")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Design, development, drug-likeness, and molecular docking studies of novel piperidin-4-imine derivatives as antitubercular agents
Authors Revathi R, Perumal RV, Pai KSR, Arunkumar G, Sriram D, Kini S
Received 17 February 2015
Accepted for publication 14 April 2015
Published 21 July 2015 Volume 2015:9 Pages 3779—3787
DOI https://doi.org/10.2147/DDDT.S83047
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Rajappan Revathi,1 Ramachandran Venkatesha Perumal,2 Karkala Sreedhara Ranganath Pai,3 Govindakarnavar Arunkumar,4 Dharmarajan Sriram,5 Suvarna Ganesh Kini1
1Department of Pharmaceutical Chemistry, Manipal College of Pharmaceutical Sciences, Manipal University, Manipal, 2Bharathi College of Pharmacy, Bharathi Nagara, 3Department of Pharmacology, Manipal College of Pharmaceutical Sciences, Manipal University, Manipal, 4Manipal Centre of Virus Research, Manipal University, Manipal, Karnataka, India; 5Pharmacy Group, Birla Institute of Technology and Science-Pilani, Hyderabad Campus, Hyderabad, India
Abstract: Tuberculosis remains one of the major grievous diseases worldwide. The emergence of resistance to antituberculosis drugs emphasize the necessity to discover new therapeutic agents for preferential tuberculosis therapy. In this study, various novel 1-(1H-benzimidazol-2-ylmethyl)piperidin-4-imine derivatives were developed and checked for favorable pharmacokinetic parameters based on drug-likeness explained by Lipinski’s rule of five. All 20 of the novel chemical entities were found to possess a favorable pharmacokinetic profile since they were not violating Lipinski’s rule of five. The title compounds were also synthesized, characterized, and tested for ex vivo antitubercular activity against Mycobacterium tuberculosis H37Rv (ATCC27294). The results revealed that four compounds (2-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]hydrazinecarbothioamide, 2-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]-N-hydroxyhydrazinecarbo-thioamide, 1-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]guanidine, and 2-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]hydrazinecarboxamide) were the most potent (minimum inhibitory concentration 6.25 µg/mL) antitubercular agents, with less toxicity (selectivity index more than 10). The molecules were also subjected to three-dimensional molecular docking on the crystal structure of enoyl-acyl carrier protein (EACP) reductase enzyme (code 1ZID, Protein Data Bank), which represents a good prediction of the interactions between the molecules and EACP reductase with minimum binding energy.
Keywords: enoyl-acyl carrier protein reductase, Schiff’s reaction, benzimidazole, Mycobacterium tuberculosis
Introduction
The microorganism Mycobacterium tuberculosis H37Rv (ATCC27294) causes the deadly disease tuberculosis, and it continues to be a global threat to human health.1 Two million people die and almost 8 million people are infected by tuberculosis every year. The increase in clinical occurrence of extensively drug-resistant and multidrug-resistant tuberculosis and increased incidence of HIV and tuberculosis coinfection lead to new clinical challenges for tuberculosis treatment.2,3 Moreover, anorexia, liver damage, nausea, and vomiting are the primary side effects caused by antitubercular drugs.4,5 So there is a constant need to design and develop novel chemical entities with high therapeutic efficacy and safety to combat this tuberculosis problem worldwide.
In M. tuberculosis H37Rv (ATCC27294), the major components in its cell wall are mycolic acids.6 In prokaryotes, the fatty acid synthesis II (FAS-II) pathway is involved in the biosynthesis of mycolic acid.7 The role of these fatty acids is essential for the survival of bacterial cells. In particular, in fatty acid elongation, the enzyme enoyl-acyl carrier protein (EACP) reductase plays a major role; it catalyzes the unsaturated fatty acids by NADH-dependent reduction and the reaction is stereospecific. Hence, an attractive antitubercular drug target is EACP reductase.8–10
Isoniazid (INH), an antitubercular drug with pyridine scaffold, prompted our research on piperidine. Moreover, six-membered nitrogen heterocycles proved to be a good candidate against M. tuberculosis H37Rv (ATCC27294),11 particularly as EACP reductase inhibitor.12 1, 4-substituted piperidine derivatives (compound 1, Figure 1), were reported to possess more potent antitubercular activity than the 1-substituted piperidine derivatives (compound 2, Figure 1).13–15 Schiff bases of INH have been reported for their antitubercular activity, in which the compound 3 (Figure 1) was found to be the most potent antitubercular agent.16,17 These reports suggest that the substitution at the fourth position as well as preparation of Schiff bases of a piperidine moiety may enhance antitubercular activity. Semicarbazones and thiosemicarbazones were reported to possess promising antitubercular activities.18 Based on these results, we included various imine substitutions (Schiff’s bases) at the 4-position of the piperidine nucleus.
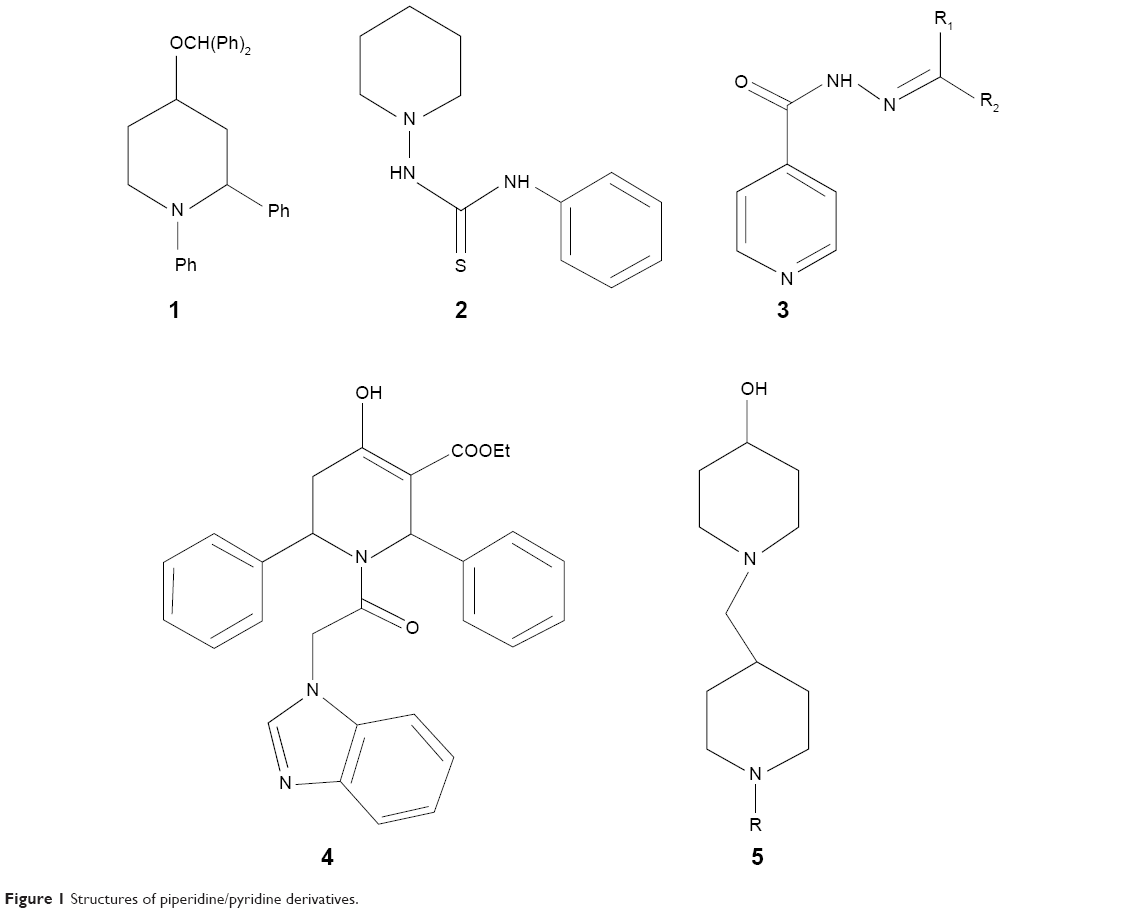 | Figure 1 Structures of piperidine/pyridine derivatives. |
N-alkyl heterocyclic-substituted piperidin-4-imine derivatives have not been much explored for their biological activities. It has been reported that tetrahydropyridol with benzimidazole19 and (piperidin-4-ylmethyl)piperidine20 possess in vitro antitubercular activity with minimum inhibitory concentration (MIC) 16 μg/mL and 4 μg/mL, respectively. Based on the evidence obtained from these two derivatives, compound 5 (Figure 1) was more potent with 2-methyl-substituted heterocycles at the first position of piperidine, whereas 2-acyl-substituted heterocycles at the first position of tetrahydropyridol (compound 4, Figure 1) were less potent against M. tuberculosis H37Rv (ATCC27294). In drug discovery, the incorporation of benzimidazole nuclei is a valuable synthetic strategy, since it exhibits antitubercular21 and broad-spectrum antibacterial22 activities. These findings prompted us to substitute the 2-methyl benzimidazole at the first position of piperidin-4-imine, under the assumption that the presence of methyl benzimidazole may increase the antitubercular activity.
Methodology
Chemistry
Toshniwal or Shital Scientific Industries apparatus were used to determine the melting points of synthesized compounds, ie, test compounds, in open capillaries without correction. Thin layer chromatography (TLC) using silica gel-coated aluminum plates (Merck, New Jersey, USA) was used to check the purity of the compounds and the blots were observed under ultraviolet light at 254 and 366 nm. A Fourier transform infrared (IR) spectrophotometer (Shimadzu, Kyoto, Japan) (cm−1) was used to record IR spectra using the KBr pellet technique. A Bruker Ascend 400 MHz spectrometer was used to record proton nuclear magnetic resonance (1H-NMR) spectra using tetramethylsilane as internal standard (chemical shifts in δ, ppm). A Shimadzu QP2010 Ultra mass spectrometer and PerkinElmer 2400 CHN elemental analyzer were used to obtain mass spectra and elemental analysis reports, respectively.
Synthesis and characterization of 1-(1H-benzimidazol-2-ylmethyl)piperidin-4-one (PB1)
To a reaction mixture of 4-piperidone hydrochloride (0.76 g, 0.005 mol) and 2-chloromethylbenzimidazole (0.8 g, 0.005 mol) in dimethyl formamide (10 mL), triethylamine (1.4 mL, 0. 01 mol) was added and heated under reflux for 7 hours. The formation of the final product was checked by TLC. The contents were brought down to room temperature and transferred into crushed ice, then refrigerated for 12 hours. The product obtained was filtered and vacuum dried after washing with water. Finally, recrystallization was carried out using methanol to yield the pure product, PB1. 1H-NMR (dimethyl sulfoxide [DMSO]-d6) δ: 2.12 (t, 4H, J=6.8 Hz, H3 and H5 piperidone), 2.50 (t, 4H, J=6.8 Hz, H2 and H6 piperidone), 3.42 (s, 2H, -N-CH2), 7.12 (d, 2H, J=8.4 Hz, H5 and H6 benzimidazole), 7.81 (d, 2H, J=8.4 Hz, H4 and H7 benzimidazole), 12.21 (s, 1H, NH). 13C nuclear magnetic resonance (13C-NMR) (DMSO-d6) δ: 41.61, 52.94 (4C, piperidone), 55.76 (CH2), 115.24, 123.07, 139.11 (6C, Ar), 141.55 (imidazole), 210.30 (C=O). IR (KBr) cm−1: 3,250 (NH); 3,041, 3,007, 2,985 (C-H); 1,704 (C=O). MS m/z: 229 (M)+. Analytical calculated for C13H15N3O: C, 68.10; H, 6.59; N, 18.33. Found: C, 67.85; H, 6.20; N, 18.66.
General method for the synthesis of PB2–PB20
To an ethanolic solution of PB1 (2.29 g, 0.01 mol) with the appropriate amine (0.01 mol), sodium acetate (0.82 g, 0.01 mol) was added with stirring and it was refluxed for 3.5 hours with constant stirring. The completion of the reaction was validated using TLC. The contents were brought down to room temperature and transferred into ice-cold water. The resulting product was separated through suction and vacuum dried after washing thoroughly with water. It was then recrystallized from the mixture of methanol and dimethyl formamide (2:8) to yield the pure compound. The methodology is depicted in Figure 2, and the physical properties are listed in Table 1.
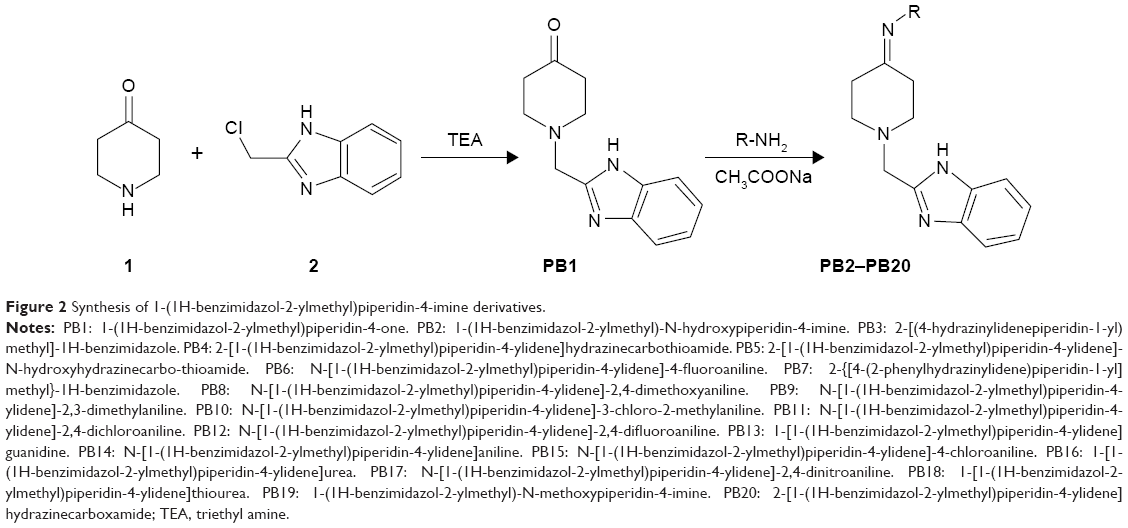 | Figure 2 Synthesis of 1-(1H-benzimidazol-2-ylmethyl)piperidin-4-imine derivatives. |
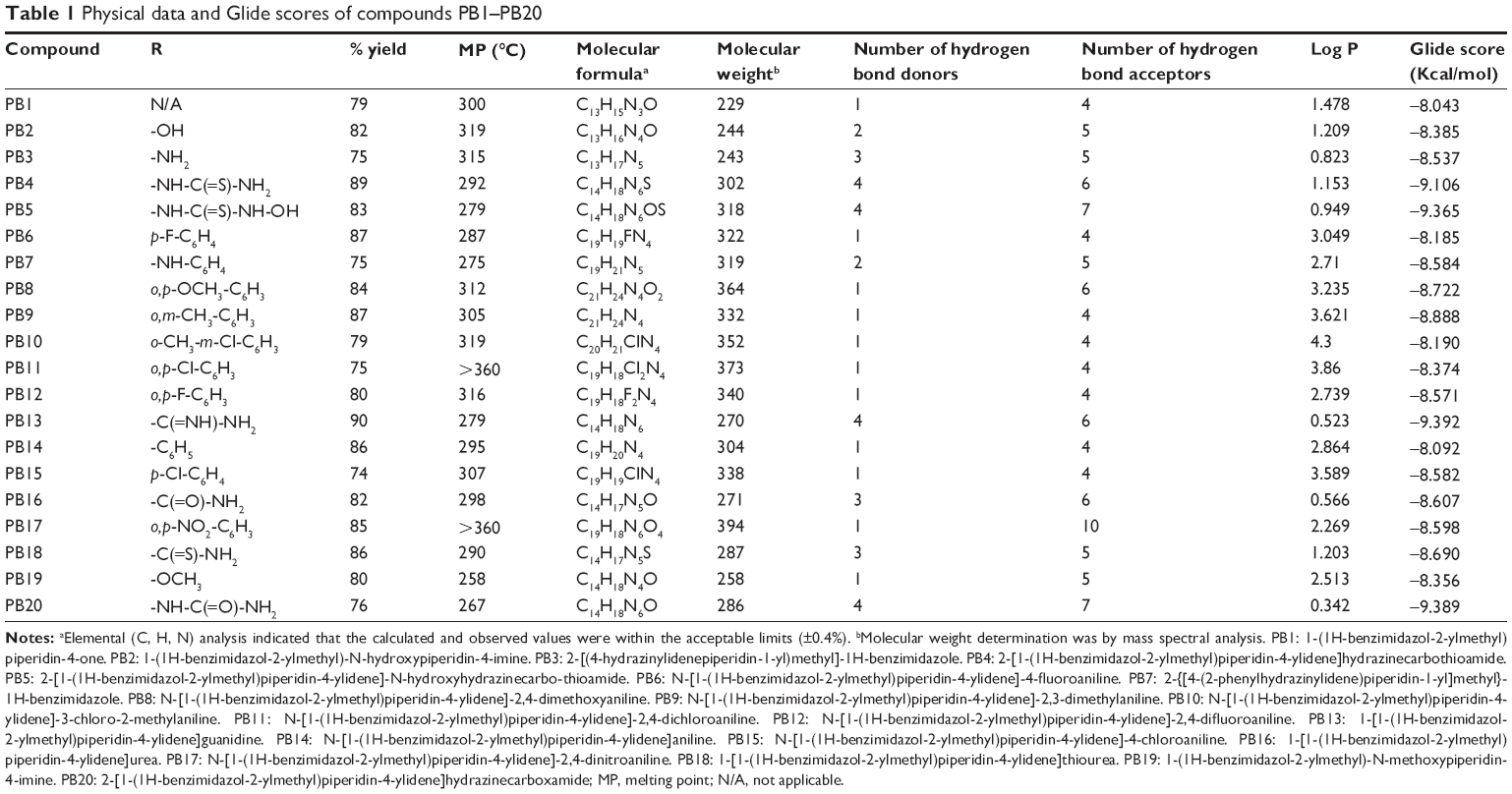 | Table 1 Physical data and Glide scores of compounds PB1–PB20 |
Characterization of 1-(1H-benzimidazol-2-ylmethyl)-N-hydroxypiperidin-4-imine (PB2)
1H-NMR (DMSO-d6) δ:1.40 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.42 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.42 (s, 2H, -N-CH2), 7.19 (d, 2H, J=8.4 Hz, H5 and H6 benzimidazole), 7.70 (d, 2H, J=8.4 Hz, H4 and H7 benzimidazole), 11.07 (s, 1H, OH), 12.30 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 25.63, 53.32 (4C, piperidine), 54.93 (CH2), 115.56, 122.76, 139.45 (6C, Ar), 142.04 (imidazole), 160.68 (C=N). IR (KBr) cm−1: 3,384 (OH); 3,295 (NH); 3,080, 2,845 (CH), 1,580 (C=N). MS m/z: 244 (M)+. Anal calcd for C13H16N4O: C, 63.91; H, 6.60; N, 22.93. Found: C, 63.61; H, 6.32; N, 23.17.
2-[1-(1H-benzimidazol-2-ylmethyl)piperidin- 4-ylidene]hydrazinecarbothioamide (PB4)
1H-NMR (DMSO-d6) δ: 1.44 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.62 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.53 (s, 2H, -N-CH2), 6.38 (s, 2H, NH2), 7.25 (d, 2H, J=8.4 Hz, H5 and H6 benzimidazole), 7.55 (d, 2H, J=8.4 Hz, H4&H7 benzimidazole), 9.11 (s, 1H, =N-NH), 12.21 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 25.07, 53.14 (4C, piperidine), 55.47 (CH2), 115.56, 122.84, 139.36 (6C, Ar), 142.04 (imidazole), 162.04 (C=N), 187.54 (C=S). IR (KBr) cm−1: 3,347, 3,277 (NH2, NH); 3,074, 2,956, 2,874 (CH), 1,633 (C=N); 1,205 (C=S). MS m/z: 302 (M)+. Anal calcd for C14H18N6S: C, 55.61; H, 6.00; N, 27.79. Found: C, 55.29, H, 5.66; N, 28.18.
2-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]-N-hydroxyhydrazinecarbo-thioamide (PB5)
1H-NMR (DMSO-d6) δ: 1.30 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.24 (t, 4H, J=6.8 Hz, H2&H6 piperidine), 3.50 (s, 2H, -N-CH2), 7.39 (d, 2H, J=8.4 Hz, H5 and H6 benzimidazole), 7.79 (d, 2H, J=8.4 Hz, H4 and H7 benzimidazole), 8.41 (s, 1H, -N-NH), 9.20 (s, 1H, -C-NH-OH), 10.26 (s, 1H, -C-NH-OH), 12.13 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 23.13, 53.36 (4C, piperidine), 55.03 (CH2), 115.76, 124.37, 139.69 (6C, Ar), 141.86 (imidazole), 161.45 (C=N), 185.48 (C=S). IR (KBr) cm−1: 3,357 (OH); 3,284 (NH), 3,057, 2,965 (CH), 1,597 (C=N) 1,186 (C=S). MS m/z: 318 (M)+. Anal calcd for C14H18N6OS: C, 52.81; H, 5.70; N, 26.39. Found: C, 52.62; H, 5.44; N, 26.21.
2-{[4-(2-phenylhydrazinylidene)piperidin-1-yl]methyl}-1H-benzimidazole (PB7)
1H-NMR (DMSO-d6) δ: 1.43 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.29 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.67 (s, 2H, -N-CH2), 7.01–7.55 (m, 9H, Ar-H), 11.19 (s, 1H, -N-NH), 12.21 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 21.16, 53.46 (4C, piperidine), 55.04 (CH2), 115.24, 116.67, 118.53, 123.07, 129.57, 139.11, 144.03 (12C, Ar), 142.47 (imidazole), 160.92 (C=N). IR (KBr) cm−1: 3,234 (NH), 3,089, 2,986, 2,904 (CH), 1,622 (C=N). MS m/z: 319 (M)+. Anal calcd for C19H21N5: C, 71.45; H; 6.63; N, 21.93. Found: C, 71.26; H, 6.44; N, 21.67.
N-[1-(1H-benzimidazol-2-ylmethyl)piperidin- 4-ylidene]-2,4-dimethoxyaniline (PB8)
1H-NMR (DMSO-d6) δ: 1.46 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.44 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.46 (s, 2H, -N-CH2), 3.75 (s, 6H, -O-CH3), 6.92–7.44 (m, 7H, Ar-H), 12.19 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 24.57, 53.47 (4C, piperidine), 54.94 (CH2), 55.93 (2C, 2OCH3), 102.02, 107.34, 114.74, 123.47, 124.45, 127.38, 139.11, 153.67, 159.94 (12C, Ar), 142.13 (imidazole), 187.13 (C=N). IR (KBr) cm−1: 3,281 (NH), 3,067, 2,913 (CH); 1,597 (C=N). MS m/z: 364 (M)+. Anal calcd for C21H24N4O2: C, 69.21; H, 6.64; N, 15.37. Found: C, 69.40; H, 6.37; N, 15.24.
N-[1-(1H-benzimidazol-2-ylmethyl)piperidin- 4-ylidene]-2,3-dimethylaniline (PB9)
1H-NMR (DMSO-d6) δ: 1.43 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.31 (s, 6H, 2CH3), 2.56 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.67 (s, 2H, -N-CH2), 6.96–7.53 (m, 7H, Ar-H), 12.22 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 17.24, 21.87 (2C, 2CH3), 24.74, 53.18 (4C, piperidine), 55.34 (CH2), 115.47, 119.75, 123.16, 127.04, 127.55, 128.94, 138.12, 139.11, 146.83 (12C, Ar), 141.55 (imidazole), 210.30 (C=O). IR (KBr) cm−1: 3,276 (NH), 3,058, 2,943, 2,892 (CH), 1,602 (C=N). MS m/z: 332 (M)+. Anal calcd for C21H24N4: C, 75.87; H, 7.28; N, 16.85. Found: C, 76.27; H, 6.97; N, 17.14.
1-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]guanidine (PB13)
1H-NMR (DMSO-d6) δ: 1.51 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.53 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.67 (s, 2H, -N-CH2), 5.43 (s, 2H, NH2), 6.05 (s, 1H, C=NH), 7.35 (d, 2H, J=8.4 Hz, H5 and H6 benzimidazole), 7.74 (d, 2H, J=8.4 Hz, H4 and H7 benzimidazole), 12.29 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 24.56, 53.06 (4C, piperidine), 55.39 (CH2), 115.91, 123.48, 139.17 (6C, Ar), 141.89 (imidazole), 161.93 (C=NH), 164.34 (C=N). IR (KBr) cm−1: 3,294, 3,216 (NH2, NH), 3,078, 2,975 (CH), 1,559 (C=N). MS m/z 270 (M)+. Anal calcd for C14H18N6: C, 62.20; H, 6.71; N, 31.09. Found: C, 61.89; H, 6.34; N, 31.46.
1-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]urea (PB16)
1H-NMR (DMSO-d6) δ: 1.62 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.61 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.62 (s, 2H, -N-CH2), 6.62 (s, 2H, NH2), 7.23 (d, 2H, J=8.4 Hz, H5 and H6 benzimidazole), 7.57 (d, 2H, J=8.4 Hz, H4 and H7 benzimidazole), 12.18 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 25.07, 53.47 (4C, piperidine), 55.25 (CH2), 116.94, 123.35, 138.05 (6C, Ar), 141.33 (imidazole), 150.02 (C=O), 164.34 (C=N). IR (KBr) cm−1: 3,307, 3,241 (NH2, NH), 3,074, 2,968 (CH), 1,684 (C=O), 1,613 (C=N). MS m/z: 271 (M)+. Anal calcd for C14H17N5O: C, 61.98; H, 6.32; N, 25.81. Found: C, 62.37; H, 5.94; N, 26.21.
1-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]thiourea (PB18)
1H-NMR (DMSO-d6) δ: 1.57 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.74 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.59 (s, 2H, -N-CH2), 5.17 (s, 2H, NH2), 7.15 (d, 2H, J=8.4 Hz, H5 and H6 benzimidazole), 7.48 (d, 2H, J=8.4 Hz, H4 and H7 benzimidazole), 12.18 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 25.05, 52.89 (4C, piperidine), 55.67 (CH2), 116.06, 123.17, 139.74 (6C, Ar), 142.07 (imidazole), 164.62 (C=N), 186.57 (C=S). IR (KBr) cm−1: 3,284, 3,247 (NH2, NH), 3,076, 3,004, 2,968 (CH), 1,238 (C=S). MS m/z: 287 (M)+. Anal calcd for C14H17N5S: C, 58.51; H, 5.96; N, 24.37. Found: C, 58.28; H, 5.59; N, 24.04.
2-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]hydrazinecarboxamide (PB20)
1H-NMR (DMSO-d6) δ: 1.49 (t, 4H, J=6.8 Hz, H3 and H5 piperidine), 2.51 (t, 4H, J=6.8 Hz, H2 and H6 piperidine), 3.74 (s, 2H, -N-CH2), 6.54 (s, 2H, NH2), 7.23 (d, 2H, J=8.4 Hz, H5 and H6 benzimidazole), 7.57 (d, 2H, J=8.4 Hz, H4 and H7 benzimidazole), 9.39 (s, 1H, =N-NH), 12.18 (s, 1H, NH benzimidazole). 13C-NMR (DMSO-d6) δ: 24.74, 53.27 (4C, piperidine), 55.11 (CH2), 116.46, 122.84, 139.04 (6C, Ar), 143.48 (imidazole), 162.11 (C=O), 164.74 (C=N). IR (KBr) cm−1: 3,294, 3,257 (NH2, NH), 3,053, 3,011, 2,965 (CH), 1,681 (C=O). MS m/z: 286 (M)+. Anal calcd for C14H18N6O: C, 58.73; H, 6.34; N, 29.35. Found: C, 59.07, H, 5.94; N, 29.71.
Antitubercular evaluation assay
Twofold serial dilutions (50.0, 25.0, 12.5, 6.25, 3.13, 1.56, and 0.78 μg/mL) of each test compound, PB1–PB20, and standard drugs were made and mixed into Middlebrook 7H11 agar medium with oleic acid, albumin, dextrose, and catalase (OADC) (Difco, Maryland, USA) growth addendum. Inoculant of M. tuberculosis H37Rv (ATCC27294) was made from fresh Middlebrook 7H11 agar slants with OADC growth addendum oriented to 1 mg/mL (wet weight) in Tween 80 (0.05%) saline diluted to 10−2 in order to get ~107 cfu/mL concentration. Five microliters of this mycobacterial inoculum was blotted into 7H11 agar tubes with varying dilutions of the test compounds and standard drugs as mentioned above. The tubes were incubated for 28 days at 37°C, then the MIC was determined.23,24 This method is same as the procedure recommended by the National Committee for Clinical Laboratory Standards for the evaluation of MIC in triplicate.25 The MIC values of the test and standard drugs are given in Figure 3.
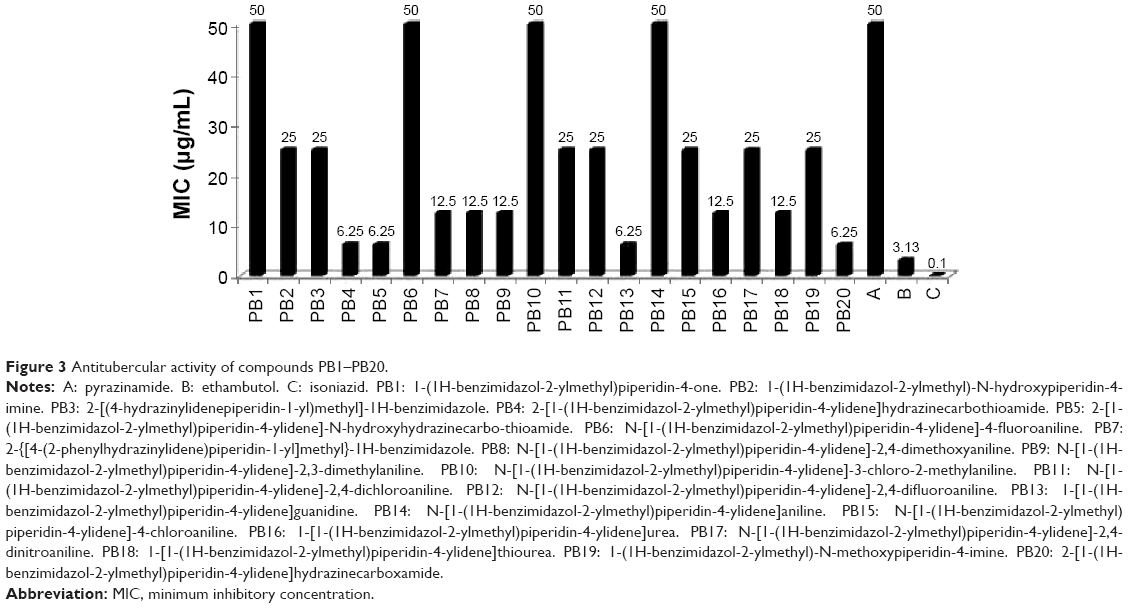 | Figure 3 Antitubercular activity of compounds PB1–PB20. |
Cytotoxicity assay
Cytotoxicity was carried out against the Vero cell line (African green monkey kidney cells), obtained from the National Centre for Cell Science (NCCS) (Pune, India). In a 96-well plate, 1×104 cells were seeded and incubated at 37°C for 24 hours in 5% CO2 atmosphere. Then, the medium from the wells was discarded once a partial monolayer was formed. Maintenance medium containing varying concentrations (62.5, 125, and 250 μg/mL) of the test compounds was added to cells. Then, every 24 hours during 48 hours’ incubation, the observations from the microscopic examination were recorded. After the treatment, the solutions were flicked off from the wells, and cells in each well were incubated for 3 hours with 50 μL of freshly prepared 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (2 mg/mL, prepared in phosphate-buffered saline) at 37°C in a 5% CO2 environment. Then the solution in each well was decanted, and the plate was washed with phosphate-buffered saline twice. Then the purple-colored crystals of formazan in each well were solubilized by adding 50 μL of isopropanol. Finally, the absorbance was recorded using a microplate reader (Bio-Tek ELX-800 MS) at 540 nm.26,27 The percentage inhibition (Figure 4) and selectivity index (SI) were calculated as per the reported methods.23,24
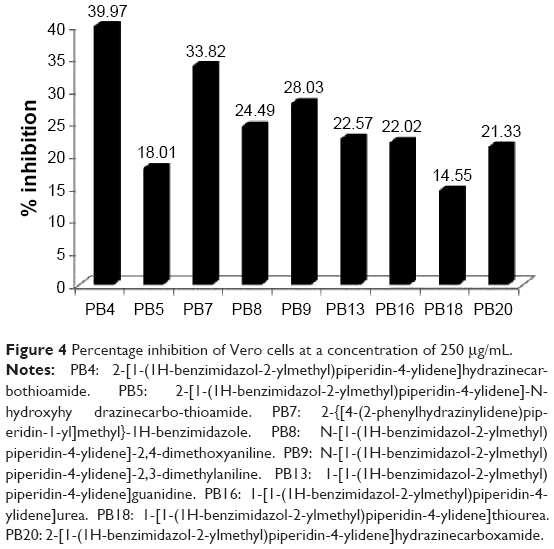 | Figure 4 Percentage inhibition of Vero cells at a concentration of 250 μg/mL. |
Docking studies
Schrödinger software was used for the docking methods. The detailed methodology for docking studies is given in the Supplementary materials. 3D structures were generated and optimized by means of OPLS-2005 using a default setting in the LigPrep. The “Refinement only” option from the preparation module in Schrödinger Maestro was utilized to prepare protein. The default box size setting in Glide was used to define the grids by centering them in the crystal structure on the ligand. The docking of the prepared ligands against the protein (code 1ZID, Protein Data Bank) was carried out. The “Extra precision” mode was used to perform the docking calculations in Glide. Schrödinger’s proprietary GlideScore was used to carry out the final scoring and rescoring of the energy-minimized poses from its scoring function.28,29 Linux OS (Red Hat Enterprise WS 5.0) was used to carry out all docking computations.
Results and discussion
Chemistry
The new chemical entities consisted of N-substituted piperidine and 2-methylbenzimidazole, and the lipophilicity was controlled by various N-substituents of piperidin-4-imine. Initially, the test compounds were evaluated for drug-likeness (Lipinski’s rule of five) using the ChemSpider database to establish a favorable pharmacokinetic (absorption, distribution, metabolism, excretion and toxicity) profile.30 All the compounds were found to be within the limits of Lipinski’s rule (number of violations =0). The compound PB1 was synthesized by the reaction of piperidin-4-one (compound 1, Figure 2) with 2-chloromethylbenzimidazole (compound 2, Figure 2) in the presence of an acid scavenger. Further, PB1 was condensed with appropriate substituted amine in the presence of sodium acetate to obtain PB2–PB20 by Schiff’s reaction, as shown in Figure 2. All the compounds are soluble in DMSO and dimethyl formamide and partially soluble in ethanol, methanol, and chloroform. Due to their solubility in organic solvents, the compounds might have good penetration in the biological membrane. Structural elucidation of the new molecules was carried out by both physical (Table 1) and spectral analysis.
In vitro antitubercular screening
The agar dilution method23,24 was used for the in vitro antitubercular screening of the title compounds against M. tuberculosis H37Rv (ATCC27294) to determine the MIC in triplicate, and the values (μg/mL) are shown in Figure 3. The compounds PB1, PB6 (N-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]-4-fluoroaniline), PB10 (N-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]-3-chloro-2-methylaniline), and PB14 (N-[1-(1H-benzimidazol-2-ylmethyl)piperidin-4-ylidene]aniline) showed equipotent activity, with MIC 50 μg/mL, and all other molecules were found to be more potent, with MIC in the range of 6.25–25 μg/mL, compared with the standard drug pyrazinamide, but less potent than the other standard drugs, INH and ethambutol. Compounds PB4, PB5, PB13, and PB20 showed the most promising antitubercular activity, with MIC 6.25 μg/mL. It has been observed that the compounds with four hydrogen bond donors were found to possess more potent antitubercular activity. In this study, no definite conclusions were drawn about the relationship between lipophilicity (log P) of the molecules and antitubercular activity, since the MIC values of antitubercular activity could not be directly correlated with log P.
In vitro cytotoxicity assay
The new analogs, which showed promising antitubercular activity (MIC ≤12.5 μg/mL), were also evaluated for in vitro cytotoxicity using MTT assay26,27 against Vero cell lines at various concentrations. The percentage inhibition of cells at 250 μg/mL concentration is presented in Figure 4. The most active compounds, PB4, PB5, PB13, and PB20, exhibited 39.9%, 18%, 22.5%, and 21.3% inhibition, respectively. The compounds were considered to be nontoxic because the SI was greater than 10.
Docking studies
The docking studies28,29 of the title compounds were carried out with EACP reductase enzyme (1ZID.pdb) by using Schrödinger software. The interaction of molecule with EACP reductase enzyme is shown in Figure 5. The docking results of the target compounds revealed that all the compounds were energetically favorable in terms of Glide dock score (Table 1). The Glide dock score of the most potent compounds, PB4, PB5, PB13, and PB20, were −9.106, −9.365, −9.392, and −9.389, respectively. The Glide dock score of the less active compounds, PB1, PB6, PB10, and PB14, were −8.043, −8.185, −8.190, and −8.092, respectively. These results indicate that the more potent compounds require less energy for good binding interaction with the receptor, whereas the less active compounds require more energy than the high active molecules. The binding interaction of the most active compound, PB13, showed that the phenyl ring and N1- of the benzimidazole binds with the amino acid residues TYR158 through π–π stacking and ALA191 through hydrogen bonding, respectively. The N- of the aliphatic side chain showed hydrogen bonding with THR196, which is similar to that of O- of the aliphatic side chain (INH) interaction with GLY96 via hydrogen bonding. These binding interactions reveal the importance of the N1- and phenyl ring of benzimidazole and also the N- of aliphatic side chain moiety for a favorable binding interaction, so that better EACP reductase inhibitory activity is expected.
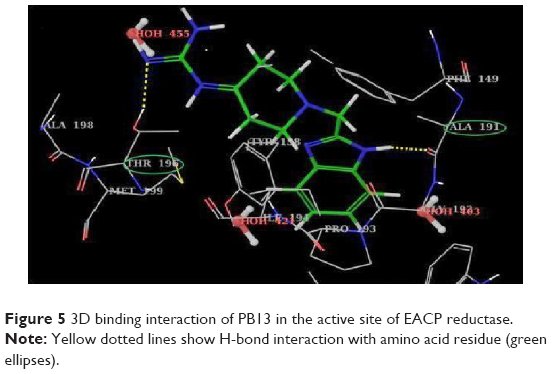 | Figure 5 3D binding interaction of PB13 in the active site of EACP reductase. |
Conclusion
In the present study, we developed novel chemical entities as potential antitubercular agents. The compounds showed good drug-likeness scores based on Lipinski’s rule. Among the 20 compounds, PB4, PB5, PB13, and PB20 exhibited potent antitubercular activity, with MIC 6.25 μg/mL, and less toxicity (SI > 10). In the in silico studies, these test compounds showed minimum binding energy with EACP reductase enzyme. So the present study provides us insight for the further development of better antitubercular agents as EACP reductase inhibitors with less or no toxicity.
Acknowledgments
We are thankful to Manipal University (Manipal, India) for providing financial assistance to one of the authors (RR) and the Manipal College of Pharmaceutical Sciences, Manipal University, for providing research facilities. We are grateful to the Birla Institute of Technology and Science-Pilani, Hyderabad Campus (Hyderabad, India), for providing antitubercular screening facilities. We gratefully acknowledge All India Council for Technical Education, Department of Biotechnology, and Department of Science and Technology (New Delhi, India) for providing research facilities to the Manipal College of Pharmaceutical Sciences through funding.
Disclosure
The authors report no conflicts of interest in this work.
References
Lienhardt C, Glaziou P, Uplekar M, Lönnroth K, Getahun H, Raviglione M. Global tuberculosis control: lessons learnt and future prospects. Nat Rev Microbiol. 2012;10(6):407–416. | ||
Grobusch MP. Drug-resistant and extensively drug-resistant tuberculosis in southern Africa. Curr Opin Pulm Med. 2010;16(3):180–185. | ||
Mainardi JL, Hugonnet JE, Gutmann L, Arthur M. Fighting resistant tuberculosis with old compounds: the carbapenem paradigm. Clin Microbiol Infect. 2011;17(12):1755–1756. | ||
Yee D, Valiquette C, Pelletier M, Parisien I, Rocher I, Menzies D. Incidence of serious side effects from first-line antituberculosis drugs among patients treated for active tuberculosis. Am J Respir Crit Care Med. 2003;167(11):1472–1477. | ||
Sun F, Chen Y, Xiang Y, Zhan S. Drug-metabolising enzyme polymorphisms and predisposition to anti-tuberculosis drug-induced liver injury: a meta-analysis. Int J Tuberc Lung Dis. 2008;12(9): 994–1002. | ||
Pasqualoto KF, Ferreira EI. An approach for the rational design of new antituberculosis agents. Curr Drug Targets. 2001;2(4):427–437. | ||
White SW, Zheng J, Zhang YM, Rock. The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. | ||
Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem. 1995;270(44):26538–26542. | ||
Lu H, Tonge PJ. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc Chem Res. 2008;41(1):11–20. | ||
Rozwarski DA, Vilchèze C, Sugantino M, Bittman R, Sacchettini JC. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J Biol Chem. 1999;274(22):15582–15589. | ||
Poce G, Cocozza M, Consalvi S, Biava M. SAR analysis of new anti-TB drugs currently in pre-clinical and clinical development. Eur J Med Chem. 2014;86:335–351. | ||
Holas O, Ondrejcek P, Dolezal M. Mycobacterium tuberculosis enoyl-acyl carrier protein reductase inhibitors as potential antituberculotics: development in the past decade. J Enzyme Inhib Med Chem. Epub 2014 Nov 10. | ||
Chauhan PM, Sunduru N, Sharma M. Recent advances in the design and synthesis of heterocycles as anti-tubercular agents. Future Med Chem. 2010;2(9):1469–1500. | ||
Weis R, Schweiger K, Faist J, et al. Antimycobacterial and H1-antihistaminic activity of 2-substituted piperidine derivatives. Bioorg Med Chem. 2008;16(24):10326–10331. | ||
Sun D, Scherman MS, Jones V, et al. Discovery, synthesis, and biological evaluation of piperidinol analogs with anti-tuberculosis activity. Bioorg Med Chem. 2009;17(10):3588–3594. | ||
Kajal A, Bala S, Kamboj S, Sharma N, Saini V. Schiff bases: a versatile pharmacophore. Journal of Catalysts. 2013;Article ID 893512:1–14. | ||
Hearn MJ, Cynamon MH, Chen MF, et al. Preparation and antitubercular activities in vitro and in vivo of novel Schiff bases of isoniazid. Eur J Med Chem. 2009;44(10):4169–4178. | ||
Banerjee D, Yogeeswari P, Bhat P, Thomas A, Srividya M, Sriram D. Novel isatinyl thiosemicarbazones derivatives as potential molecule to combat HIV-TB co- infection. Eur J Med Chem. 2011;46(1): 106–121. | ||
Aridoss G, Amirthaganesan S, Kumar NA, et al. A facile synthesis, antibacterial, and antitubercular studies of some piperidin-4-one and tetrahydropyridine derivatives. Bioorg Med Chem Lett. 2008; 18(24):6542–6548. | ||
Bogatcheva E, Hanrahan C, Nikonenko B, et al. Identification of SQ609 as a lead compound from a library of dipiperidines. Bioorg Med Chem Lett. 2011;21(18):5353–5357. | ||
Kazimierczuk Z, Andrzejewska M, Kaustova J, Klimesova V. Synthesis and antimycobacterial activity of 2-substituted halogenobenzimidazoles. Eur J Med Chem. 2005;40(2):203–208. | ||
He Y, Yang J, Wu B, Risen L, Swayze EE. Synthesis and biological evaluations of novel benzimidazoles as potential antibacterial agents. Bioorg Med Chem Lett. 2004;14(5):1217–1220. | ||
Hall L, Jude KP, Clark SL, et al. Evaluation of the Sensititre MycoTB plate for susceptibility testing of the Mycobacterium tuberculosis complex against first- and second-line agents. J Clin Microbiol. 2012; 50(11):3732–3734. | ||
Addla D, Jallapally V, Gurram D, Yogeeswari P, Sriram D, Kantevari S. Rational design, synthesis and antitubercular evaluation of novel 2-(trifluoromethyl)-phenothiazine-[1,2,3]triazole hybrids. Bioorg Med Chem Lett. 2014;24(1):233–236. | ||
NCCLS (National Committee for Clinical Laboratory Standards). Wayne, Pa: NCCLS; 1997. Performance Standards for Antimicrobial Disk Susceptibility Tests, 6th ed. Approved Standard M2-A6. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89(2):271–277. | ||
Asgaonkar KD, Mote GD, Chitre TS. QSAR and molecular docking studies of oxadiazole-ligated pyrrole derivatives as enoyl-ACP (CoA) reductase inhibitors. Sci Pharm. 2013;82(1):71–85. | ||
Mathew B, Mathew GE, Sonia G, Kumar A, Charles NP, Kumar P. Design of 1-(furan-2-yl)-N-(5-substituted phenyl-1,3,4-thiadiazol-2-yl) methanimine derivatives as enoyl-ACP reductase inhibitors: synthesis, molecular docking studies and anti-tubercular activity. Bangladesh J Pharmacol. 2013;8:242–248. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.