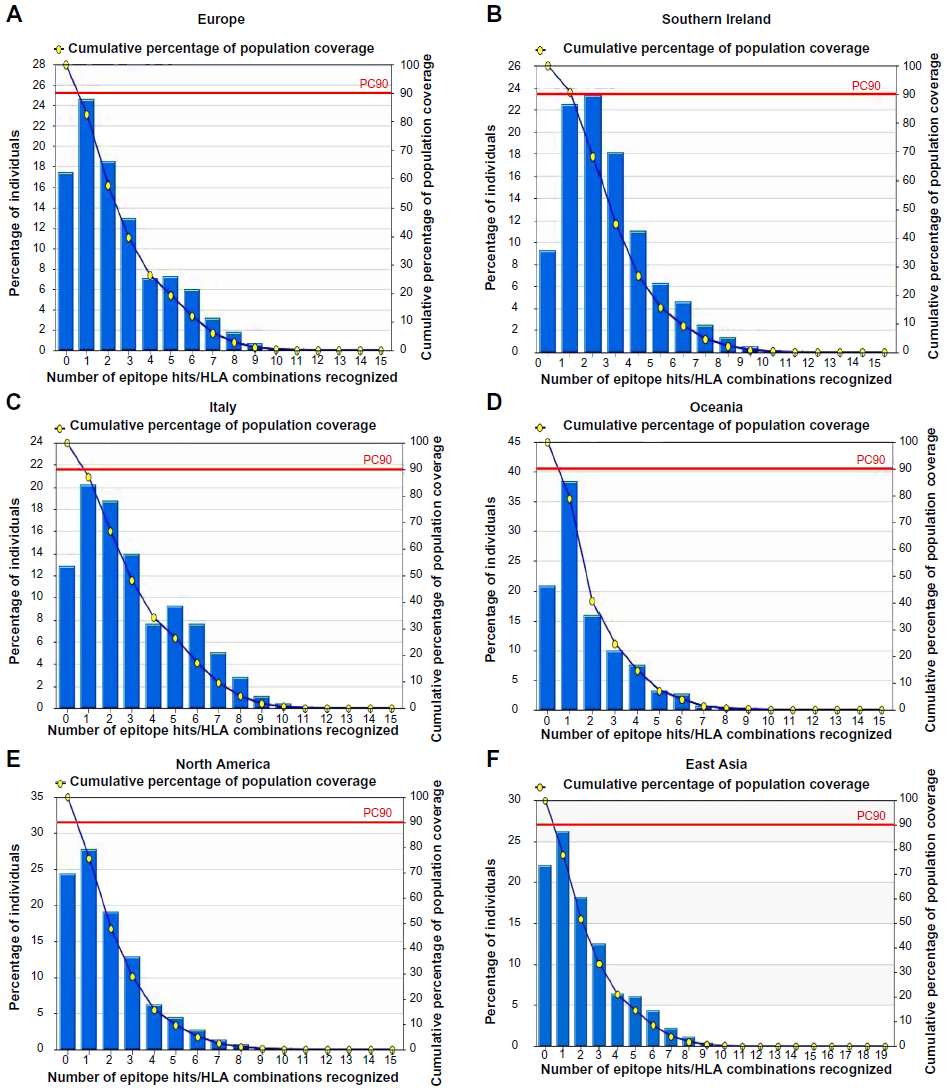 | Figure 2 Population coverage, based on MHC-I restriction data. Different HCoV-affected regions were selected for evaluation of the population coverage of the proposed epitopes. |
Allergenicity assessment
The sequence-based allergenicity prediction was precisely calculated using the AllerHunter tool, and the predicted queried epitope allergenicity score was 0.02 (sensitivity =93.0%, specificity =79.4%).
Molecular docking analysis
The predicted epitope bound in the groove of the HLA-B*15:01 with an energy of −17.662 kcal/mol. The docking interaction was visualized with the PyMOL molecular graphics system, version 1.5.0.4 (Schrödinger, LLC, Portland, OR, USA), shown in Figure 3.
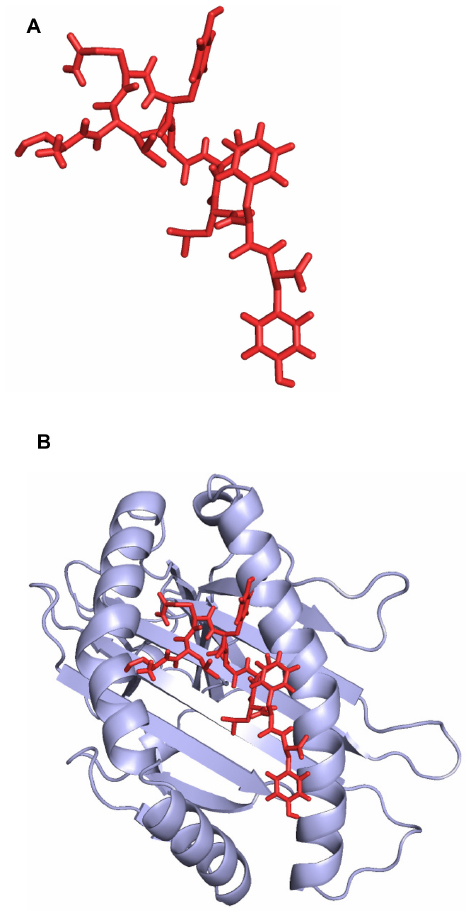 | Figure 3 HLA-B*15:01 and epitope KSSTGFVYF interaction analysis. (A) The three dimensional structure of the epitope KSSTGFVYF. (B) The epitope KSSTGFVYF binds in the groove of the HLA-B*15:01. |
B cell epitope identification
Here, we predicted amino acid scale-based methods for the identification of potential B-cell epitopes. According to this procedure we used different analysis methods for the prediction of a continuous B cell epitope.
The Kolaskar and Tongaonkar32 antigenicity prediction method analyzed antigenicity on the basis of the physiochemical properties of amino acids and abundances in experimentally known epitopes. The average antigenic propensity of the protein was 1.058, with maximum of 1.240 and minimum of 0.920. The antigenic determination threshold for the protein was 1.00; all values greater than 1.00 were potential antigenic determinants. We found that seven epitopes satisfied the threshold value set prior to the analysis, and they had the potential to express the B cell response. The results are summarized in Table 3 and Figure 4.
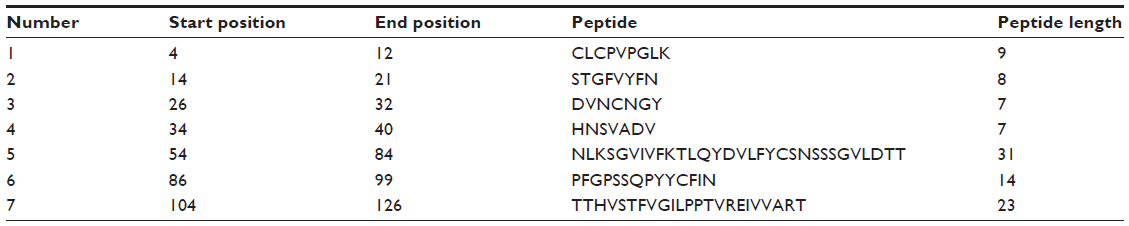 | Table 3 Kolaskar and Tongaonkar antigenicity analysis |
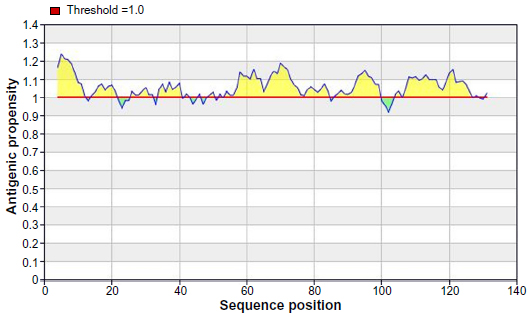 | Figure 4 Kolashkar and Tongaonkar antigenicity prediction of the most antigenic protein, B2KKT9. |
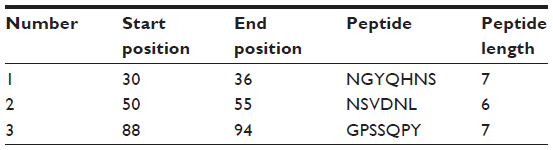
To be a potent B cell epitope, it must have surface accessibility. Hence Emini surface accessibility prediction was obtained. The region 88 to 94 amino acid residues were more accessible. This is described in Figure 5 and Table 4.
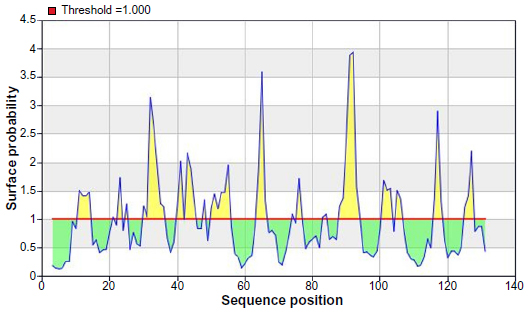 | Figure 5 Emini surface accessibility prediction of the most antigenic protein, B2KKT9. |
 | Table 4 Emini surface accessibility prediction of the peptides |
The beta turns are often accessible and considerably hydrophilic in nature. These are two properties of antigenic regions of a protein.38 For this reason, Chou and Fasman beta-turn prediction was done. The region 73–95 (approximately 73–79 and 88–95) was considered as a β-turns region (Figure 6).
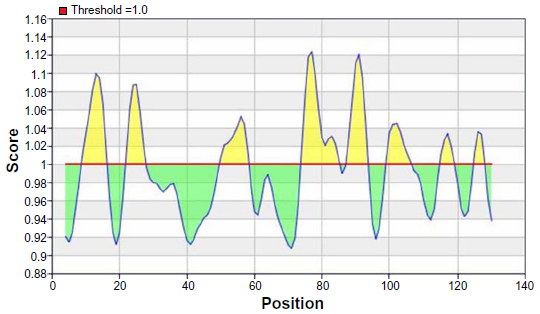 | Figure 6 Karplus and Schulz flexibility prediction of the most antigenic protein, B2KKT9. |
From the experimental evidence, it has been found that the flexibility of the peptide is correlated to antigenicity.39 Hence, the Karplus and Schulz34 flexibility prediction method was implemented. In this prediction method, the region of 75–95 was found to be the most flexible (Figure 7). Finally, we launched the Bepipred linear epitope prediction tool. This program is based on a Hidden Markov model, the best single method for predicting linear B-cell epitopes. The result of analysis with this method is summarized in Figure 8 and Table 5. By cross-referencing all the data, we predicted that the peptide sequences from 88–94 amino acids are capable of inducing the desired immune response as B cell epitopes.
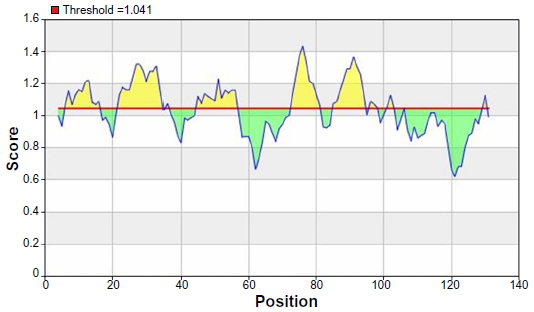 | Figure 7 Chou and Fasman beta-turn prediction of the most antigenic protein, B2KKT9. |
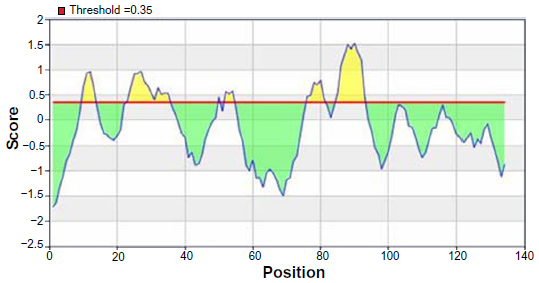 | Figure 8 Bepipred linear epitope prediction of the most antigenic protein, B2KKT9. |
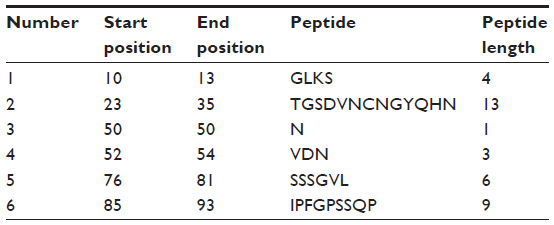 | Table 5 Bepipred linear epitope prediction |
Discussion
The development of a new vaccine in a timely fashion is very crucial for defending the ever rising global burden of disease.40–44 With the advancement of sequence-based technology, now we have enough information about the genomics and proteomics of different viruses. As a result, with the help of various bioinformatics tools, we can design peptide vaccines based on a neutralizing epitope. For example, the design of an epitope-based vaccine against rhinovirus,45 dengue virus,46 chikungunya virus,47 Saint Louis encephalitis virus,48 etc has already been suggested. Though epitope-based vaccine design has become a familiar concept, in the case of HCoV there has not yet been much work done. The HCoV is an RNA virus, which tends to mutate more frequently than the DNA viruses.49 These types of mutation mostly occur at the outer membrane protein, ie, at the spike protein.50 These types of mutation increase the sustainability of the HCoVs, by ensuring their escape from both the cell-mediated and humoral immune responses.51 Despite this, spike proteins have the most potential as a target for vaccine design because of their ability to induce a faster and longer-term mucosal immune response than that of the other proteins52 and for this reason, has gained much popularity with researchers.53,54 From this aspect, a universal HCoV vaccine needs to be designed, in order to overcome the adverse effects of this viral infection.
At present, vaccines are mostly based on B cell immunity. But recently, vaccine based on T cell epitope has been encouraged as the host can generate a strong immune response by CD8+ T cell against the infected cell.55 With time, due to antigenic drift, any foreign particle can escape the antibody memory response; however, the T cell immune response often provides long-lasting immunity. Here, we predicted both B cell and T cell epitopes for conferring immunity in different ways, but other recent studies about HCoV represented the T cell epitope only, and we want to express our greater findings here.56 There are several criteria that need to be fulfilled by a vaccine candidate epitope, and our predicted epitope fulfilled all the criteria. The initial criterion is the conservancy of the epitopes, which was measured by the IEDB conservancy analysis tool. Among the five potential T cell epitopes, all possessed the same conservancy, of 64.29%. We also found similar conservancy of the B cell epitope, which was 64.29%. Having the same conservancy for all the epitopes, the KSSTGFVYF epitope possessed the highest amount of interactions with the HLA alleles. A very recent study showed a highly conserved sequence in RNA directed RNA polymerase of HCoVs;56 nevertheless, our discovery of a spike protein with 64.29% conservancy among the 56 spike proteins has drawn much attention, and we consider this too as a epitope candidate for vaccine development.
Population coverage is another important factor in the development of a vaccine. For the all predicted epitopes, the cumulative percentage of population coverage was measured. We found the highest population coverage in South Ireland, which was 90.73%, followed by Italy and North America, with 87.13% and 75.68% coverage, respectively. The HCoV was first found in Europe,57,58 hence, we also observed the overall coverage in Europe and found this to be 82.59%. Oceania’s region covered 79.08%. We also recorded 80.31% population coverage for the East Asian region, considered as one of the hot spots of this viral infection. It has been reported that in Hong Kong and the People’s Republic of China, during 2001–2002 there were about 587 cases (among these, 26 children) of acute respiratory disease caused by different types of HCoV infection. Specifically, in Hong Kong, each year this virus caused about 224 hospitalizations per 100,000 population aged ≤6 years.59
However, allergenicity is one of the prominent obstacles in vaccine development. Today, most vaccines stimulate the immune system into an “allergic” reaction,60 through induction of type 2 T helper T (Th2) cells and immunoglobulin E (IgE). The AllerHunter score value is the probability that a particular sequence is a cross-reactive allergen. However, the threshold for prediction of allergen cross-reactivity is adjusted such that a sequence is predicted as a cross-reactive allergen if its probability is >0.06. Here, our proposed epitope’s allergenicity score was 0.02, and thus it was considered as a nonallergen. According to the FAO/WHO evaluation scheme of allergenicity prediction, a sequence is potentially allergenic if it either has an identity of at least six contiguous amino acids or >35 percent sequence identity over a window of 80 amino acids when compared to known allergens. Hence, our query epitopes did not fulfill the criteria for the FAO/WHO evaluation scheme of allergenicity prediction and was classified by this scheme as a nonallergen.
However, our predicted in silico results were based on diligent analysis of sequence and various immune databases. This type of study has recently received experimental validation,61 and for this reason, we have suggested that the proposed epitope would be able to trigger an efficacious immune response as a peptide vaccine in vivo.
Conclusion
Our study has shown that integrated computational approaches could be used for predicting vaccine candidates against pathogens such as HCoV, with previously described, validated procedures.
In this way, in silico studies save both time and costs for researchers and can guide the experimental work, with higher probabilities of finding the desired solutions and with fewer trial and error repeats of assays.
Disclosure
The authors report no conflicts of interests in this work.
References
González JM, Gomez-Puertas P, Cavanagh D, Gorbalenya AE, Enjuanes L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch Virol. 2003;148(11):2207–2235. | |
Siddell SG. The Coronaviridae. New York, NY: Plenum Press; 1995. | |
Drexler JF, Gloza-Rausch F, Glende J, et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. 2010;84(21):11336–11349. | |
Cavanagh D, Mawditt K, Welchman Dde B, Britton P, Gough RE. Coronaviruses from pheasants (Phasianus colchicus) are genetically closely related to coronaviruses of domestic fowl (infectious bronchitis virus) and turkeys. Avian Pathol. 2002;31(1):81–93. | |
Parry J. WHO investigates China’s fall in SARS cases. BMJ. 2003; 326(7402):1285. | |
Yoo D, Deregt D. A single amino acid change within antigenic domain II of the spike protein of bovine coronavirus confers resistance to virus neutralization. Clin Diagn Lab Immunol. 2001;8(2):297–302. | |
Poland GA, Ovsyannikova IG, Jacobson RM. Application of pharmacogenomics to vaccines. Pharmacogenomics. 2009;10(5):837–852. | |
Flower DR. Bioinformatics for Vaccinology. Chichester: John Wiley & Sons, Ltd; 2008. | |
Bourdette DN, Edmonds E, Smith C, et al. A highly immunogenic trivalent T cell receptor peptide vaccine for multiple sclerosis. Mult Scler. 2005;11(5):552–561. | |
López JA, Weilenman C, Audran R, et al. A synthetic malaria vaccine elicits a potent CD8(+) and CD4(+) T lymphocyte immune response in humans. Implications for vaccination strategies. Eur J Immunol. 2001;31(7):1989–1998. | |
Knutson KL, Schiffman K, Disis ML. Immunization with a HER-2/neu helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer patients. J Clin Invest. 2001;107(4):477–484. | |
Petrovsky N, Brusic V. Computational immunology: The coming of age. Immunol Cell Biol. 2002;80(3):248–254. | |
Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes – a framework for modelling, testing, and applications. Methods. 2004;34(4):436–443. | |
Peters B, Bulik S, Tampe R, Van Endert PM, Holzhütter HG. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J Immunol. 2003;171(4):1741–1749. | |
Bhasin M, Raghava GP. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci. 2004;13(3):596–607. | |
Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57(1–2):33–41. | |
Apweiler R, Bairoch A, Wu CH, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32(Suppl 1):D115–D119. | |
Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. | |
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8:4. | |
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8:424. | |
Buus S, Lauemøller SL, Worning P, et al. Sensitive quantitative predictions of peptide-MHC binding by a ‘Query by Committee’ artificial neural network approach. Tissue Antigens. 2003;62(5):378–384. | |
Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. 2005;6:132. | |
Tenzer S, Peters B, Bulik S, et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci. 2005; 62:1025–1037. | |
Bui HH, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007; 8(1):361. | |
Muh HC, Tong JC, Tammi MT. AllerHunter: a SVM-pairwise system for assessment of allergenicity and allergic cross-reactivity in proteins. PLoS One. 2009;4(6):e5861. | |
Liao L, Noble WS. Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J Comput Biol. 2003;10(6):857–868. | |
Chen YPP, Wong L, editors. A Better Gap Penalty for Pairwise-SVM: Proceedings of the 3rd Asia-Pacific Bioinformatics, Singapore, January 17–21, 2005. Singapore: Scientific Publishing Co; 2005. | |
Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40(Web Server issue):W288–W293. | |
Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49(11):3315–3321. | |
Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. | |
Nair DT, Singh K, Siddiqui Z, Nayak BP, Rao KV, Salunke DM. Epitope recognition by diverse antibodies suggests conformational convergence in an antibody response. J Immunol. 2002;168(5):2371–2382. | |
Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1–2):172–174. | |
Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55(3):836–839. | |
Karplus PA, Schulz GE. Prediction of chain flexibility in proteins. Naturwissenschaften. 1985;72(4):212–213. | |
Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2(1):2. | |
Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu Rev Biochem. 1978;47:251–276. | |
Rini JM, Schulze-Gahmen U, Wilson IA. Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science. 1992;255(5047):959–965. | |
Rose GD, Gierasch L, Smith JA. Turns in peptides and proteins. Adv Protein Chem. 1985;37:1–109. | |
Novotný J, Handschumacher M, Haber E, et al. Antigenic determinants in proteins coincide with surface regions accessible to large probes (antibody domains). Proc Natl Acad Sci U S A. 1986;83(2):226–230. | |
Marshall SJ. Developing countries face double burden of disease. Bull World Health Organ. 2004;82(7):556. | |
De Groot AS, Rappuoli R. Genome-derived vaccines. Expert Rev Vaccines. 2004;3(1):59–76. | |
Korber B, LaBute M, Yusim K. Immunoinformatics comes of age. PLoS Comput Biol. 2006;2(6):e71. | |
Fauci AS. Emerging and re-emerging infectious diseases: influenza as a prototype of the host-pathogen balancing act. Cell. 2006;124(4):665–670. | |
Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov. 2007;6(5):404–414. | |
Lapelosa M, Gallicchio E, Arnold GF, Arnold E, Levy RM. In silico vaccine design based on molecular simulations of rhinovirus chimeras presenting HIV-1 gp41 epitopes. J Mol Biol. 2009;385(2):675–691. | |
Chakraborty S, Chakravorty R, Ahmed M, et al. A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In Silico Biol. 2010;10(5–6):235–246. | |
Islam R, Sakib MS, Zaman A. A computational assay to design an epitope-based peptide vaccine against chikungunya virus. Future Virol. 2012;7(10):1029–1042. | |
Hasan MA, Hossain M, Alam MJ. A computational assay to design an epitope-based Peptide vaccine against Saint Louis encephalitis virus. Bioinform Biol Insights. 2013;7:347–355. | |
Twiddy SS, Holmes EC, Rambaut A. Inferring the rate and time-scale of dengue virus evolution. Mol Biol Evol. 2003;20(1):122–129. | |
Manzin A, Solforosi L, Petrelli E, et al. Evolution of hypervariable region 1 of hepatitis C virus in primary infection. J Virol. 1998;72(7):6271–6276. | |
Christie JM, Chapel H, Chapman RW, Rosenberg WM. Immune selection and genetic sequence variation in core and envelope regions of hepatitis C virus. Hepatology. 1999;30(4):1037–1044. | |
Ma C, Li Y, Wang L, et al. Intranasal vaccination with recombinant receptor-binding domain of MERS-CoV spike protein induces much stronger local mucosal immune responses than subcutaneous immunization: Implication for designing novel mucosal MERS vaccines. Vaccine. 2014;32(18):2100–2108. | |
Yang ZY, Kong WP, Huang Y, et al. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428(6982):561–564. | |
Agnihothram S, Gopal R, Yount BL, et al. Platform strategies for rapid response against emerging coronaviruses: MERS-CoV serologic and antigenic relationships in vaccine design. J Infect Dis [serial on the Internet]. Nov 2013 [cited May 16, 2014]. Available from: http://jid.oxfordjournals.org/content/early/2013/11/18/infdis.jit609. Accessed July 12, 2014. | |
Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78(15):8312–8321. | |
Sharmin R, Islam AB. A highly conserved WDYPKCDRA epitope in the RNA directed RNA polymerase of human coronaviruses can be used as epitope-based universal vaccine design. BMC Bioinformatics. 2014;15:161. | |
van der Hoek L, Pyrc K, Jebbink MF, et al. Identification of a new human coronavirus. Nat Med. 2004;10(4):368–373. | |
Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pöhlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci U S A. 2005;102(22):7988–7993. | |
Chiu SS, Chan KH, Chu KW, et al. Human coronavirus NL63 infection and other coronavirus infections in children hospitalized with acute respiratory disease in Hong Kong, China. Clin Infect Dis. 2005;40(12):1721–1729. | |
McKeever TM, Lewis SA, Smith C, Hubbard R. Vaccination and allergic disease: a birth cohort study. Am J Public Health. 2004;94(6):985–989. | |
Khan MK, Zaman S, Chakraborty S, et al. In silico predicted mycobacterial epitope elicits in vitro T-cell responses. Mol Immunol. 2014;61(1):16–22. |
Supplementary materials
 | Figure S1 Phylogenetic tree, showing the evolutionary divergence among the different membrane proteins of human coronaviruses.
|
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at