")
Back to Journals » International Journal of Nanomedicine » Volume 11
Design of a novel theranostic nanomedicine: synthesis and physicochemical properties of a biocompatible polyphosphazene–platinum(II) conjugate
Authors Avaji P, Park JH, Lee HJ, Jun YJ, Park KS, Lee KE, Choi S, Lee HJ, Sohn YS
Received 5 November 2015
Accepted for publication 14 January 2016
Published 2 March 2016 Volume 2016:11 Pages 837—851
DOI https://doi.org/10.2147/IJN.S99917
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Prakash G Avaji,1,2,* Jung Hyun Park,1,* Hyun Jeong Lee,1 Yong Joo Jun,2 Kyung Su Park,3 Kyung Eun Lee,3 Soo-Jin Choi,4 Hwa Jeong Lee,1 Youn Soo Sohn2
1Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul, Republic of Korea; 2C & Pharm, Ewha Womans University, Seoul, Republic of Korea; 3Advanced Analysis Center, Korea Institute of Science and Technology, Seoul, Republic of Korea; 4Department of Food Science and Technology, Seoul Women’s University, Seoul, Republic of Korea
*These authors contributed equally to this work
Abstract: To develop a theranostic nanomedicine involving the antitumor-active moiety (dach)Pt(II) (dach: trans-(±)-1,2-diaminocyclohexane) of oxaliplatin (OX), a new biocompatible polyphosphazene carrier polymer was designed by grafting with a methoxy poly(ethylene glycol) (MPEG) to increase duration of circulation in the blood and with aminoethanol (AE) as a spacer group. The antitumor (dach)Pt moiety was conjugated to the carrier polymer using cis-aconitic acid (AA) as a linker, resulting in a polymer conjugate formulated as [NP(MPEG550)(AE-AA)Pt(dach)]n, named “Polyplatin” (PP). PP was found to self-assemble into very stable polymeric nanoparticles with a mean diameter of 55.1 nm and a critical aggregation concentration of 18.5 mg/L in saline. PP could easily be labeled with a fluorescence dye such as Cy5.5 for imaging studies. The time-dependent ex vivo image studies on organ distributions and clearance of Cy-labeled PP have shown that PP accumulated in the tumor with high selectivity by the enhanced permeability and retention effect but was cleared out from all the major organs including the liver in about 4 weeks postinjection. Another time-dependent bioimaging study on distribution and clearance of PP in mouse kidney using laser ablation inductively coupled plasma mass spectroscopy has shown that PP accumulates much less in kidney and is more rapidly excreted than monomeric OX, which is in accord with the very low acute toxicity of PP as shown by its high LD50 value of more than 2000 mg/kg. The pharmacokinetic study of PP has shown that it has a much longer half-life (t1/2β) of 13.3 hours compared with the 5.21 hours of OX and about a 20 times higher area under the curve value of 42,850.8 ng h/mL compared with the 2,320.4 ng h/mL of OX. The nude mouse xenograft trials of PP against the gastric MKN-28 tumor cell line exhibited remarkably better tumor efficacy compared with OX at the higher tolerated dose, with lower systemic toxicity.
Keywords: polyphosphazene, anticancer agent, theranostics, nanomedicine, oxaliplatin
Introduction
Among the major platinum(II)-based chemotherapeutic agents such as cisplatin, carboplatin, and oxaliplatin (OX),1–5 the third-generation OX exhibits better efficacy and lower toxicity compared with cisplatin and carboplatin as well as no cross-resistance with cisplatin.6,7 However, OX exhibits neurotoxicity, leading to chemotherapy-induced peripheral neuropathy8,9 along with milder but similar side effects associated with cisplatin, which limit its clinical use.
Therefore, tremendous efforts have been made during the last decades to overcome such drug toxicities along with improved efficacy of platinum drugs using polymer therapy10,11 by exploring new polymeric platinum drugs based on various nanostructures including polymeric micelles, dendrimers, nanotubes, or nanocapsules.7,12 In particular, extensive research has been focused in recent years on the development of polymeric conjugate drugs by incorporating the antitumor-active moiety of OX in many different kinds of polymers. In the early stage, the active moiety (dach)Pt(II) was conjugated simply to the poly(ethylene glycols) (PEG) using a peptide spacer13 or to the hydrophilic N-(2-hydroxypropyl)methacrylamide using a peptide–aminomalonate spacer (AP5346), which is still in clinical Phase II trials.14,15 Kataoka's group16 reported the first micellar polymeric (dach)Pt conjugate prepared by conjugation of the (dach)Pt moiety to the amphiphilic poly(ethylene glycol)-b-poly(glutamic acid) block copolymer. Their initial micellar (dach)Pt conjugate was subjected to optimization to obtain the improved clinical candidate compound (NC-4016),17 which entered a Phase I clinical trial very recently.18 There are other reports on micellar block copolymers conjugated with the (dach)Pt moiety.19,20
We have also shown in our previous reports21,22 that polyphosphazenes conjugated with the active moiety of OX exhibited excellent tumor selectivity by enhanced permeability and retention (EPR) effect.23 Most recently, we have also reported a micellar polyphosphazene-(dach)Pt conjugate compound,24 which self-assembled into polymeric micelles in distilled water. However, it was later found that this polymeric conjugate did not form polymeric micelles in saline or buffer solutions, thus hampering its clinical application. The stability of nanostructures is one of the most essential requirements for passive tumor targeting by EPR effect. The bonding mode between the platinum(II) cation of the antitumor-active (dach)Pt(II) moiety and the carboxylate or amine group of the carrier polymer is also important, because this determines the releasing properties of the active moiety from the polymer in physiological conditions.10,25
It was critically pointed out in recent reviews26,27 that translation of nanomedicines into clinical use has been very limited because of their safety or for pharmacodynamic reasons despite great efforts in nanotechnology during last decades, and the paradigm for cancer therapy based on conventional nanomedicine is changing to theranostic nanomedicine, combining diagnostic and therapeutic functions into a single agent for more efficient personalized therapy.28,29 Fortunately, we have found very recently that the present drug carrier polyphosphazene bearing a long circulating PEG and a multifunctional side group could be a new platform of theranostic agents for cancer therapy, since both a hydrophobic anticancer drug and an imaging agent could easily be introduced to the polyphosphazene carrier polymer. In this study, we will show the theranostic and safety aspects of the polyphosphazene-(dach)Pt conjugate drug by performing imaging studies on its time-dependent biodistribution and clearance from major organs.
Materials and methods
Instruments and measurements
1H NMR (proton nuclear magnetic resonance) and proton-decoupled 31P NMR spectra were recorded using Varian 500 MHz NMR spectrophotometer (Varian, Inc., Palo Alto, CA, USA). Phosphoric acid was used as an internal standard for 31P NMR spectra. The cryo-transmission of electron microscopy (TEM) of Polyplatin (PP) (20 mg/mL) was measured as a thin aqueous film supported on a holey carbon grid at −170°C using the Tecnai F20 (FEI, Eindhoven, the Netherlands). The infrared spectra were measured using FT/IR-6100 Fourier transform infrared spectrophotometer. Elemental analysis was performed by means of Carlo Ebra-EA1108 (Carlo Ebra Reagents, Milan, Italy). The particle size distribution of the polyphosphazene carrier polymer and PP were measured by dynamic light scattering method using Malvern Zetasizer instrument (Nano-ZS; Malvern Instruments, Malvern, UK). The critical aggregation concentration (CAC) of nanoparticulate PP was measured in saline by the fluorescence pyrene method.30
Synthesis of the polyphosphazene intermediate, [NP(MPEG550)(AE)]n
Poly(dichlorophosphazene), prepared based on the previously published method,31 was reacted with sodium salt of methoxy poly(ethylene glycol) (MPEG550) to obtain a PEGylated polyphosphazene solution according to the procedure detailed in the literature.24 Meanwhile, the sodium salt of 2-aminoethanol (AE) prepared by reaction of AE (1.30 g, 21.3 mmol) with sodium hydride (0.61 g, 25.4 mmol) in dry tetrahydrofuran (50 mL) for 5 hours at 0°C was filtered and washed with diethyl ether. The sodium salt of AE dissolved in dimethyl sulfoxide (50 mL) was added to the PEGylated polyphosphazene solution. Finally, the mixed solution was stirred at 50°C for 24 hours. The reaction mixture was filtered and the filtrate was concentrated using a rotary evaporator. The concentrated solution was dialyzed for 1 day in methanol and 1 day in distilled water using cellulose membrane (molecular weight cutoff [MWCO]: 3.5 KDa), and then subjected to freeze-drying to obtain the pure intermediate. Yield: 70%. Elemental analysis (%): Calculated for C27H57N2O14P·H2O: C, 47.45; H, 8.64; N, 4.10. Found: C, 47.38; H, 8.61; N, 3.95. 1H NMR (CDCl3, ppm): δ 3.37 (s, 3H, methoxy proton of MPEG), 3.45–3.53 (m, 4H, methylene (CH2)2 protons of AE), 3.54–3.81 (brm, 46H, MPEG-ethylene –O–CH2–CH2– protons), 3.83–4.10 (brm, 2H, MPEG–methylene –P–O–CH2– protons). 31P NMR (CDCl3, ppm): −2.66 (O–P–O). Lower critical solution temperature (LCST): >100°C.
Synthesis of the polyphosphazene carrier polymer, [NP(MPEG550)(AE)(AA)]n
To obtain a polyphosphazene drug carrier polymer, cis-aconitic acid (AA) anhydride (2.35 g, 15.05 mmol) was slowly added to a solution of [NP(MPEG550)(AE)]n (1.0 g, 1.51 mmol) dissolved in 0.5M NaHCO3 buffer solution (pH 9.0, 50 mL) at 4°C, and 0.5 M Na2CO3 solution was added to maintain the reaction mixture at pH 9.0,32 which was stirred for 5 hours at 4°C. The final reaction mixture was dialyzed in distilled water using a cellulose membrane (MWCO: 3.5 KDa). The dialyzed solution was subjected to freeze-drying to obtain the pure carrier polymer [NP(MPEG550)(AE)(AA)]n. Yield: 78%. 1H NMR (CDCl3, ppm): δ 3.41 (s, 3H, methoxy protons of MPEG), 3.49–3.53 (m, 6H, methylene protons of cis-aconitate and methylene protons of AE protons), 3.57–3.83 (brm, 46H, PEG-ethylene –O–CH2–CH2– protons), 3.83–4.05 (brm, 2H, PEG-methylene –P–O–CH2– protons), 5.92 (s, 1H, –C=CH– vinylic proton of the cis-aconitate). 31P NMR (CDCl3, ppm): δ 3.95 (O–P–O). LCST: >100°C.
Synthesis of polyphosphazene-Pt conjugate PP, [NP(MPEG550)(AE)(AA)Pt(dach)]n
To a solution of polyphosphazene carrier polymer [NP(MPEG550)(AE)(AA)]n (1 g, 1.22 mmol) dissolved in methanol (10 mL), a solution of Ba(OH)2·8H2O (0.46 g, 1.46 mmol) in the same solvent (20 mL) was added to obtain the barium salt of the carrier polymer. The reaction mixture was stirred for 5 hours at room temperature and then subjected to evaporation by a rotary evaporator. To the residue dissolved in distilled water (10 mL), a solution of (dach)Pt(SO4) (0.59 g, 1.46 mmol, pH 7.2) in distilled water was slowly added until the reaction mixture became neutral at pH 7.2, and the reaction mixture was stirred for further 3 hours at room temperature. The reaction solution was filtered to remove barium sulfate, and the filtrate was dialyzed for 1 day in distilled water by using cellulose membrane (MWCO: 3.5 kDa). The dialyzed solution was subjected to freeze-drying to finally obtain the polyphosphazene-(dach)Pt conjugate [NP(MPEG550)(AE)(AA)Pt(dach)]n, named “Polyplatin”. Yield: 74%. Elemental analysis (%): Calculated for C39H73N4O19PPt·H2O; C, 40.83; H, 6.54; N, 4.88. Found: C, 40.54; H, 6.34; N, 4.62. 1H NMR (D2O, ppm): δ 1.04–1.20 (brm, 4H, cyclohexane-C-4, C-5 protons), 1.44 (brs, 2H, cyclohexane-C-3 protons), 1.82–1.93 (brm, 2H, cyclohexane-C-6 protons), 2.02–2.52 (brm, 2H, cyclohexane C-1, C-2 protons), 3.26 (s, 3H, methoxy proton of MPEG), 3.41–3.45 (m, 6H, methylene protons of cis-aconitate and methylene protons of AE moiety), 3.47–3.81 (brm, 46H, MPEG-ethylene –O–CH2–CH2– protons), 3.95–4.21 (brm, 2H, MPEG-methylene –P–O–CH2– protons), 4.69 (s, 1H, –C=CH– protons of cis-aconitate). 31P NMR (D2O, ppm): δ −4.53 (O–P–O). LCST: >100°C.
Preparation of Cy5.5-labeled PP for imaging study
In the previous synthetic process for the carrier polymer [NP(MPEG550)(AE)(AA)]n a little less equivalent of AA was reacted with the AE group of the polyphosphazene intermediate [NP(MPEG550)(AE)]n to save some unreacted terminal amine group for Cy5.5 labeling for imaging study. To a suspension of Cy5.5 mono acid (1 mg, 9.69×10−4 mmol), polyphosphazene drug carrier or its (dach)Pt(II) conjugate (60 mg, 0.06 mmol) in anhydrous DMF (5 mL) was added a coupling agent N, N, N′, N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (7 mg, 1.8×10−3 mmol) and triethyl amine (0.5 mL), and finally the reaction mixture was stirred at room temperature for 18 hours. The reaction mixture was diluted with ethanol (50 mL) and centrifuged in Vivaspin Polyethersulfone Ultrafiltration Spin Columns (MWCO: 50 kDa; GE Healthcare Bio Sciences Corp., Piscataway, NJ, USA) with capacity of 6 mL at 4,000 rpm and finally washed with water and saline several times.
Biodistribution and clearance studies of Cy5.5-labeled PP by ex vivo imaging
For biodistribution study, BALB/C nude mice (5 weeks old, 15–20 g) were allowed to adapt for 4 days and were then inoculated subcutaneously with non-small-cell lung carcinoma A549 cells (1×107 cells suspended in serum-free Roswell Park Memorial Institute 1640 media) in the right flank region. After 2 weeks, when the tumor reached 250–300 mm3, the Cy5.5-labeled PP dissolved in saline (60 mg/kg) was intravenously (IV) injected into the tail vein of the tumor-bearing mice. The animals were sacrificed at 12, 24, 48, and 72 hours after drug administration. Blood samples were collected by cardiac puncture with a syringe. Tumor, muscle, liver, kidney, spleen, lung, and blood were harvested from animals and stored at −80°C for analysis.
For clearance study, we used the same BALB/C nude mice without tumor inoculation. After adaptation in the same aforementioned way, five times higher dose of the Cy5.5-labeled PP (300 mg/kg) compared with the dose used in the biodistribution study was injected IV into the tail vein of 30 mice. The animals were sacrificed at predetermined times postinjection, with short intervals during the first week and then once every week thereafter. The fluorescent images of dissected organs and tumors were obtained using 12-bit CCD camera (Kodak Image Station 4000MM Instrument; Carestream Health, Inc., Rochester, NY, USA) equipped with a special C-mount lens and Cy5.5 bandpass emission filter (680–720 nm Omega optical).
All animal experiments were approved by the Institutional Animal Care and Use Committee of Ewha Womans University, Republic of Korea (IACUC Number 14-048).
Bioimaging of Pt in kidney of mouse by laser ablation inductively coupled plasma mass spectroscopy
To examine the time-dependent biodistribution and clearance of platinum metal of PP, the majority of which is distributed in kidneys right after IV injection, kidneys of the mice were dissected at 2, 12, and 24 hours postinjection according to the procedure detailed in the “Biodistribution and clearance studies of Cy5.5-labeled PP by ex vivo imaging” section and then sectioned into 100 μm units using the Cryostat, followed by drying at room temperature. In addition, the kidney samples of the mice treated with OX (as reference) at the same dose as PP based on (dach)Pt content were prepared for comparison. These kidney samples were subjected to Pt distribution mapping using femtosecond laser ablation inductively coupled plasma mass spectroscopy.33
In vitro drug release study
The time-dependent release of the antitumor-active moiety (dach)Pt from PP was examined under sink condition against different buffers (pH 5.4 and 7.4) at 37°C. Each 5 mL (2 mg/mL) of the solution was placed in a cellulose tubing membrane with MWCO of 2,000 Da and suspended in 30 mL of respective buffers under gentle magnetic stirring at 100 rpm. At predetermined time intervals, 1 mL samples were withdrawn from the dialysate over 10 days. The amount of released platinum was determined using inductively coupled plasma mass spectrometry.
Other in vivo animal studies
Pharmacokinetic study of PP and OX (as reference) was performed following our previous protocol,24 using male Sprague–Dawley rats weighing 260–280 g (7 weeks old) purchased from Orient Bio, Seongnam, Republic of Korea. Acute toxicity study of PP was carried out according to our previous method,34 given in the Organisation for Economic Co-operation and Development guideline 423, using male ICR mice aged 6 weeks and weighing 20–22 g. Finally, in vivo nude mouse xenograft trials to evaluate the in vivo antitumor activity of PP and OX (as reference) were performed against a gastric tumor cell line (MKN-28) according to our previous procedure,24 using BALB/C nude mice (5 weeks old, 15–20 g).
Results and discussion
Synthesis and characterization of the polyphosphazene carrier polymer and PP
The polyphosphazene carrier polymer and its (dach)Pt(II) conjugate PP were prepared by the reaction illustrated in Figure 1. Poly(dichlorophosphazene) (2) was prepared from hexachlorocyclotriphosphazene (1) by the reported method.31 The drug carrier polymer, [NP(MPEG550)(AE)(AA)]n (5), was prepared by stepwise nucleophilic substitutions of the chloropolymer (2) with hydrophilic MPEG550, then with the sodium salt of AE as a spacer group to obtain the intermediate polymer (4), and finally with AA as a linker for platination. All the substituted intermediates (3)–(6) and the final polyphosphazene-(dach)Pt conjugate PP were characterized by 1H and 31P NMR spectroscopy (Figures S1 and S2), and, in particular, the carrier polymer (5) and the final polymer conjugate PP (7) were subjected to elemental analysis in addition to their 1H and 31P NMR studies. The final PP was obtained as a light yellow solid soluble in water and polar organic solvents. PP was found to self-assemble into very stable nanoparticles both in distilled water and saline, as demonstrated in the following sections.
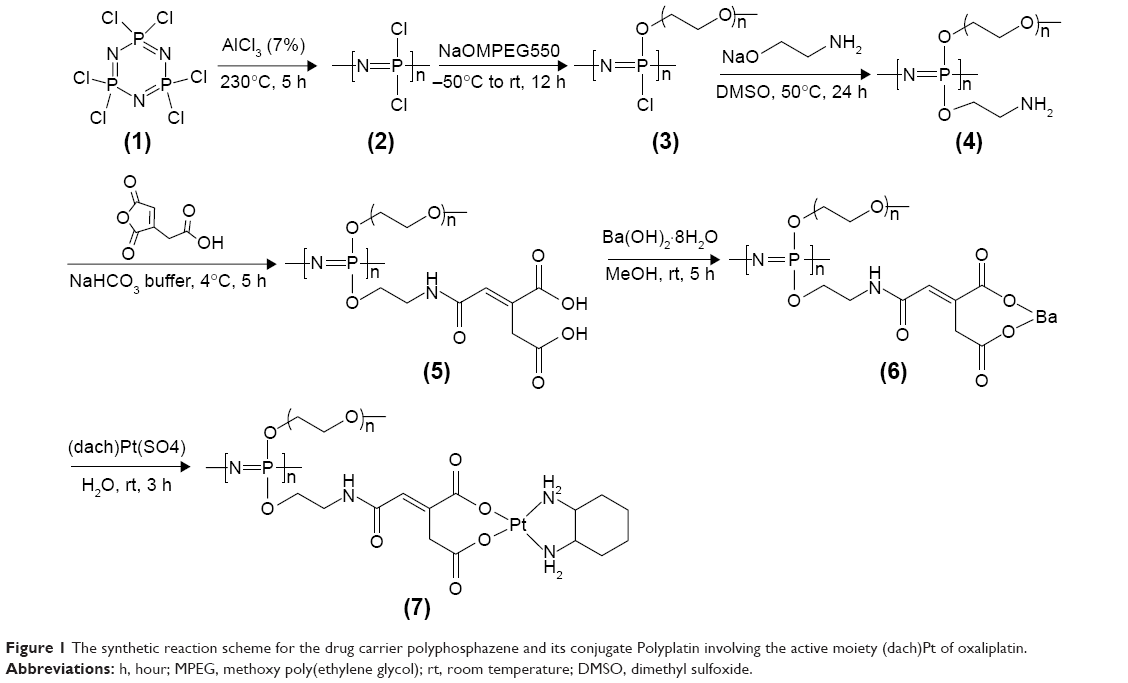 | Figure 1 The synthetic reaction scheme for the drug carrier polyphosphazene and its conjugate Polyplatin involving the active moiety (dach)Pt of oxaliplatin. |
Physicochemical properties and bonding mode of PP
To examine the morphology of the carrier polymer and PP, we measured their dynamic light scattering (DLS) pattern, since the tumor targeting properties (via EPR effect) are strongly dependent on the particle size and shape of the polymer conjugate in aqueous solution.23 The results of the DLS measurements of the carrier polymer and PP are displayed in Figure 2. The polyphosphazene carrier polymer exhibited a hydrodynamic volume of a mean diameter of 7.89 nm (Figure 2A), but its polymeric conjugate PP was presumed to self-assemble into nanoparticles with a mean diameter of 55.1 nm (Figure 2B). Therefore, to further confirm the exact morphology of PP, we measured its cryo-TEM image in saline, illustrated in Figure 2C. Surprisingly, as seen in the figure, the self-assembled nanoparticles are not spheres with smooth surface but coarse aggregates probably due to the heavy (dach)Pt(II) moiety, which makes it difficult to define their exact morphology. However, we found that these nanoparticles are very stable aggregates with a low CAC of 18.5 mg/mL in saline (measured by the fluorescence pyrene method), as shown in Figure 2D.30
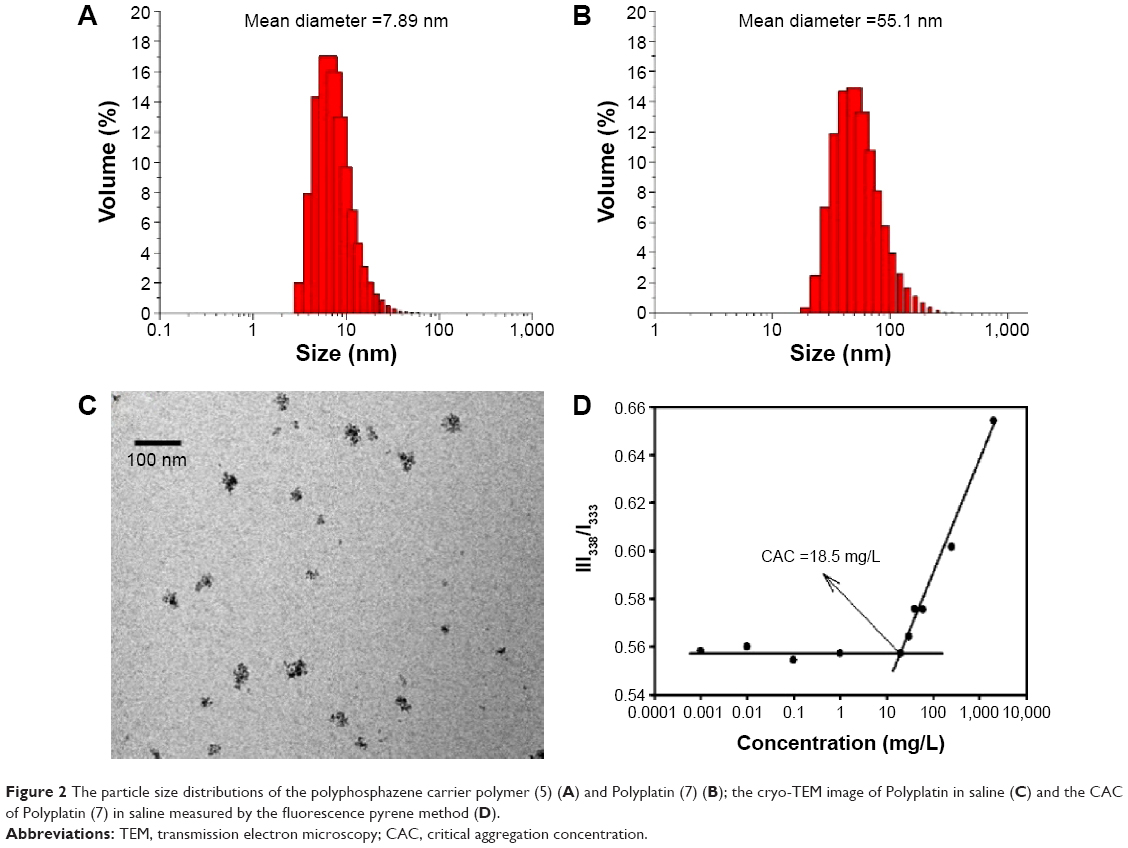 | Figure 2 The particle size distributions of the polyphosphazene carrier polymer (5) (A) and Polyplatin (7) (B); the cryo-TEM image of Polyplatin in saline (C) and the CAC of Polyplatin (7) in saline measured by the fluorescence pyrene method (D). |
One of the most important properties to be considered for clinical applications of the nanoparticulate polymer conjugate in addition to the aforementioned particle size for EPR effect is the solution stability of the conjugate nanoparticles in physiological conditions, since the conjugate nanoparticles should remain undestroyed during its circulation in the blood even after dilution by injection. As mentioned, the present conjugate nanoparticles behave like polymeric micelles; the low CAC value (18.5 mg/L) of the present polymer conjugate may be accepted as a measure of its solution stability in physiological conditions in contrast to no micelle formation of our previous conjugate in saline.24
We further studied the morphology and solution stabilities of PP in aqueous solution by time-dependent NMR and DLS measurements using the NMR sample solution of 5% PP in D2O and the DLS sample solution of 0.5% PP in saline, respectively. Both sample solutions were left in the dark at room temperature for more than 3 months, but no change was observed in the proton and phosphorous NMR spectra and particle size distributions.
Another important property required for the amphiphilic polymer conjugates for IV injection is the lower critical solution temperature (LCST), since all the amphiphilic polymers in aqueous solution exhibit an LCST at which the polymers precipitate from their solution when the solution temperature is raised.35 Therefore, the polymer conjugate for IV injection must have a remarkably higher LCST than the body temperature (37°C) in order to avoid precipitation of the injected polymer drug in the blood system. The LCST of the present PP measured for 5% solution was higher than 100°C, which is safe for IV injection.
Finally, the bonding mode of the platinum(II) cation to the carboxylate anions of the linker AA connected to the polymer spacer is also very important for releasing kinetics of the antitumor moiety (dach)Pt(II) from the polymer conjugate, which is closely related with both drug efficacy and toxicity. However, in general, because of structural complexity, direct evidence on the exact bonding mode of the (dach)Pt(II) moiety in the polymer conjugates was rarely reported. In most of the polymeric (dach)Pt(II) conjugates reported,6,8,17 the (dach)Pt(II) cation was presumed to be linearly bonded to two separate carboxylate anions linked to the polymer backbone except for the (O, N) chelation by amino acid.10
In this study, to confirm the bonding mode of the active (dach)Pt moiety to the linker AA connected to the spacer group AE of the polymer, we have simulated two different local environments of (dach)Pt(II) bound to two different carboxylate anions by preparing a monomeric complex of a linearly bonded [(dach)Pt(OOCCH3)2] and a chelated monomeric complex [(dach)Pt(AA)(AE)] (O, O-chelation model) to compare their carboxylate vibrational modes with those of PP [NP(MPEG550)(AE)(AA)Pt(dach)]n in their infrared spectra (Figure S3). To compare their carboxylate carbonyl vibrational patterns, their IR spectra in the region of 1,200–1,800 cm−1 were directly compared, as shown in Figure 3.
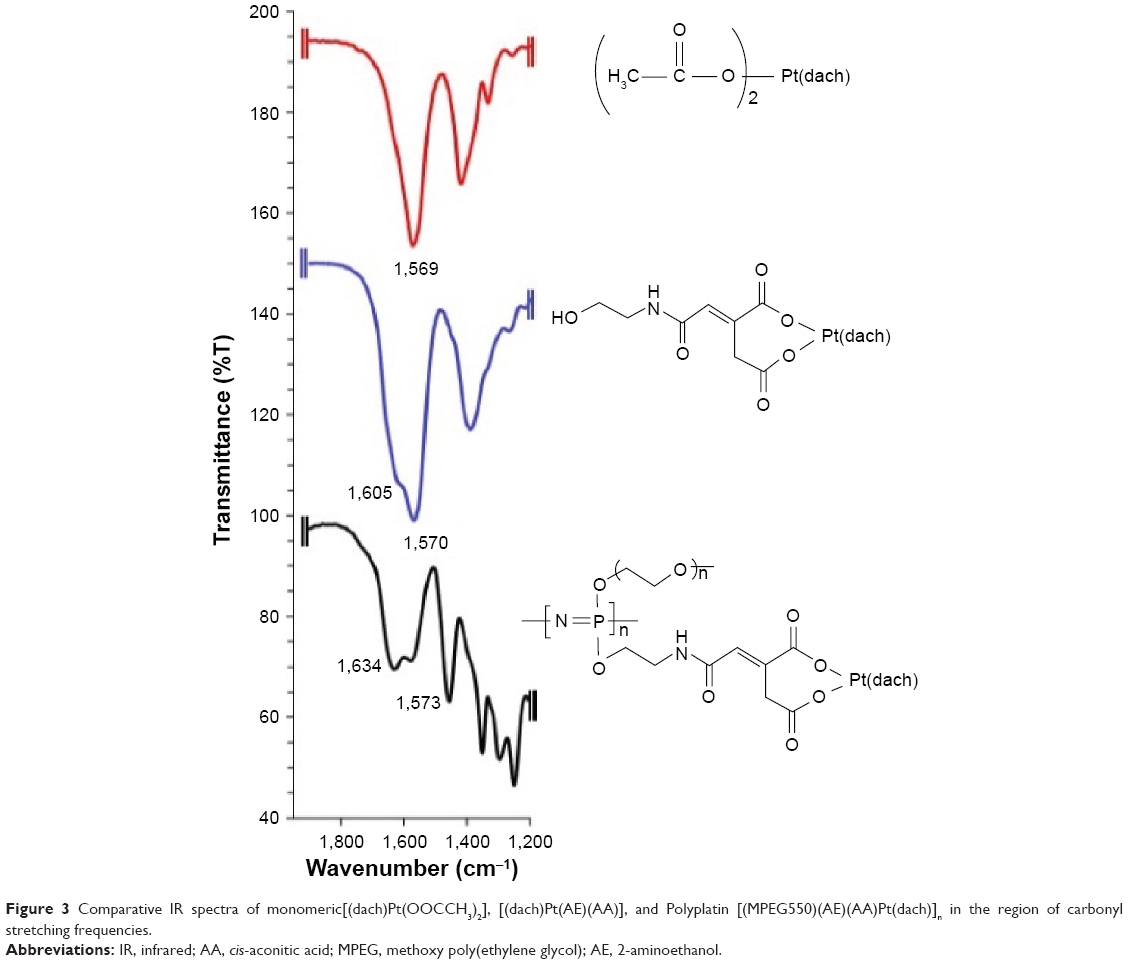 | Figure 3 Comparative IR spectra of monomeric[(dach)Pt(OOCCH3)2], [(dach)Pt(AE)(AA)], and Polyplatin [(MPEG550)(AE)(AA)Pt(dach)]n in the region of carbonyl stretching frequencies. |
As seen in the figure, the linear diacetatoplatinum(II) complex showed one single carbonyl stretching vibration at 1,569 cm−1, while the monomeric chelated complex [(dach)Pt(AA)(AE)] exhibited two carbonyl stretching bands at 1,605 and 1,570 cm−1, which were similarly observed at 1,634 and 1,573 cm−1 in the present polymer conjugate PP. Therefore, it may be concluded that the (dach)Pt moiety is chelated by two carboxylate anions of AA linked to the polymer conjugate PP. AA is known to be involved in the citric acid cycle and used as acid-cleavable linker for polymer–drug conjugates.36 Therefore, the present PP was rationally designed based on passive tumor targeting and acid-releasing kinetics in the microenvironment of tumor cells.
Time-dependent biodistribution and clearance of Cy5.5-labeled PP by image study
The fluorescence intensities of the major organs including tumor, muscle, lung, kidney, spleen, liver, and blood harvested at 12, 24, 48, and 72 hours postinjection from the mice injected with Cy-labeled PP were measured using Kodak Image Station 4000MM, and the results for the major organs are shown in Figure 4A as well as that for plasma in Figure 4B. It is seen from the reference color barcode below the photo in Figure 4A that PP accumulated dominantly in the tumor tissue by EPR effect23 and reached its maximum concentration at 48 hours postinjection. The plasma concentration of PP in Figure 4B is shown to decrease slowly, indicating longer circulation of PP in the blood. It is further clear from the quantitative diagram of the time-dependent fluorescence intensities of the major organs in Figure 4C that PP concentration in the tumor was remarkably high compared with that in all other organs, and particularly, the tumor selectivity in terms of the tumor-to-tissue ratio was outstanding, reaching the maximum value of 12 at 48 hours postinjection, as shown in Figure 4D.
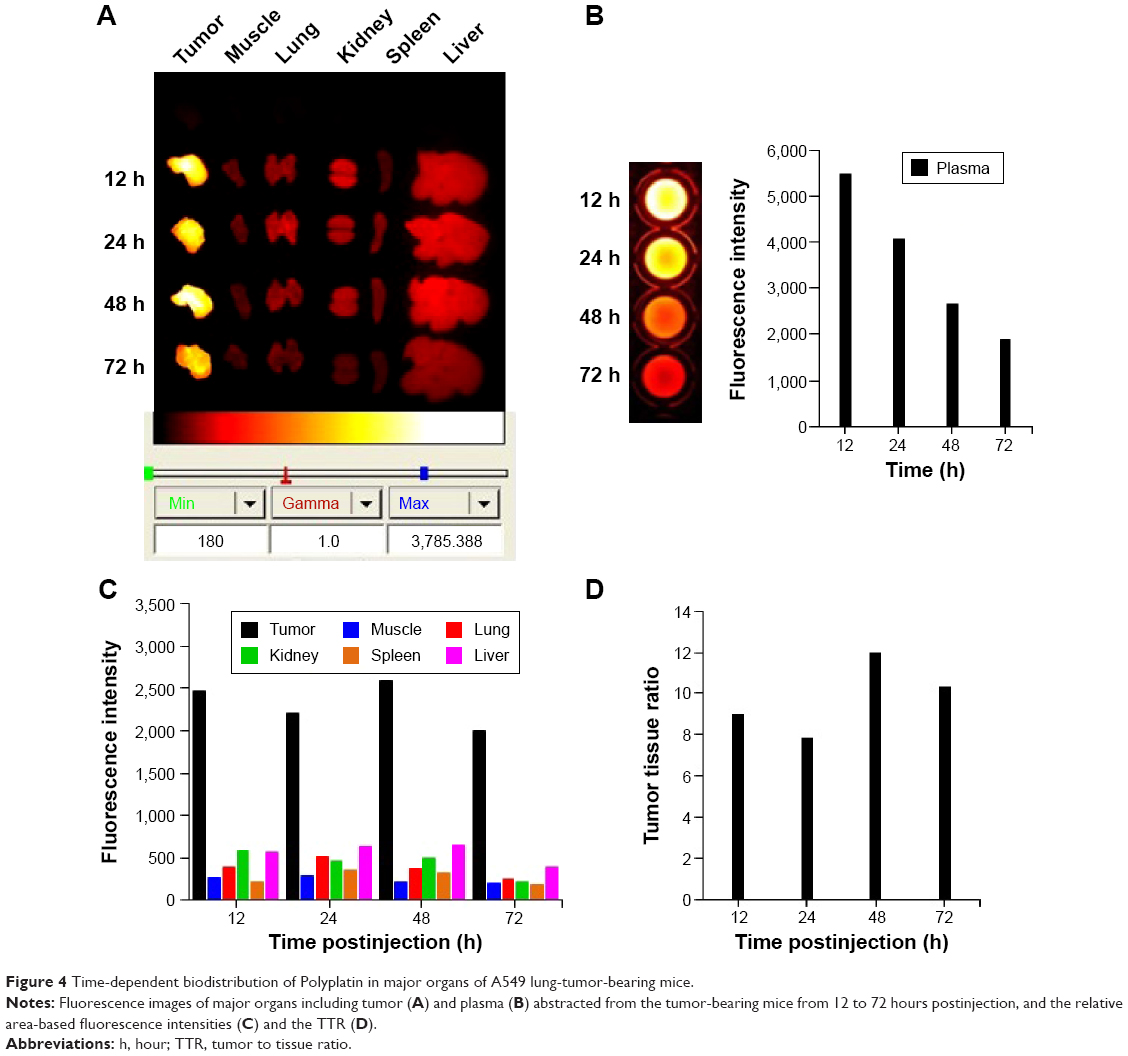 | Figure 4 Time-dependent biodistribution of Polyplatin in major organs of A549 lung-tumor-bearing mice. |
We have also performed clearance study of Cy-labeled PP, since one of the most critical safety factors of polymeric nanomedicine is its clearance from the human organs. We injected 30 BALB/C mice without tumors a five times higher dose of Cy-labeled PP (300 mg/kg) compared with the dose (60 mg/kg) used for the biodistribution study. The time-dependent fluorescence intensities of the major organs dissected from the mice sacrificed at scheduled times after injection were measured until the fluorescence intensity of each organ dropped below that of each blank organ. All the time-dependent clearance rates of PP from the major organs are illustrated collectively in Figure 5 (data of each organ in Figure S4). It is seen in the figure that at the early stage after PP injection, kidney was the main route for its excretion, but the PP accumulated in the liver and spleen for 2 days after its injection was then rapidly excreted thereafter. In any case, PP was cleared out from all the major organs including the liver in about 4 weeks after injection, probably due to the hydrolytic degradation of the polyphosphazene backbone.37
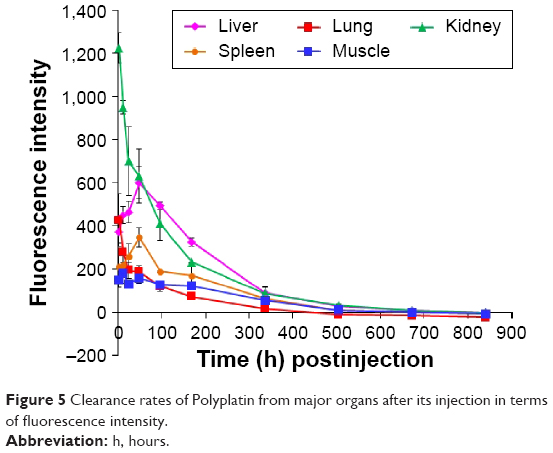 | Figure 5 Clearance rates of Polyplatin from major organs after its injection in terms of fluorescence intensity. |
Since nephrotoxicity is a common adverse effect of platinum-based anticancer drugs and since even the present polymeric conjugate drug PP exhibited dominant initial distribution in kidney as shown in Figure 5, we have examined directly the time-dependent images of platinum metal of PP and OX (as reference) distributed in the kidney by means of a most recently developed technique, the laser ablation inductively coupled plasma mass spectrometry, and the results are shown in Figure 6. From the color barcode, it can be seen that the platinum image at 2 hours postinjection OX showed dominant but a little heterogeneous distribution around the kidney, while that of PP exhibited much less and relatively homogeneous distribution of platinum metal overall in kidney. In addition, it was observed from the platinum images at 12 and 24 hours postinjection that the present PP was excreted from the kidney much more rapidly compared with OX, which is in accord with the results of the acute toxicity study of PP to be presented later.
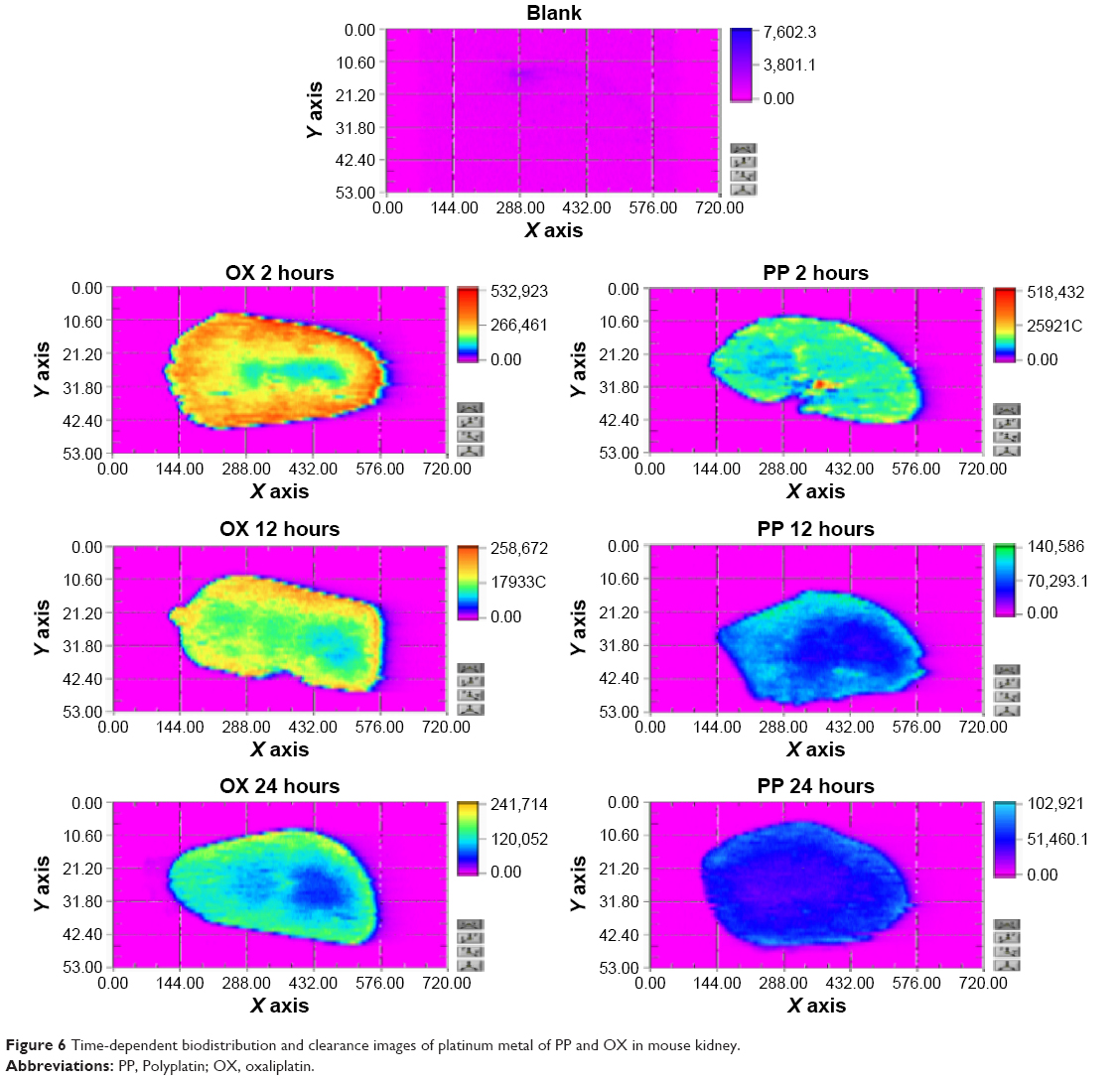 | Figure 6 Time-dependent biodistribution and clearance images of platinum metal of PP and OX in mouse kidney. |
Pharmacokinetics of PP
To compare the pharmacokinetic behavior of the present polymeric PP with the monomeric OX (as reference), we performed pharmacokinetic studies in male Sprague–Dawley rats using the standard protocol.24 The important pharmacokinetic parameters derived from the time-dependent plasma concentrations of PP and OX (as reference) (Figure S5) are listed in Table 1. Although the time-dependent plasma concentration curves of PP and OX both exhibited biphasic kinetic patterns like most other small molecular platinum(II) drugs, there are remarkable differences in their pharmacokinetic parameters (listed in Table 1).
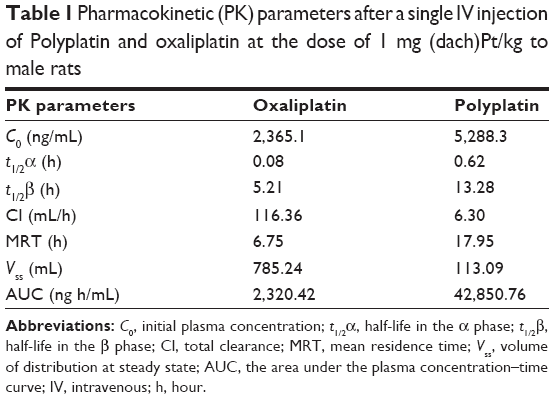 | Table 1 Pharmacokinetic (PK) parameters after a single IV injection of Polyplatin and oxaliplatin at the dose of 1 mg (dach)Pt/kg to male rats |
It is seen from the table that the present polymeric PP exhibited more than twice longer half-life of the β phase (t1/2 β) and more than 18 times larger area under the curve value (ng h/mL), reflecting the total time of drug exposure in the body and bioavailability, compared with the monomeric OX. Such a large area under the curve value of PP compared with that of monomeric OX is presumed to be ascribed to about 18 times longer clearance time of PP (6.3 mL/h) compared with OX (116.36 mL/h), probably due to the long blood circulation time of the PEGylated polyphosphazene carrier polymer.
Acute toxicity of PP
ICR mice were injected with PP in saline at doses of 5, 50, 300, and 2,000 mg/kg of PP to obtain its LD50 value and evaluate acute toxicity by IV injection. After injection, the mortality and changes in the body weight of the mice were recorded daily for a total of 14 days. No mortality was observed even at the highest dose of 2,000 mg/kg, and there was no considerable difference in body weight change depending on the dose injected (Figure S6). Therefore, the LD50 value of PP was estimated to be higher than 2,000 mg/kg, which implies very low toxicity of PP.
In vitro release of the (dach)Pt(II) moiety from PP
To investigate the pH-dependent releasing behavior of the active (dach)Pt moiety from PP in physiological condition, in vitro drug releasing experiments were performed by dialysis of PP in pH 5.4 and pH 7.4 phosphate buffer solutions at 37°C using a cellulose tubing membrane with MWCO of 2,000 Da. The time-dependent platinum content released from PP in a sustained and pH-sensitive manner was measured using inductively coupled plasma atomic absorption spectrometry, and the results are displayed in Figure 7. The proton NMR study on the released products indicated that (dach)Pt(II) moiety released from the conjugate formed the major part (>70%) along with small amounts of fragmented conjugate oligomers. The accumulated amount of platinum released from PP at pH 5.4 for 24 hours was approximately 25% of the total loaded (dach)Pt(II) and that for 10 days was 98%, while at pH 7.4, the accumulated amount of released (dach)Pt(II) was 18% and 72% for 24 hours and 10 days, respectively.
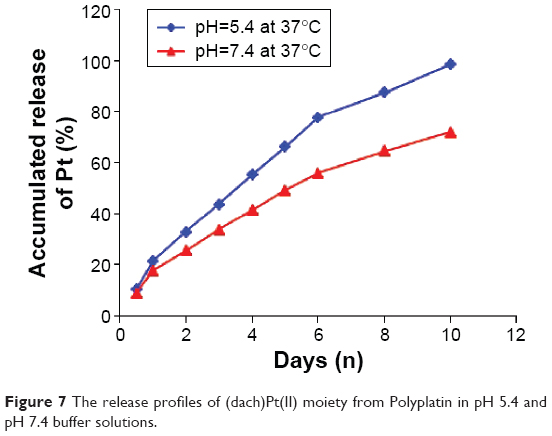 | Figure 7 The release profiles of (dach)Pt(II) moiety from Polyplatin in pH 5.4 and pH 7.4 buffer solutions. |
Such data indicate that our rational design of PP by introduction of AA as an acid cleavable linker for platination was appropriate, since (dach)Pt(II) moiety is better released from PP in the acidic condition, similar to the tumor microenvironment (as expected). Such a drug-releasing behavior seems to be related with the pH values mainly because the carboxylate-to-platinum(II) bond in the polyphosphazene-Pt(II) conjugate PP is more hydrolysable in acidic solutions than in neutral solutions.22 Such a fact is beneficial to delivery at acidic pH that prevails in the tumor microenvironment and can contribute to intratumoral site-specific release of the active moiety (dach)Pt(II) in cancerous cells.
In vivo nude mouse xenograft trials
To compare the drug efficacy of PP with that of the monomeric OX (as reference), in vivo xenograft trials of PP and OX have been performed using BALB/C nude mice (5 weeks old, 15–20 g) against the gastric tumor cell line (MKN-28). Both PP and OX in distilled water were administered to mice IV according to the standard schedule of triple injections on days 1, 5, and 9, and the results are displayed in Figure 8.
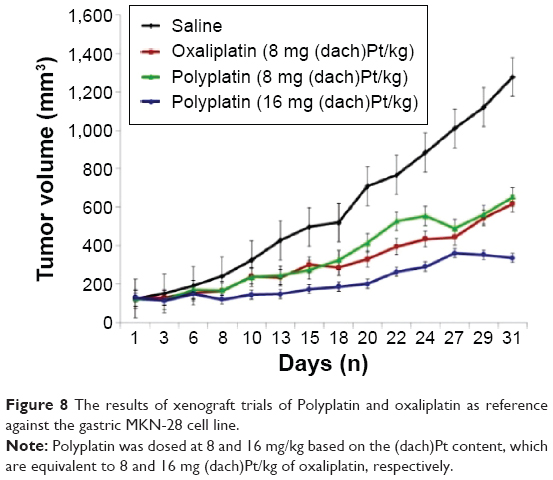 | Figure 8 The results of xenograft trials of Polyplatin and oxaliplatin as reference against the gastric MKN-28 cell line. |
To compare the antitumor activity of PP with that of OX, the dose of PP was determined based on the same content of (dach)Pt moiety of OX. The (dach)Pt content in OX is about 78%, and the known optimal dose of 10 mg/kg of OX corresponds to 7.8 mg (dach)Pt/kg. Therefore, in this experiment, the tumor growth inhibition effect of PP was compared with OX at the dose of 8 mg (dach)Pt/kg in the polymer conjugate. As is seen in Figure 8, PP has shown antitumor activity comparable to OX at the optimal dose of 8 mg (dach)Pt/kg, but much better activity at the double dose of 16 mg (dach)Pt/kg without any death of the five test mice. However, at the double dose of 16 mg (dach)Pt/kg of OX, all the mice were dead right after the second treatment, which is not shown in the figure, probably because of overdose of OX (LD50 of OX is known to be 14 mg (dach)Pt/kg).
The body weight changes of the mice treated with PP and OX (as reference) were observed (Figure S7). The mice treated with PP showed nearly no body weight changes both at low and high doses even during the drug injection period (days 1, 5, and 9), while mice treated with OX showed a significant weight loss of 5%–6% during the drug injection period. In conclusion, PP exhibited remarkably higher antitumor activity with lower systemic toxicity, which is in accord with all the aforementioned physicochemical and animal data.
Conclusion
A novel theranostic nanomedicine was prepared by conjugating the antitumor (dach)Pt moiety of OX to a newly designed polyphosphazene carrier polymer bearing MPEG550 and AE as side groups. This polyphosphazene-(dach)Pt conjugate named “Polyplatin” was found to self-assemble into very stable nanoparticles with a mean diameter of 55.1 nm and a low CAC of 18.5 mg/L, which is ideal for passive tumor targeting. Furthermore, PP was easily labeled with a fluorescence dye, Cy5.5, for various imaging studies. The time-dependent ex vivo imaging studies of the Cy-labeled PP have shown that PP IV injected into mice exhibited not only high tumor selectivity by EPR effect but also clearance from all the major organs in about 4 weeks postinjection. In addition, time-dependent bioimaging study of Pt distributed in the kidney of mouse injected with PP and OX (as reference) clearly showed that PP accumulated much less in the kidney and was excreted much more rapidly than OX. Such a result is in accord with the extremely low acute toxicity of PP, showing a very high value of LD50 (>2,000 mg/kg), indicating very low toxicity of PP. The nude mouse xenograft trials of PP and OX (as reference) against the gastric MKN-28 tumor cell line have shown that PP exhibited remarkably better antitumor efficacy with low systemic toxicity compared with OX. Therefore, PP is a strong candidate as a theranostic agent for clinical studies.
Acknowledgments
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (HI11C0532), by a National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MOE) (2014R1A1A2055876), and by C & Pharm.
Disclosure
The authors report no conflicts of interests in this work.
References
Highly MS, Calvert AH. Clinical experience with cisplatin and carboplatin. In: Kelland LR, Farrell NP, editors. Platinum-Based Drugs in Cancer Therapy. Totowa, NJ: Humana Press; 2000:171–194. | ||
Gordon M, Hollander S. Review of platinum anticancer compounds. J Med. 1993;24:209–265. | ||
Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci. 2007;334:115–124. | ||
Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. | ||
Grothey A, Goldberg RM. A review of oxaliplatin and its clinical use in colorectal cancer. Expert Opin Pharmacother. 2004;5:2159–2170. | ||
Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–476. | ||
Callari M, Aldrich-Wright JR, de Souza PL. Polymers with platinum drugs and other macromolecular metal complexes for cancer treatment. Prog Polym Sci. 2014;39:1614–1643. | ||
Wang R, Hu X, Wu S, et al. Biological characterization of folate-decorated biodegradable polymer–platinum(II) complex micelles. Mol Pharm. 2012;9:3200–3208. | ||
Pasetto LM, D’Andrea MR, Rossi E, Monfardini S. Oxaliplatin-related neurotoxicity: how and why? Crit Rev Oncol Hematol. 2006;59:159–168. | ||
Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed. 2006;45:1198–1215. | ||
Kabanov AV, Okano T. Challenges in polymer therapeutics: state of the art and prospects of polymer drugs. In: Maeda H, editor. Polymer Drugs in the Clinical Stage: Advantages and Prospects. New York, NY: Kluwer Academic/Plenum Publishers. Adv Exp Med Biol. 2003;519:1–27. | ||
Oberoi HS, Nukolova NV, Kabanov AV, Bronich TK. Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Deliver Rev. 2013;65:1667–1685. | ||
Furin A, Guiotto A, Baccichetti F, et al. Synthesis, characterization and preliminary cytotoxicity assays of poly(ethylene glycol)malonato-Pt-DACH conjugates. Eur J Med Chem. 2003;38:739–749. | ||
Sood P, Thurmond KB, Jacob JE, et al. Synthesis and characterization of AP5346, a novel polymer-linked diaminocyclohexyl platinum chemotherapeutic agent. Bioconjug Chem. 2006;17:1270–1279. | ||
Rice JR, Gerberich JL, Nowotnik DP, Howell SB. Preclinical efficacy and pharmacokinetics of AP5346, a novel diaminocyclohexane-platinum tumor-targeting drug delivery system. Clin Cancer Res. 2006;12:2248–2254. | ||
Cabral H, Nishiyama N, Okazaki S, Koyama H, Kataoka K. Preparation and biological properties of dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt)-loaded polymeric micelles. J Control Release. 2005;101:223–232. | ||
Cabral H, Nishiyama N, Kataoka K. Optimization of (1,2-diamino-cyclohexane) platinum(II)-loaded polymeric micelles directed to improved tumor targeting and enhanced antitumor activity. J Control Release. 2007;121:146–155. | ||
Rafi M, Cabral H, Kano MR, et al. Polymeric micelles incorporating (1,2-diaminocyclohexane) platinum(II) suppress the growth of orthotopic scirrhous gastric tumors and their lymph node metastasis. J Control Release. 2012;159:189–196. | ||
Xiao H, Zhou D, Liu S, et al. Delivery of active DACH-Pt anticancer species by biodegradable amphihpilic polymers using thiol-ene radical addition. Macromol Biosci. 2012;12:367–373. | ||
Paraskar A, Soni S, Roy B, Papa AL, Sengupta S. Rationally designed oxaliplatin-nanoparticle for enhanced antitumor efficacy. Nanotechnology. 2012;23:075103. | ||
Song R, Jun YJ, Kim JI, Jin C, Sohn YS. Synthesis, characterization, and tumor selectivity of a polyphosphazene-platinum(II) conjugate. J Control Release. 2005;105:142–150. | ||
Jun YJ, Kim JI, Jun MJ, Sohn YS. Selective tumor targeting by enhanced permeability and retention effect. Synthesis and antitumor activity of polyphosphazene-platinum(II) conjugates. J Inorg Biochem. 2005;99:1593–1601. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. | ||
Avaji PG, Joo HI, Park JH, Park KS, Jun YJ, Lee HJ, Sohn YS. Synthesis and properties of a new micellar polyphosphazene-platinum(II) conjugate drug. J Inorg Biochem. 2014;140:45–52. | ||
Kim YS, Song R, Chung HC, Jun MJ, Sohn YS. Coordination modes vs antitumor activity: synthesis and antitumor activity of novel platinum(II) complexes of N-substituted amino dicarboxylic acids. J Inorg Biochem. 2004;98:98–104. | ||
Duncan R. Polymer therapeutics: top 10 selling pharmaceuticals – what next? J Control Release. 2014;190:371–380. | ||
Venditto VJ, Szoka FC Jr. Cancer nanomedicines: so many papers and so few drugs! Adv Drug Deliv Rev. 2013;65:80–88. | ||
Ryu JH, Koo H, Sun IC, et al. Tumor-targeting multi-functional nanoparticles for theragnosis: new paradigm for cancer therapy. Adv Drug Deliv Rev. 2012;64:1447–1458. | ||
Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62:1064–1079. | ||
Inoue T, Chen G, Nakamae K, Hoffman AS. An AB block copolymer of oligo(methyl methacrylate) and poly(acrylic acid) for micellar delivery of hydrophobic drugs. J Control Release. 1998;51:221–229. | ||
Sohn YS, Cho YH, Beak H, Jung OS. Synthesis and properties of low molecular weight polyphosphazenes. Macromolecules. 1995;28:7566–7568. | ||
Sanjoh M, Hiki S, Lee Y, et al. pDNA/poly(L-lysine) Polyplexes functionalized with a pH-sensitive charge-conversional poly(aspartamide) derivative for controlled gene delivery to human umbilical vein endothelial cells. Macromol Rapid Commun. 2010;31:1181–1186. | ||
Moreno-Gordaliza E, Giesen C, Lazaro A, et al. Elemental bioimaging in kidney by LA-ICP-MS as a tool to study nephrotoxicity and renal protective strategies in cisplatin therapies. Anal Chem. 2011;83:7933–7940. | ||
Jadhav VB, Jun YJ, Song JH, et al. A novel micelle-encapsulated platinum(II) anticancer agent. J Control Release. 1998;51:221–229. | ||
Sohn YS, Kim JI, Song R, Jeong B. The relationship of thermosensitive properties with the structure of organo-phosphazenes. Polymer. 2004;45:3081–3084. | ||
Yoo HS, Lee EA, Park TG. Doxorubicin-conjugated biodegradable polymeric micelles having acid-cleavable linkages. J Control Release. 2002;82:17–27. | ||
Allcock HR, Pucher SR, Scopelianos AG. Poly[(amino acid ester)phosphazenes] as substrate for the controlled release of small molecules. Biomaterials. 1994;15:563–569. |
Supplementary materials
 | Figure S1 1H NMR spectra of the polyphosphazene carrier polymer[NP(PEG550)(AE)(AA)]n (A) and its (dach)Pt(II) conjugate [NP(MPEG550)(AE)(AA)Pt(dach)]n (B). |
 | Figure S2 31P NMR spectra of the half PEGylated polymer [NP(MPEG550)Cl]n (A), carrier polymer [NP(MPEG550)(AE)(AA)]n (B), and its Pt conjugate [NP(MPEG550)(AE)(AA)Pt(dach)]n (C). |
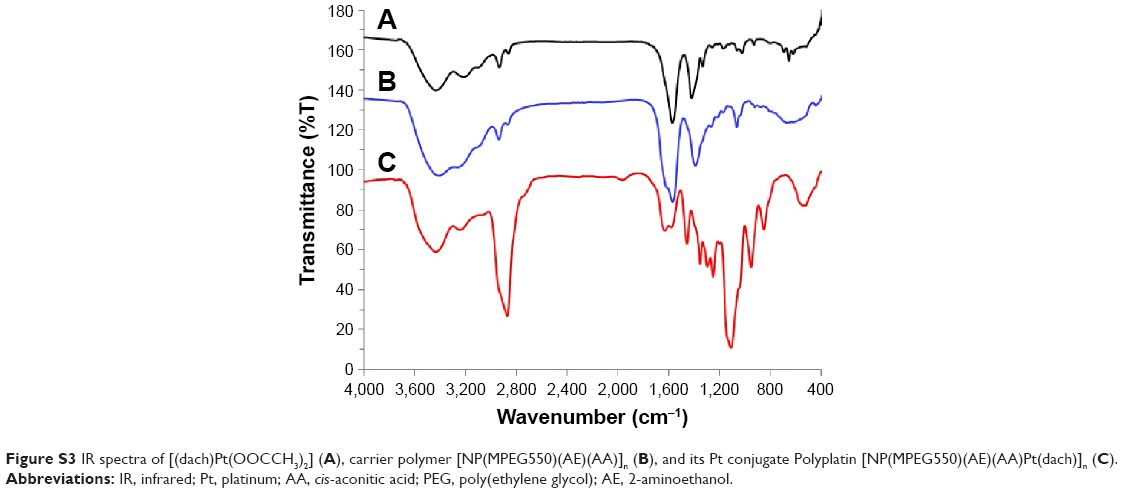 | Figure S3 IR spectra of [(dach)Pt(OOCCH3)2] (A), carrier polymer [NP(MPEG550)(AE)(AA)]n (B), and its Pt conjugate Polyplatin [NP(MPEG550)(AE)(AA)Pt(dach)]n (C). |
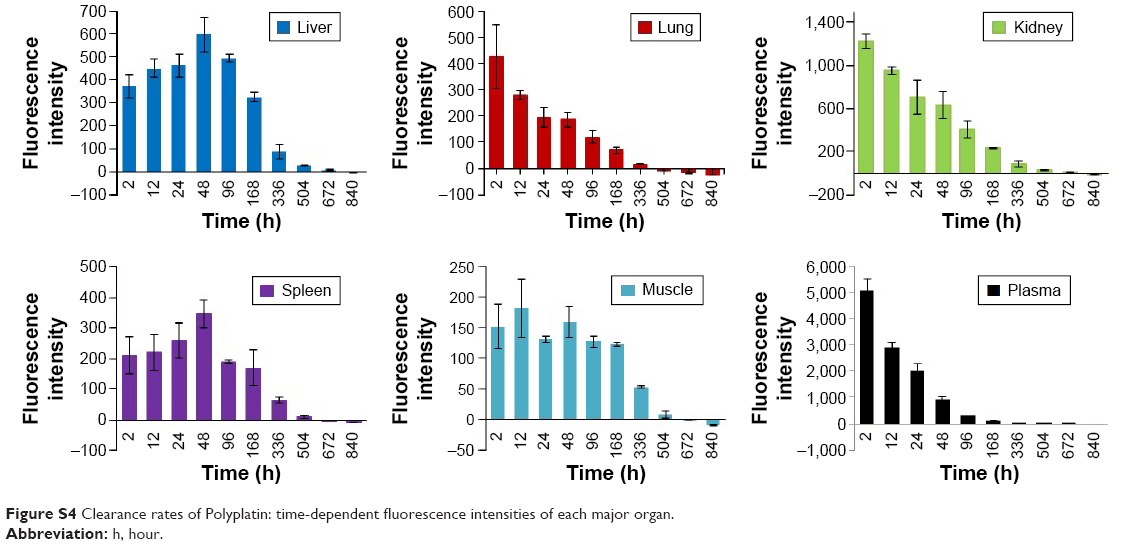 | Figure S4 Clearance rates of Polyplatin: time-dependent fluorescence intensities of each major organ. |
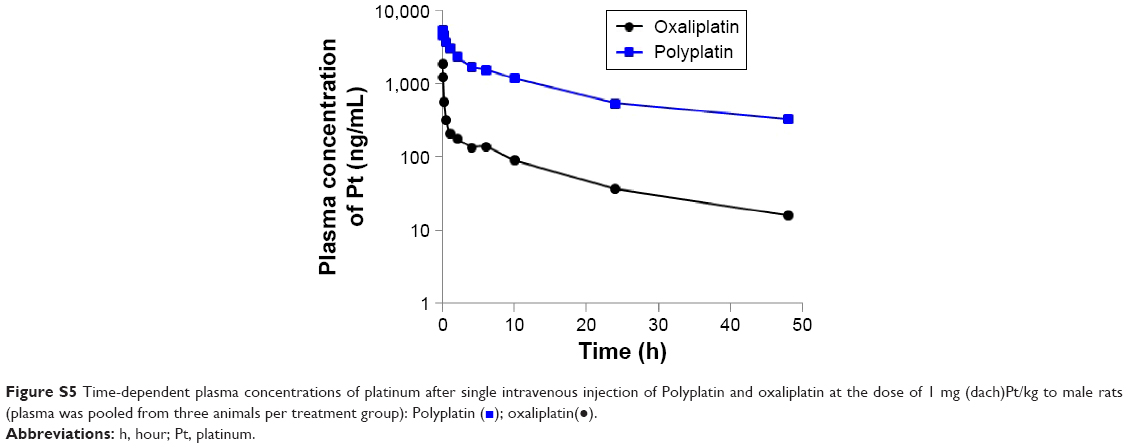 | Figure S5 Time-dependent plasma concentrations of platinum after single intravenous injection of Polyplatin and oxaliplatin at the dose of 1 mg (dach)Pt/kg to male rats (plasma was pooled from three animals per treatment group): Polyplatin ( |
 ); oxaliplatin(•).
); oxaliplatin(•).
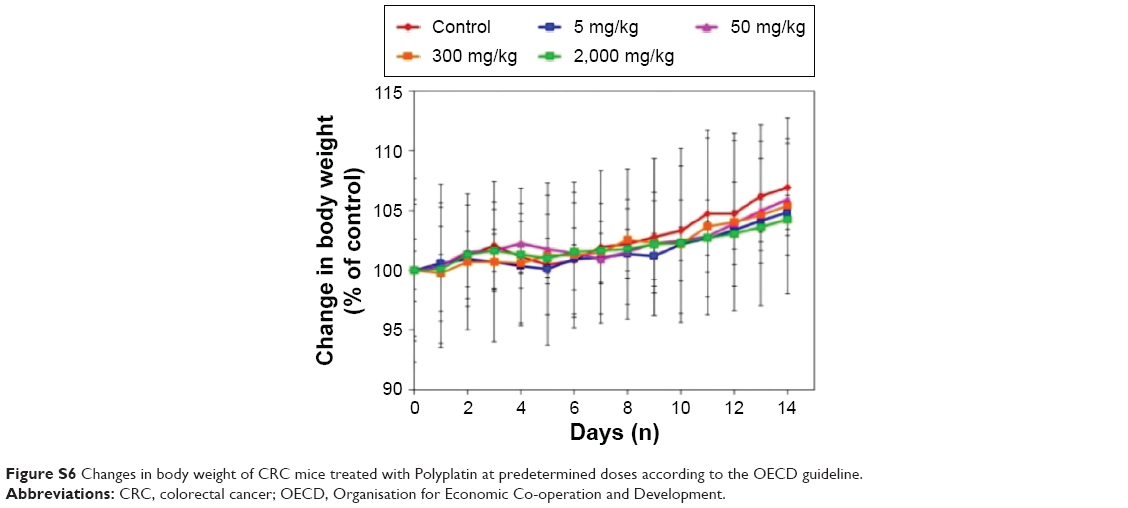 | Figure S6 Changes in body weight of CRC mice treated with Polyplatin at predetermined doses according to the OECD guideline. |
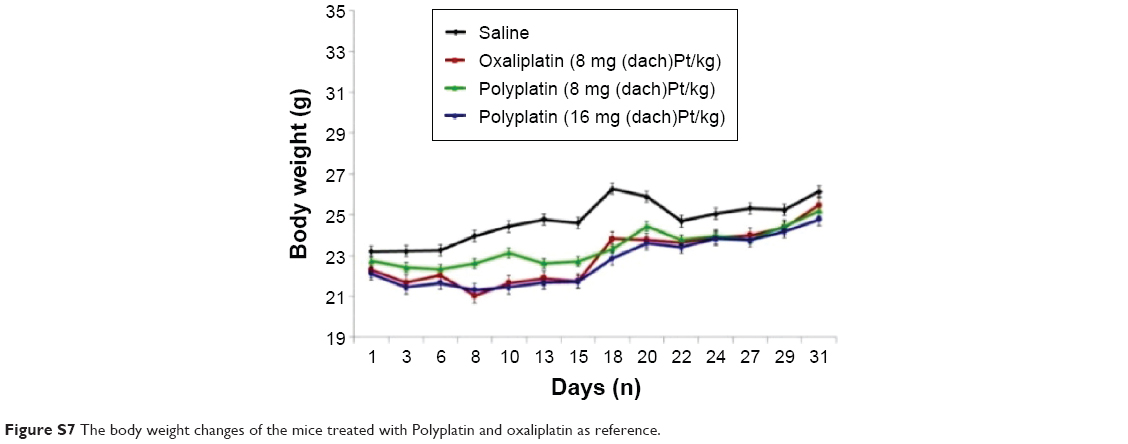 | Figure S7 The body weight changes of the mice treated with Polyplatin and oxaliplatin as reference. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.