")
Back to Journals » International Journal of Nanomedicine » Volume 11
Design and statistical modeling of mannose-decorated dapsone-containing nanoparticles as a strategy of targeting intestinal M-cells
Authors Vieira A , Chaves L, Pinheiro M , Ferreira D, Sarmento B , Reis S
Received 23 January 2016
Accepted for publication 19 March 2016
Published 3 June 2016 Volume 2016:11 Pages 2601—2617
DOI https://doi.org/10.2147/IJN.S104908
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Alexandre CC Vieira,1,* Luíse L Chaves,1,* Marina Pinheiro,1 Domingos Ferreira,2 Bruno Sarmento,3–5 Salette Reis1
1UCIBIO, REQUIMTE, Chemistry Department, Faculty of Pharmacy, 2Laboratory of Pharmaceutical Technology, Department of Drug Sciences, Faculty of Pharmacy, 3I3S, Institute for Research and Innovation in Health, 4INEB – Institute of Biomedical Engineering, University of Porto, Porto, 5CESPU, Institute of Research and Advanced Formation in Health Sciences and Technology, University Institute of Health Sciences, Gandra, Portugal
*These authors contributed equally to this work
Abstract: The aim of the present work was to develop and optimize surface-functionalized solid lipid nanoparticles (SLNs) for improvement of the therapeutic index of dapsone (DAP), with the application of a design of experiments. The formulation was designed to target intestinal microfold (M-cells) as a strategy to increase internalization of the drug by the infected macrophages. DAP-loaded SLNs and mannosylated SLNs (M-SLNs) were successfully developed by hot ultrasonication method employing a three-level, three-factor Box–Behnken design, after the preformulation study was carried out with different lipids. All the formulations were systematically characterized regarding their diameter, polydispersity index (PDI), zeta potential (ZP), entrapment efficiency, and loading capacity. They were also subjected to morphological studies using transmission electron microscopy, in vitro release study, infrared analysis (Fourier transform infrared spectroscopy), calorimetry studies (differential scanning calorimetry), and stability studies. The diameter of SLNs, SLN-DAP, M-SLNs, and M-SLN-DAP was approximately 300 nm and the obtained PDI was <0.2, confirming uniform populations. Entrapment efficiency and loading capacity were approximately 50% and 12%, respectively. Transmission electron microscopy showed spherical shape and nonaggregated nanoparticles. Fourier transform infrared spectroscopy was used to confirm the success of mannose coating process though Schiff’s base formation. The variation of the ZP between uncoated (approximately –30 mV) and mannosylated formulations (approximately +60 mV) also confirmed the successful coating process. A decrease in the enthalpy and broadening of the lipid melting peaks of the differential scanning calorimetry thermograms are consistent with the nanostructure of the SLNs. Moreover, the drug release was pH-sensitive, with a faster drug release at acidic pH than at neutral pH. Storage stability for the formulations for at least 8 weeks is expected, since they maintain the original characteristics of diameter, PDI, and ZP. These results pose a strong argument that the developed formulations can be explored as a promising carrier for treating leprosy with an innovative approach to target DAP directly to M-cells.
Keywords: Box–Behnken design, leprosy, drug delivery, targeting, oral route, solid lipid nanoparticle
Introduction
The oral route is still considered the most convenient and safest route of drug administration with higher patient compliance, lesser complications, and lower cost, in comparison with parenteral drug delivery.1–5 Besides all the advantages of the oral route, factors like low aqueous solubility of drugs, limited gastrointestinal (GI) absorption, rapid metabolism of drug, and low mucosal permeability play a major role in the disappointing in vivo results.5–8 The intestinal mucosa plays an effective role as a physical barrier that covers the surfaces of the GI tract, allowing selective absorption of nutrients, electrolytes, and fluids, at the same time protecting the host from environmental pathogens.1,4,7,9 The first barrier encountered by such drugs is the mucus layer, which functions as a diffusional and enzymatic barrier.10 After oral administration, a drug must infiltrate through the unstirred layer of mucus before reaching the surface of the intestinal epithelium, the second barrier.9 The protective role of the intestinal epithelium is mainly due to the presence of organized immune-inductive tissues, namely, the Peyer’s patches (PP), which can trigger immune responses after antigenic stimulation.11 This epithelium is characterized by the presence of microfold cells (M-cells), which take up the antigens and microorganisms derived from the intestinal lumen and then deliver them to the underlying immune system of mucosa.9,10 Following their transcytosis across the follicle-associated epithelia of PP, the antigens move to the intraepithelial pocket beneath the M-cell basolateral membrane, which contains several populations of lymphocytes and mononuclear phagocytes, a heterogeneous population of macrophages, and classical dendritic cells.12 These unique features make M-cells attractive targets for oral drug and vaccine delivery studies.11
Many attempts have been made to deliver nonspecific and specific drugs targeting specialized M-cells.13 A specific targeting effect can be achieved by attaching specific ligands on the surface of drug delivery systems, in order to target the receptors that are expressed at the apical region of M-cells.10 Significant advances have been made in identifying “M-cell–specific surface marker” receptors,12 although the most investigated family of M-cell targeting molecules is the C-type lectins. Among the C-type lectin receptors, there are some prototypes that recognize specific carbohydrates and certain endogenous glycoproteins, including mannose.10,14–16 Once recognized by mannose receptors, the M-cells increase the uptake process.15,17 Besides its specific conjugation with the C-type lectin receptors, mannose has the additional advantage to act as a mucoadhesive, as it may enhance the residence time of drug substances in the gut.18,19 In this context, mannosylated nanocarriers obtained by the decoration of particulates with mannose or its derivatives have been considered promising non-live vectors.4,10,13,20 Moreover, M-cells may be exploited by some pathogenic microorganisms, such as Salmonella, Yersinia, Typhimurium (Salmonella typhimurium), Mycobacterium sp., and retrovirus, as a portal for the host organism.11,12,21 M. leprae is an intracellular pathogen and the endocytic/phagocytic pathway may represent an interesting approach to allow targeted drug delivery intracellularly. In this context, M. leprae is the causative agent of leprosy, a chronic infectious disease.8,22 M. leprae is phagocytized by numerous cell types; monocyte-derived macrophages are one of the most abundant host cells to come in contact with the mycobacteria.23 The backbone of current treatment of leprosy is a multidrug therapy consisting of dapsone (DAP), clofazimine, and rifampicin as first-line drugs.24,25 Although multidrug therapy shows good effectiveness, many patients experience reactive episodes, potentially serious events that may occur during or after treatment;26 for example, hemolytic anemia, hepatitis, and agranulocytosis are more frequently reported during DAP treatment.27 Despite its therapeutic potential, DAP’s low solubility was found to result in a low therapeutic index and high microbial resistance.8 Hence, some efforts have been made to improve the treatment efficacy and reduce the duration of treatment.27
Several studies have reported on the use of mannose coating of different nanocarriers such as liposomes,28,29 dendrimers,30–32 nanoemulsions,33 nanogels,34 solid lipid nanoparticles (SLNs),35 and nanostructured lipid carriers36 to target specific cells containing mannose receptors, although some studies have reported on the use of this sugar to target the mannose receptors present on M-cell surface to enhance cellular uptake.13,18 This transport is performed by fluid-phase or receptor-mediated endocytosis, wherein the content is transported in vesicles across the cytoplasm and suffers exocytosis in the basolateral membrane.11 On the other hand, it reaches the underlying lymphoid tissues and the content may be transported to the mesenteric lymph nodes and may reach the systemic circulation through lymphatic vessels.11 In addition, glycoconjugates similar to the mannose residues present have been found to promote a high interaction with mucosa tissue of the GI tract.17,37 It has previously been described that nanoparticles decorated with D-mannosamine may enhance the absorption of the particles within PP, in particular, on the M-cells by specific interaction with mannose receptors by receptor-mediated endocytosis on the M-cells’ apical membranes.13,18
SLNs are one of the most promising particulate delivery systems to improve the oral bioavailability of hydrophobic drugs.3,38 After oral administration, these nanosystems may interact with the gut surface. The lipid core of SLNs has been reported to stimulate chylomicron formation and facilitate lymphatic uptake by the M-cells.3,13,37,39,40
The purpose of this investigation was to develop mannosylated SLNs (M-SLNs) loaded with DAP, a first-line drug used in the treatment of leprosy, to form M-SLN-DAP, in order to promote bioadhesion of the systems, increasing the residence time in the gut mucosa, combined with specific binding to mannose receptors expressed on M-cells. The SLNs were developed and optimized through an experimental design and statistical modeling, and the mannosylation was carried out from the optimized SLNs. All the obtained formulations were further characterized.
Materials and methods
Materials
DAP was purchased from CHEMOS GmbH (Regenstauf, Germany). The solid lipids, cetyl palmitate, Compritol® 888 ATO, Precirol® ATO 5, and Gelucire® 43/01, were kindly provided by Gattefossé (Saint-Priest, France). Softisan® 142 was purchased from Cremer Care (Hamburg, Germany), Glycerol Tripalmitate® from Alfa Aesar (Haverhill, MA, USA), Witepsol® E85 from Sasol (Johannesburg, South Africa), and stearic acid was from Merck & Co., Inc. (Whitehouse Station, NJ, USA). Tween® 80, D-(+)-mannose, and stearylamine were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). All other chemicals used in the study were of analytical grade.
Methods
Preformulation studies
The interaction of DAP was evaluated in four different lipids commonly used to formulate SLNs. Known amounts of physical mixtures of lipid and DAP (1:1 w/w) were heated at 100°C and then left at room temperature (25°C) until solidification. The obtained melted mixtures were further analyzed by differential scanning calorimetry (DSC) (DSC 200 F3 Maia®; Netzsch-Gerätebau GmbH, Selb-Germany), in order to investigate the lipid–drug interaction phenomena.
Subsequently, four lipids were chosen to produce pilot nanoparticles, as given in the “Production of SLNs” section, at fixed amounts of lipid (200 mg) and surfactant (60 mg), and process parameters (70% of sonicator amplitude during 5 minutes) (VibraCell VCX 130 equipped with a VC 18 probe, Sonics & Materials Inc., Newtown, CT, USA), in order to evaluate the influence of the type of lipid on entrapment efficiency (EE) and loading capacity (LC).
Production of SLNs
DAP-loaded SLNs and placebo SLNs were prepared by hot ultrasonication method. Briefly, the lipid phase containing the lipid and DAP was heated to 75°C until complete drug solubilization in the lipid matrix. The aqueous phase containing the surfactant (Tween 80) in double-deionized water was previously heated in a water bath at the same temperature. The aqueous phase (6 mL) was poured into the lipid phase and homogenized using a probe sonicator (VCX130; Sonics & Materials, Inc., Newtown, CT, USA), varying the amplitude frequency according to the experimental design, for 5 minutes. The nanosuspension obtained was rapidly cooled in an ice bath. Placebo SLNs were prepared in a similar way without the drug.
Experimental design
A three-level, three-factor Box–Behnken design was applied to maximize the experimental efficiency, requiring a minimum number of experiments, in order to optimize SLNs and study the effects of independent variables on dependent variables (Table 1). Independent variables were the amount of drug (X1), the sonicator amplitude (X2), and the amount of surfactant (X3). Other parameters, that is, the amount of solid lipid, the sonication time, and the volume of aqueous phase, were set at fixed levels. The established dependent variables were: Y1 = mean particle diameter, Y2 = polydispersity index (PDI), Y3 = zeta potential (ZP), Y4 = EE, and Y5 = LC. For each factor, the lower (−1), medium (0), and higher values (+1) were chosen on the basis of tested lower and upper values for each variable, according to preformulation studies and literature research. The data were analyzed using analysis of variance (ANOVA).
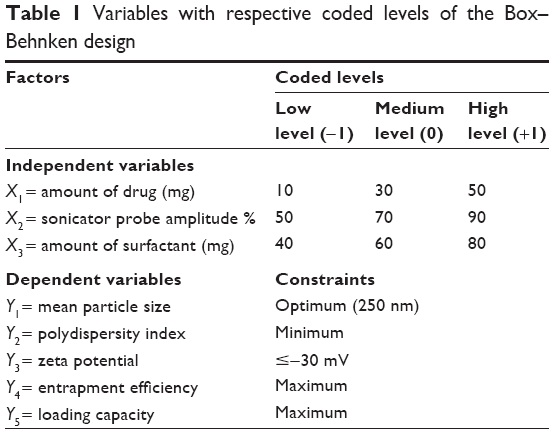 | Table 1 Variables with respective coded levels of the Box–Behnken design |
The polynomial equation generated from the experimental design is given below:
Yn = b0 + b1X1 + b2X2 + b3X3 + b12X1X2 + b13X1X3+ b23X2X3 + b11X12 + b22X22 + b33X32 | (1) |
where Yn is the dependent variable, b0 is the intercept, X1, X2, and X3 are the coded levels of independent variables, and b1–b33 are the regression coefficients computed from the observed experimental values of the respective response (Yn); the terms X1X2 and Xi2 (i = 1, 2, or 3) represent the interaction and quadratic terms, respectively. The polynomial equation was statistically evaluated by finding the statistical significance of coefficients using ANOVA (P-values ≤0.05). Best-fitting experimental model (linear, two-factor interaction, quadratic, and cubic models) was statistically evaluated according multiple correlation coefficient (R2) provided by STATISTICA 10 software.
Optimization and validation of SLNs
In order to establish the reliability of the developed model, optimum variables were used to prepare a checkpoint SLN formulation. The observed results were compared with the predicted values to calculate the predicted error. The graphical and numerical analyses were performed by STATISTICA 10 to obtain the optimum values of the variables based on the criteria of desirability (Table 1).
Synthesis of M-SLNs
M-SLNs were obtained as given in the “production of SLNs” section, with some modifications. Stearylamine (SA) was added in lipid phase at a concentration of 2% w/w of the lipid, and all the subsequent steps were exactly carried out as described earlier for obtaining a modified SLN (M-SLN). The mannose coating was carried out according to the method of Jain et al.41 Briefly, a known volume of a solution of D-mannose (50 mM) in acetate buffer (pH 4) was added to the SLN, and the mixture was maintained in agitation with magnetic stirring at room temperature for 48 hours to achieve maximum conjugation.41 Mannosylated nanoparticles (M-SLN) were subjected to dialysis (Cellu. Sep®Cellu.Sep® T3 with a nominal molecular weight cut off of 12,000–14,000. Seguin, TX – USA). in high volume of double-distilled water for 30 minutes, in order to remove unreacted mannose or other impurities.
Particle diameter, polydispersity (PDI), and zeta potential (ZP) analysis
The particle diameter, PDI, and ZP of all SLN nanosuspensions were analyzed using a ZetaPALS zeta potential analyzer (Brookhaven Instruments, Holtsville, NY, USA). All samples were diluted with double-distilled water to reach a suitable concentration before measurement. All analyses were carried out with an angle of 90° at room temperature.
Entrapment efficiency (EE) and loading capacity (LC)
The EE (%) of DAP within SLNs was determined by the indirectly method, as previously described.42 Briefly, a known aliquot of nanoparticles was properly diluted and subjected to centrifugation with Amicon® Ultra-4 Centrifugal Filter Devices (Millipore, Billerica, MA, USA) The nonentrapped drug was quantified by ultraviolet spectroscopy at 291 nm.40,42 A standard curve was used to determine the concentration of DAP and the results are expressed as mean ± standard deviation (n=3).
Transmission electron microscopy (TEM) analysis
The morphology of the optimized SLNs and M-SLN was observed by TEM (TEM Jeol JEM-1400; JEOL Ltd., Tokyo, Japan). Images were obtained after one drop of nanoparticle suspension was placed over a grid followed by negative staining with uranyl acetate and subjecting it to an accelerating voltage of 60 kV.
Fourier transform infrared spectroscopy (FTIR)
The freeze-dried optimized SLNs and M-SLNs with and without DAP and pure DAP were evaluated using an FTIR spectrophotometer (Frontier™; PerkinElmer Inc., Waltham, MA, USA) equipped with a horizontal attenuated total reflectance sampling accessory with a diamond crystal. The samples were transferred directly to the attenuated total reflectance compartment, and the result was obtained by combining the 16 scans. The spectra were recorded between 4,000 and 600 cm−1 with a resolution of 4 cm−1.
Differential scanning calorimetry
DSC measurements were performed using DSC 200 F3 Maia (Netzsch). Approximately 10 mg of binary mixtures or an equivalent amount of DAP was weighted in an aluminum pan and sealed. For the nanoparticles, known amounts of the selected formulations were weighed and sealed in aluminum pans similarly. The reference pan was left empty. Heating curves for the drug and the mixtures of drug and lipid were recorded with a heating rate of 10°C/min from 25°C to 200°C. Then they were cooled to 25°C under liquid nitrogen, with a cooling rate of 40°C/min. The onset, melting point (peak maximum), and melting enthalpy (ΔH) were calculated using the (NETZSCH Proteus® Software – Thermal Analysis – Version 6.1) software provided for the DSC equipment.
In vitro release study
The release profiles of DAP from SLN- and M-SLN-loaded nanoparticles were investigated by simulating the release of drug through the GI tract and its movement from the stomach to the small intestine, changing the buffer solution from pH 1.2 (firsts 2 hours) to 6.8 (during 3 hours) and 7.4 (until 10 hours), respectively. The influence of the acidic environment of the stomach on the nanoparticles was also investigated by evaluating only the passage of nanoparticles in physiological conditions of the small intestine, that is, subjecting the nanoparticles only to pH 6.8 and 7.4.
In vitro release studies were performed using the dialysis bag method in sink conditions.8 The formulations were first poured into the dialysis bag (Cellu. Sep®Cellu.Sep® T2 with a nominal molecular weight cut off of 6,000–8,000) with the two ends fixed by thread and were placed in preheated dissolution media. The suspension was stirred at 37°C using a heating and magnetic stirring plate (RT 15 Power IKAMAG Multiposition Magnetic Stirrer, Staufen, Germany) at 350 rpm. At fixed time intervals, 200 μL of sample was withdrawn and the same volume of fresh buffer was added according to the conditions. At specified time periods, the dialysis bags were withdrawn from one medium and poured into the subsequent buffer solution. The samples were read in an ultraviolet 96-well plate reader at 291 nm. All the analytical conditions were previously optimized, and standard curves were obtained in triplicate.
Stability studies
In order to evaluate the stability of optimized DAP-loaded SLN and M-SLN nanoparticles, a brief study was carried out by evaluating the particle diameter, PDI, and ZP for a period of 60 days while they were stored at room temperature, in comparison to the first day of production, as described earlier.
Statistical analysis
All experiments/determinations were performed in triplicate. Comparison of diameter, PDI, ZP, and EE was performed using two-tailed, Student’s t-test and assuming equal variance of both groups (95% confidence interval). Suitability analysis of the equation regressions from the results of Box–Behnken design was performed by a one-way ANOVA using STATISTICA 10 software.
Results and discussion
Preformulation studies
Assessment of the interactions of the drug in the lipid matrix was the first step to formulate suitable nano-lipid carriers, since it is one of the most important factors for determining drug EE of the SLNs.43–45
Different methods are reported in the studies of drug excipient compatibility and stability. However, DSC has proven to be an important tool during preformulation of pharmaceuticals, since it permits evaluation of the interactions among the formulation components according to shift, appearance, or disappearance of the peaks (exothermic or endothermic), and/or variations in enthalpy values in the thermal curves of drug–excipient mixture.46 More specifically, DSC analysis may provide information about the value of the enthalpy of mixing between the drug and lipid candidates during SLN design, indicating the amount of energy required to achieve mutual solubility, taking into account the differences in polarity of different components, and allowing selection of the most promising lipid candidates for loading drugs.47 When the enthalpy involved in the melting peak of the drug decreases, it means that the presence of the lipid causes a decrease in crystallinity of the drug and, consequently, leads to a higher interaction at the molecular level. The smaller the energy involved, the greater is the interaction.46 Several interactions occur between the drug and the lipid, including hydrogen bonding, electrostatic, and hydrophobic.47 Eight lipids with different physicochemical characteristics were evaluated as shown in Table 1.
The thermograms of pure DAP and the physical mixtures are shown in Figure 1, and the DSC parameters are depicted in Table 2. DAP shows an endothermic event at 179°C and 75 J/g of enthalpy involved (ΔH). Based on the results obtained from the DSC analysis of the binary mixtures, it was possible to infer that all the lipids caused a decrease of approximately 50% of the ΔH of DAP melting point. The values ranged from 34.08 (Glycerol Tripalmitate) to 41.32 J/g (cetyl palmitate). In addition, all the lipids did not alter the temperature of DAP melting point. Therefore, all the studied lipids were found to be suitable in terms of stability to produce SLNs and presented great potential to load DAP. In this way, it was decided to select four lipids based on phase transition temperature to produce pilot batches in order to evaluate mainly the EE, besides the diameter, PDI, and ZP values.
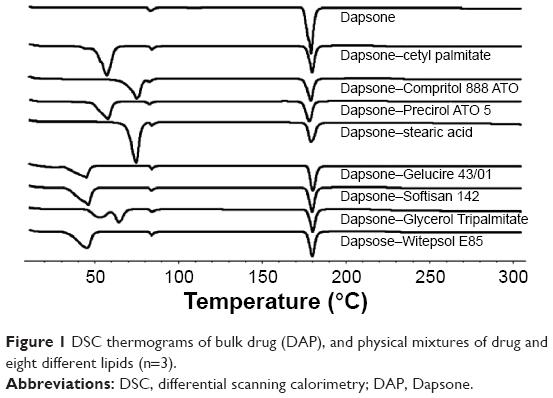 | Figure 1 DSC thermograms of bulk drug (DAP), and physical mixtures of drug and eight different lipids (n=3). |
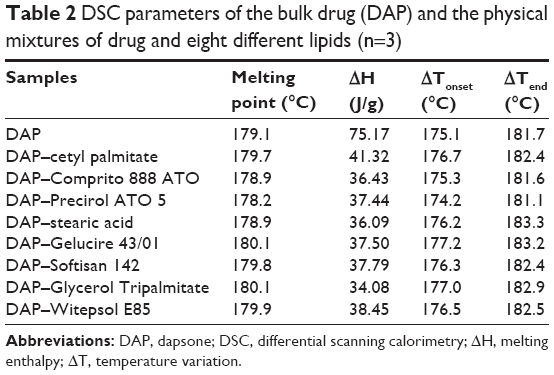 | Table 2 DSC parameters of the bulk drug (DAP) and the physical mixtures of drug and eight different lipids (n=3) |
A prerequisite for the SLNs to be successfully used as drug carriers is to remain solid at normal human body and room temperatures. For this purpose, it is recommended that the lipids have phase transition above 40°C.48 In addition, for production purposes, it is ideal that lipids resist physical stress after the freeze-drying process.
Thus, the lipids with higher phase transition temperature were selected to develop pilot batches, that is, cetyl palmitate, Compritol 888 ATO, Precirol ATO 5, and stearic acid. The pilot SLNs were obtained as given in the “Preformulation studies” section, and all the variables were maintained constant.
The obtained data are shown in Table 3. As could be seen, the secondary parameters studied (diameter, PDI, and ZP) were in good agreement with the desired profile, although cetyl palmitate showed a higher value of EE and seemed to be the best choice for the subsequent design.
 | Table 3 Characterization of obtained pilot SLNs regarding particle diameter, PDI, ZP, EE, and LC (n=3) |
Experimental design
The preliminary study is also important to identify the critical factors that directly affect the main parameters of evaluation, that is, diameter, PDI, and EE, during development of SLNs.45 Some of the variables studied were amount of lipid, amount and type of surfactant, time and amplitude of the sonication probe, amount of drug, and volume of the aqueous phase. Based on the data obtained, three critical factors were considered to affect the responses: amount of drug, amount of surfactant, and amplitude of the sonication probe. All other parameters were maintained constant, as described in the “Preformulation studies” section. These three variables were thoroughly studied.
The use of experimental design allows reducing the number of experiments, the amount of material, and the time consumed.49,50 Optimization and characterization of the preparation procedure of nanoparticles was performed using a Box–Behnken experimental design. This response surface methodology was selected because, among other advantages, it allows avoiding extreme combinations of the studied factors. In this last case, the formation of nanoparticles could have been compromised. Also, the Box–Behnken design requires the performance of a minimum number of experiments when three factors are considered, as compared to others, namely, the central composite design.51
The Box–Behnken experimental design allowed us to obtain 13 different formulations, with a triplication of the central point, resulting in a total of 15 formulations. The replication of the central point enables to estimate the experimental error.8,52 The results of the five selected responses (Y1–Y5) are shown in Table 4.
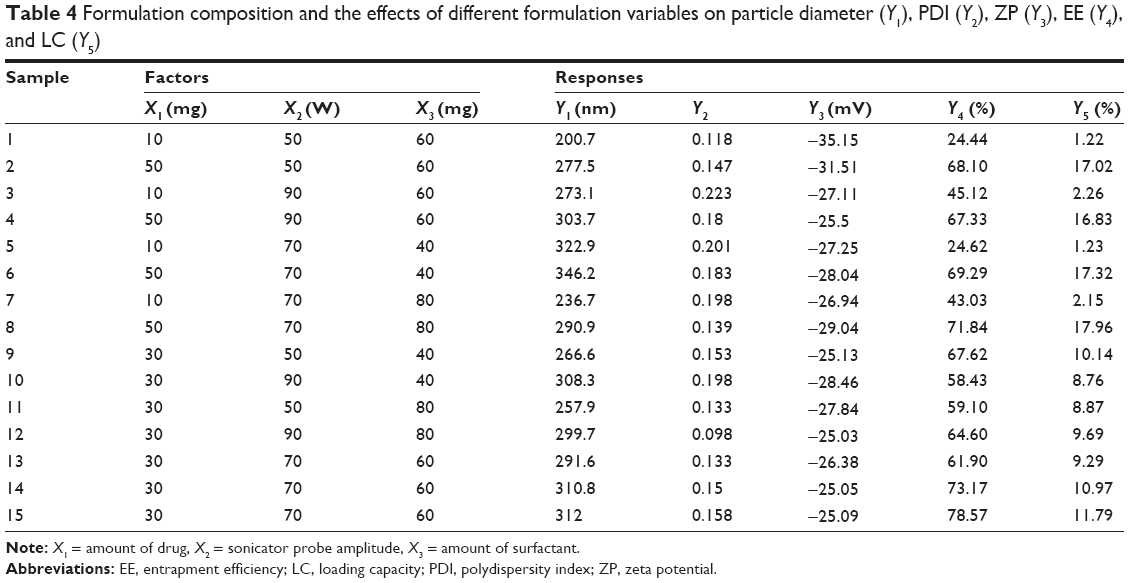 | Table 4 Formulation composition and the effects of different formulation variables on particle diameter (Y1), PDI (Y2), ZP (Y3), EE (Y4), and LC (Y5) |
Based on the data obtained, the fitting models of polynomial equations involving the main effects and interaction factors for diameter, PDI, ZP, EE, and LC were statistically analyzed by calculating the P-values with a 95% confident level. (Table 3).
The intercept corresponds to the mean of the responses. The magnitude of coefficients in polynomial equation has either a positive sign, indicating synergistic effect, or a negative sign, indicating antagonistic effect.43,45 Interaction terms or quadratic relationships are represented by more than one factor or higher order terms in regression equations, respectively. Nonlinearity between factors and responses is also suggested, that is, the variance of one factor can produce a different degree of response at different levels or more than one factor is varied.8,51,52 The polynomial equations were statistically validated using ANOVA and were considered statistically significant when F was higher than Fcritic for all responses. Results of the statistical analysis are summarized in Table 4.
The values of the obtained responses varied from 200.7 to 346.2 nm for the diameter, from 0.098 to 0.223 for PDI, from −35.15 to −25.05 mV for ZP, from 24.44% to 78.57% for EE, and from 1.22% to 17.96% for LC.
Effect of the amount of drug
The effect of the amount of drug (X1) in the dependent parameters may be inferred from the coefficients shown in Tables 5 and 6. For the diameter, EE, and LC, the effects were statistically significant, when as the P-values were ≤0.05, with a 95% confidence interval.
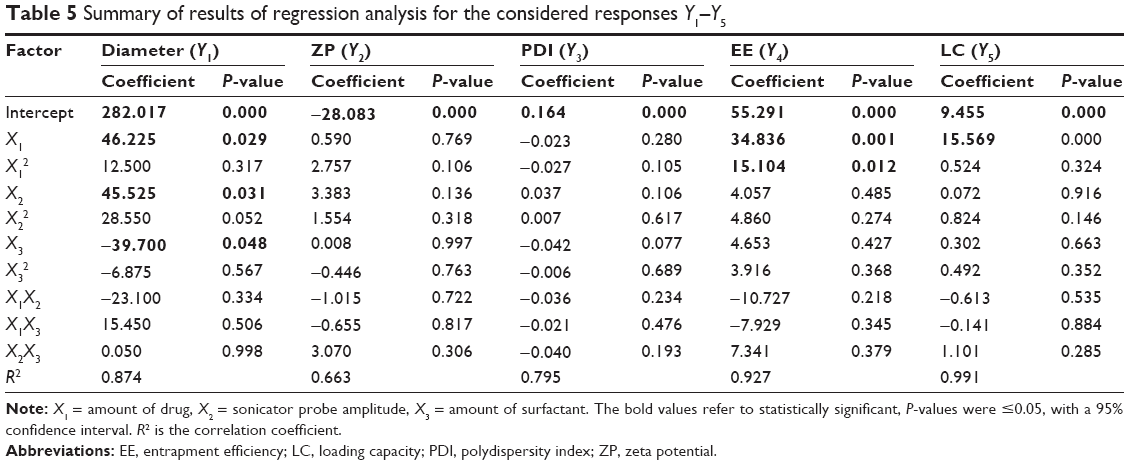 | Table 5 Summary of results of regression analysis for the considered responses Y1–Y5 |
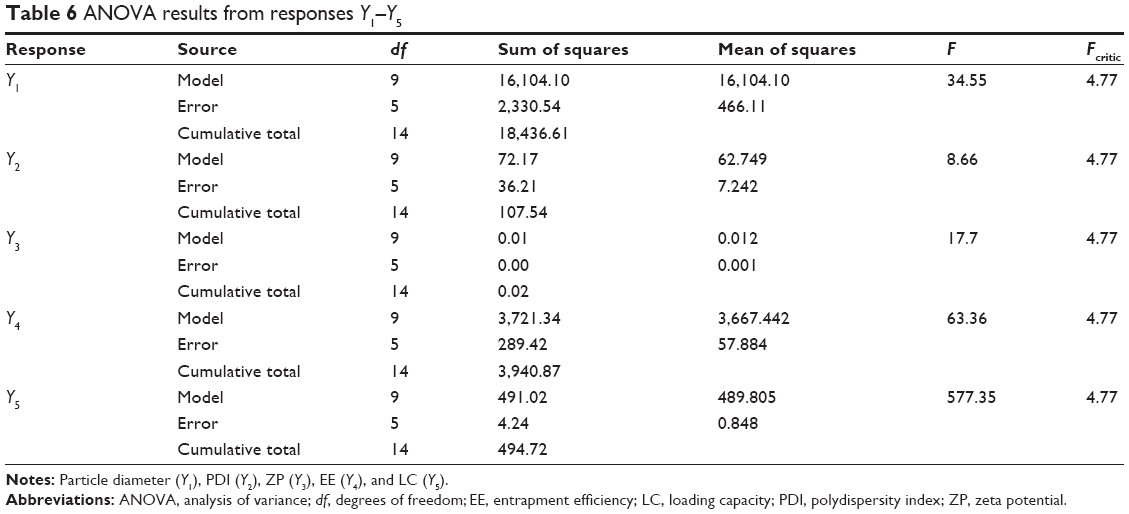 | Table 6 ANOVA results from responses Y1–Y5 |
As mentioned earlier, a positive sign means a synergic effect, that is, the value of the responses increases with an increase in the level of the independent variable; the opposite is true with a negative sign. For the three responses influenced by the amount of drug, in all cases, the increasing amount of drug led to an enhancement in the responses.
The increase in particle diameter accompanying an increase in EE and LC may be correlated with the saturation of the external media with the drug, due to its low solubility in aqueous phase. Thus, the lipid matrix seems to be enriched with the drug in a linear way, as can be observed from the linear coefficient of the LC (X1), leading to an increase in particle diameter. On the other hand, the lipid:drug ratio seems to present a nonlinear relationship, as the quadratic coefficient of EE is also significant.
Effect of the sonicator probe amplitude
The effect of the sonicator probe amplitude (X2) in the dependent variables is shown in Tables 5 and 6. This factor only positively influenced the diameter of the nanoparticles (Y1), since the P-values were ≤0.05, with a 95% confidence interval. It means that this response increases with an increase in amplitude of the sonicator probe.
The possible explanation for this phenomenon may be due to the increase in the energy on the surface of the nanoparticles, which may disrupt the outer core of lipid, causes the drug molecule to reach the external phase and form aggregates, resulting in a higher diameter and PDI. In addition, these results are supported by other researchers who found that this enhancement in particle diameter happens when the amplitude goes beyond a given sonication energy threshold, leading to formation of aggregates, which is generally known as “sonication-induced aggregate formation”.45,53
Effect of the amount of surfactant
The effect of the amount of surfactant (X3) was only statistically significant (P-value ≤0.05, with a 95% confidence interval) for the particle diameter (Y1), as expressed in Tables 5 and 6. The coefficient of the regression analysis presented a negative sign, which means that the response decreases with an increase in the level of the variable X3, that is, higher amounts of surfactant generate smaller particles.
It is already known that higher amounts of surfactant may promote formation and stabilization of smaller particles due to the decrease in interfacial tension between the lipid and the aqueous phase51 and, consequently, controls the aggregation of lipid particle.45
Optimization and validation of production of SLNs
Based on the polynomial models, the results of three-dimensional response surface analysis were plotted (Figure 2), representing the effect of significant independent factors on each observed response.8 The correlation coefficients of the obtained models are presented in Table 7.
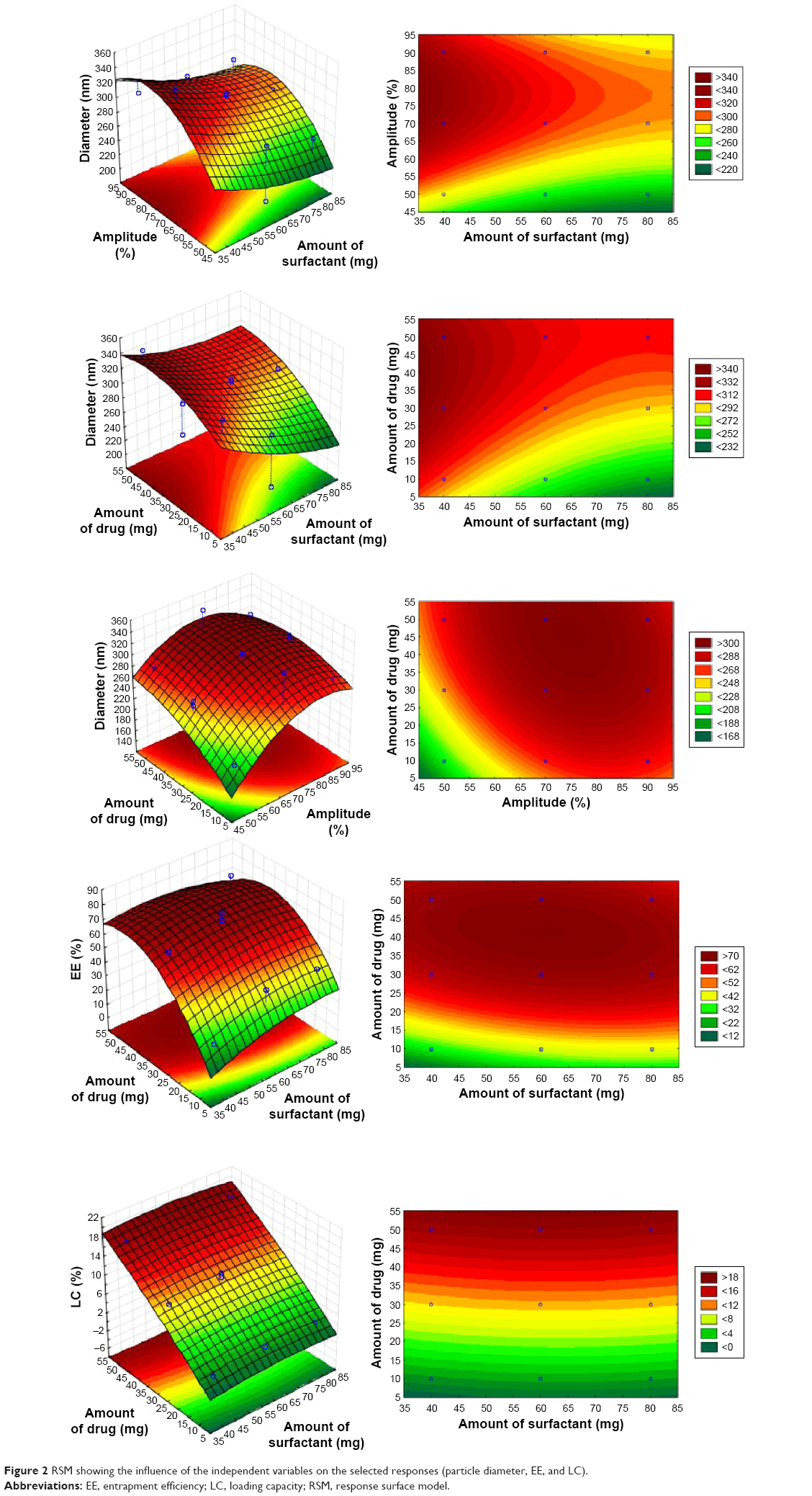 | Figure 2 RSM showing the influence of the independent variables on the selected responses (particle diameter, EE, and LC). |
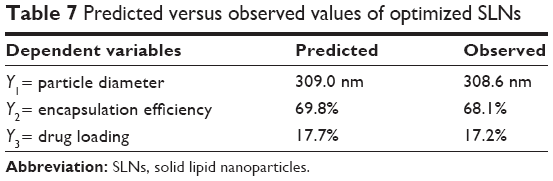 | Table 7 Predicted versus observed values of optimized SLNs |
The qualities of the models were evaluated by plotting correlation plots (predicted vs experimental data) and residual plots (residual vs experimental data) (Figure 3). The obtained models for the dependent variables showed high predictability coefficients with few outliers.8 Desirability function is a way of overcoming the difficulty of multiple, or sometimes opposing, responses in which each response is associated with its own partial desirability function. The point possessing the highest value for desirability is termed as optimum.54
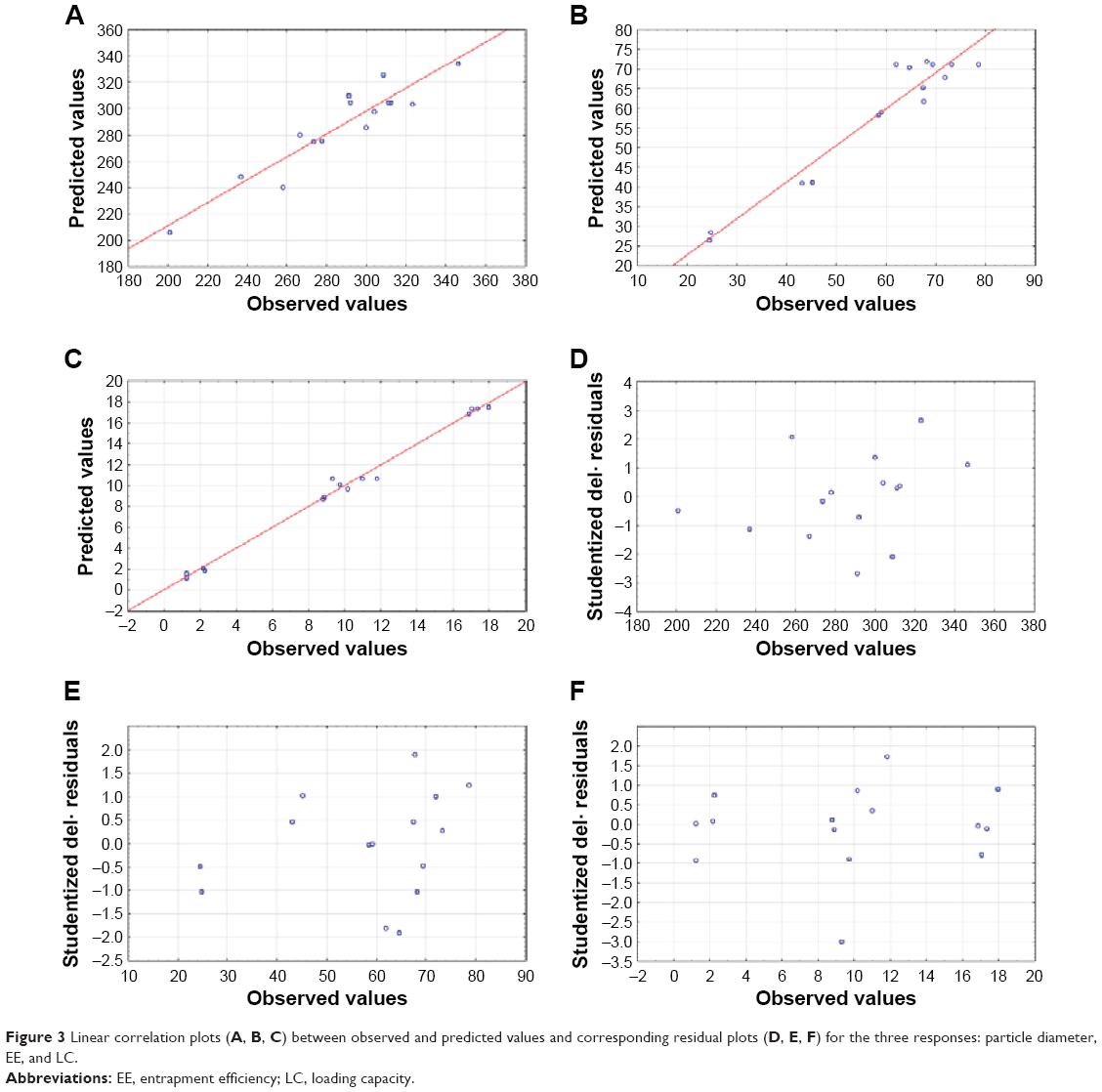 | Figure 3 Linear correlation plots (A, B, C) between observed and predicted values and corresponding residual plots (D, E, F) for the three responses: particle diameter, EE, and LC. |
In order to validate the models, the formulation given as the best was selected as the checkpoint. The values obtained for the amount of lipid, surfactant, and drug were 200, 70, and 50 mg, respectively. The predicted values for the three models were reasonably similar to experimental data (Table 7). In addition, the residual plots provided a better visual comprehension of the capability of the models, since all the data were widely scattered in predicting the responses, as all the points were tightly distributed along the horizontal axis (Figure 3). Thus, they can be judged as suitable models, with no trend in the data. These data reflect greater robustness.
Synthesis and characterization of M-SLNs
M-SLNs were produced adopting the method to obtain nonfunctionalized nanoparticles. The intermediate step in the production of M-SLNs was based on the addition of SA, which is a lipophilic cationic molecule with a primary amine group, into the lipid matrix,6 which generates positively charged nanoparticles due to the presence of amino groups on the surface of SLNs.38
The reaction of unprotected sugars with long-chain fatty amine has been described.41 In this procedure, the reaction was achieved by ring opening of the unprotected anomeric hydroxyl group of mannose with the amino groups present on the surface of the nanoparticles in acetate buffer (pH 4.0), resulting in the formation of Schiff’s base (–N=CH–).13,41 A deeper discussion regarding this linkage and the equilibrium is given in the FTIR spetroscopy detction, wherein FTIR analysis is discussed.
The values of particle diameter, PDI, and ZP of the M-SLN-DAP obtained and those of respective unloaded nanoparticles, together with the optimized SLNs are given in Table 8. It was observed that both unloaded SLNs and M-SLNs presented inferior values of particle diameter (283.5 and 228.6 nm, respectively), while the loaded nanoparticles showed slightly higher diameter (308.6 and 333.2 nm, respectively). This is expected and points to a rearrangement of the lipid matrix caused by the entrapment of the drug. All the PDI values were approximately 0.2, which is considered a good value for SLN, as it proves the homogeneous distribution of the particles.55 The ZP values of SLNs and SLN-DAP were −28.11 and −28.45, respectively. When mannose was added, the ZP changed to positive, being the obtained values for M-SLN and M-SLN-DAP +61.83 and +65.66 respectively, providing evidence of mannose coating. However, a higher ZP is considered to provide stronger electrostatic repulsion and, thus, better stability of nanoparticles.19
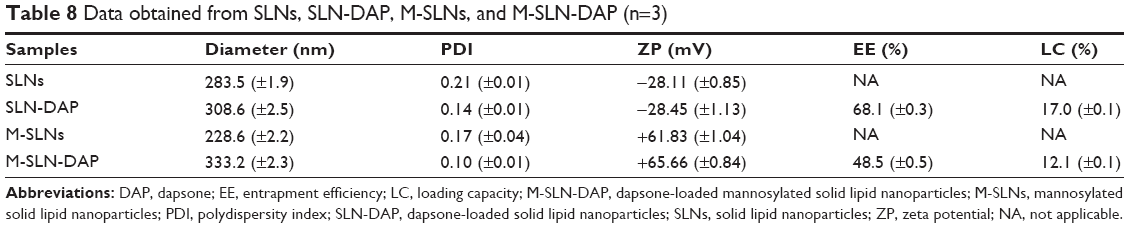 | Table 8 Data obtained from SLNs, SLN-DAP, M-SLNs, and M-SLN-DAP (n=3) |
The EE and LC of M-SLN-DAP were lower, compared to those of SLN-DAP. This may be explained by a small drug release during the functionalization process. Although the mannosylation process decreased the EE and LC, the drug loading was still high, being the nanoparticles able to charge high amounts of drug.
The positive charge of ZP observed for M-SLNs compared to SLNs suggests the successful mannose coating on the surface of the nanoparticles. This phenomenon has already been described earlier.18 Surface charge of the nanoparticles plays an important role in the nonspecific interactions with the surrounding media. Positively charged surfaces enhance interactions with the mucus, and the ammonium groups may strengthen adhesiveness and, thus, increase the residency time by forming electrostatic interactions with the mucus and subsequent binding to M-cells within PP.18 Moreover, the presence of basic or acidic functional groups leads to pH-dependent behaviors that are useful for targeting different regions of the GI tract.10,18
TEM analysis
In order to evaluate the morphological characteristics of SLN and M-SLN, loaded and unloaded, TEM studies were performed (Figure 4). The images show that the particles obtained were spherical in shape, which is typical of SLN systems.56 Moreover, the images demonstrated that the nanoparticles presented a narrow diameter distribution with particle diameter near 200 nm, which was in accordance with the DLS measurement. The presence of regular particles and the absence of particle aggregation corroborate the PDI results, showing only one population.
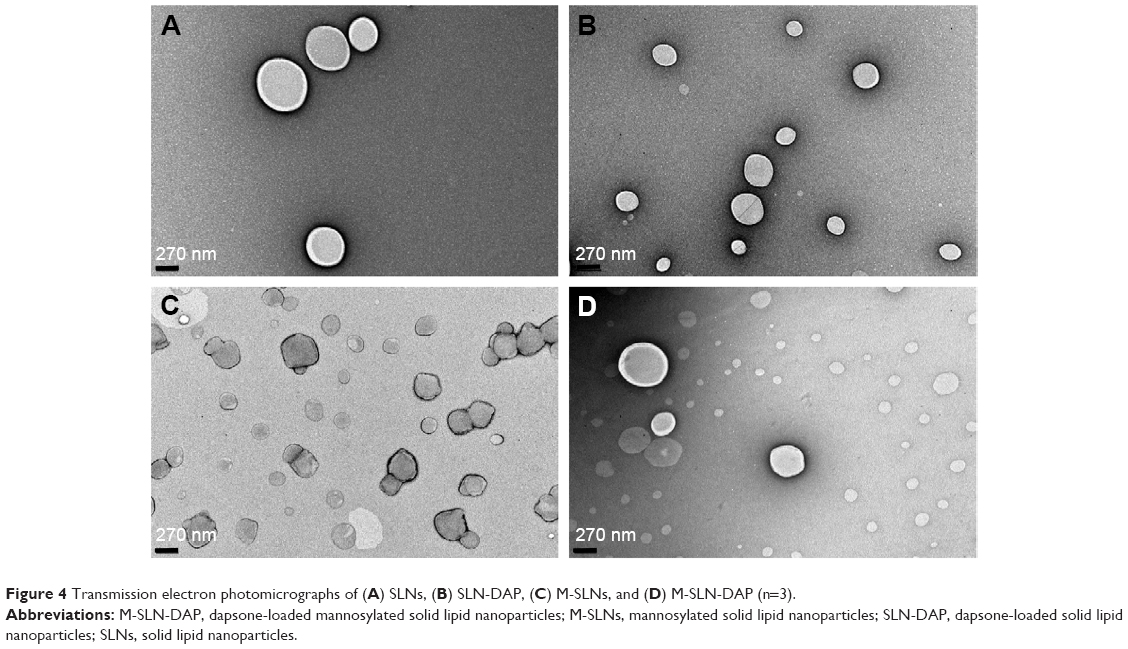 | Figure 4 Transmission electron photomicrographs of (A) SLNs, (B) SLN-DAP, (C) M-SLNs, and (D) M-SLN-DAP (n=3). |
From the images, it may be concluded that the drug incorporation and the mannose coat did not affect the particle shape and the diameter to a large extent.
FTIR spectroscopy
Figure 5 shows the spectra of SLN, SLN-DAP, M-SLN, and M-SLN-DAP, respectively.
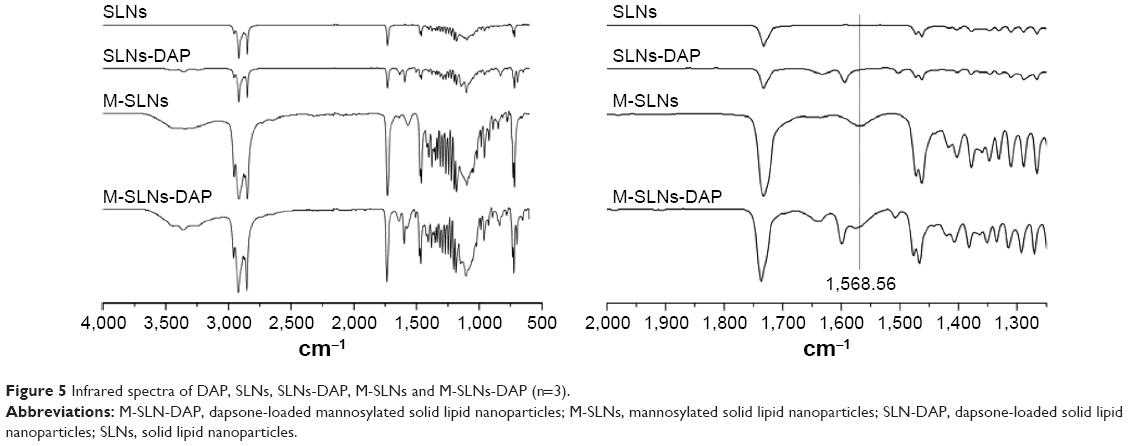 | Figure 5 Infrared spectra of DAP, SLNs, SLNs-DAP, M-SLNs and M-SLNs-DAP (n=3). |
Regarding mannosylation, the mannose coating was done by ring-opening reactions followed by the reaction of aldehyde groups of mannose in acidic buffer solution (pH 4.0) with the amino groups of the SA, leading to the formation of Schiff’s base.41 It is already reported that Schiff’s bases may remain in an equilibrium stage by reduction of the secondary amine, especially in lower pH. In the spectra of M-SLNs, the presence of N–H bending of secondary amines at 1,558.56 cm−1 and C=N stretch at 1,410.01 cm−1 are observed,13,41,57,58 confirming the formation of the Schiff’s base and the linkage between mannose and amine termination present in SA in the SLNs.
Differential scanning calorimetry
DSC thermal analysis can be utilized to study and characterize the matrix state consisting of polymorphism, drug entrapment, and component interactions.18
In Figure 6, the DSC thermograms of DAP, cetyl palmitate, the physical mixtures (without any process), and both SLNs (a) and M-SLNs (b) loaded and unloaded with DAP are given.
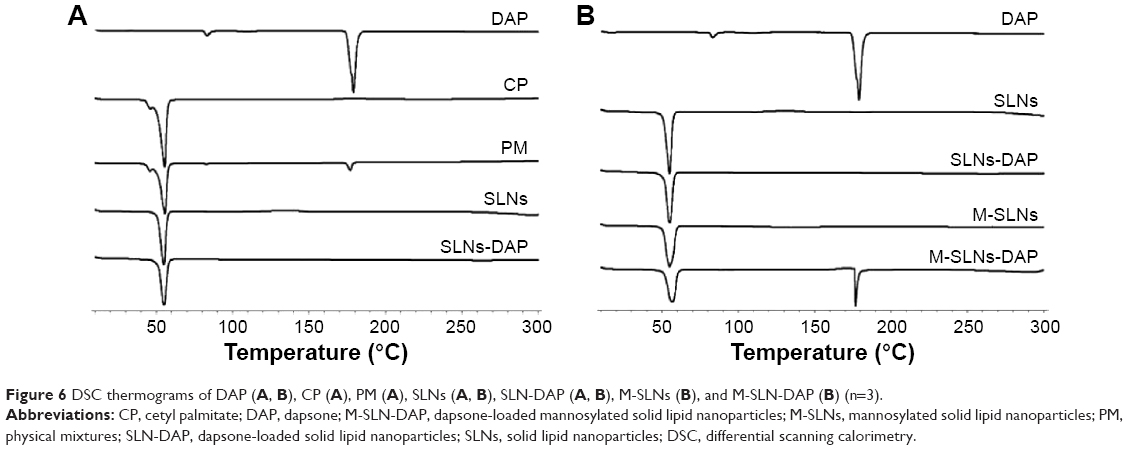 | Figure 6 DSC thermograms of DAP (A, B), CP (A), PM (A), SLNs (A, B), SLN-DAP (A, B), M-SLNs (B), and M-SLN-DAP (B) (n=3). |
The differences between the thermograms of the nanoparticles and the bulk lipid, as well as the physical mixture, show that there was a change in the crystallinity of the lipid matrix during the process. Lipid molecules may show different polymorphic forms: α, β′, and the most stable β form.59 Depending on the chemical nature of the lipid and the parameters established for the production of lipid nanoparticles, different fractions of α and β′ modification will be obtained, as they were achieved through cooling process60,61 (see “Production of SLNs” section). In addition, none of the SLN-DAP thermograms show endothermic peak at 179.1°C relative to DAP melting point, which may be attributed to the amorphous state of the entrapped drug.
On the other hand, it is possible to observe an endothermic event in the thermograms of M-SLN-DAP (176°C). This event may be explained by the melting of the nonentrapped drug, which was higher compared to SLN-DAP (Table 9).
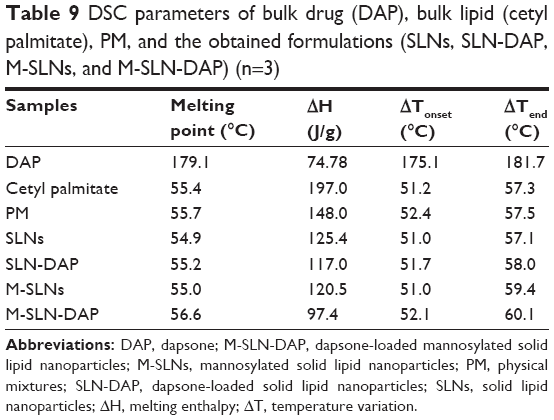 | Table 9 DSC parameters of bulk drug (DAP), bulk lipid (cetyl palmitate), PM, and the obtained formulations (SLNs, SLN-DAP, M-SLNs, and M-SLN-DAP) (n=3) |
The enthalpies of all SLN formulations are much lower than for bulk lipid, which could be related to the presence of interactions between solid lipid and surfactants, or to the increase in surface area of the nanoparticles.56 Changes in crystallinity of the lipid have been already reported.62 Changes in polymorphic state of the lipid from crystalline to amorphous may provide the drug accommodation and higher drug entrapment.45
In vitro release study
The in vitro release profile of DAP from SLN and M-SLN was evaluated by the diffusion of DAP through the dialysis membrane for 8 hours in different pH conditions simulating the GI tract.
Figure 7 shows the obtained profiles, in which the amounts of DAP released from the functionalized and nonfunctionalized SLNs were plotted as a function of time in two different conditions. Figure 7A simulates the passage of the nanoparticles in the acidic environment of the stomach (pH 1.2), while Figure 7B shows the release of DAP, assuming that the first contact of the SLNs is with the intestinal environment. In both conditions, SLN and M-SLN showed overlapping profiles, proving that the presence of mannose on the surface of the nanoparticles did not interfere with the release of the drug.63
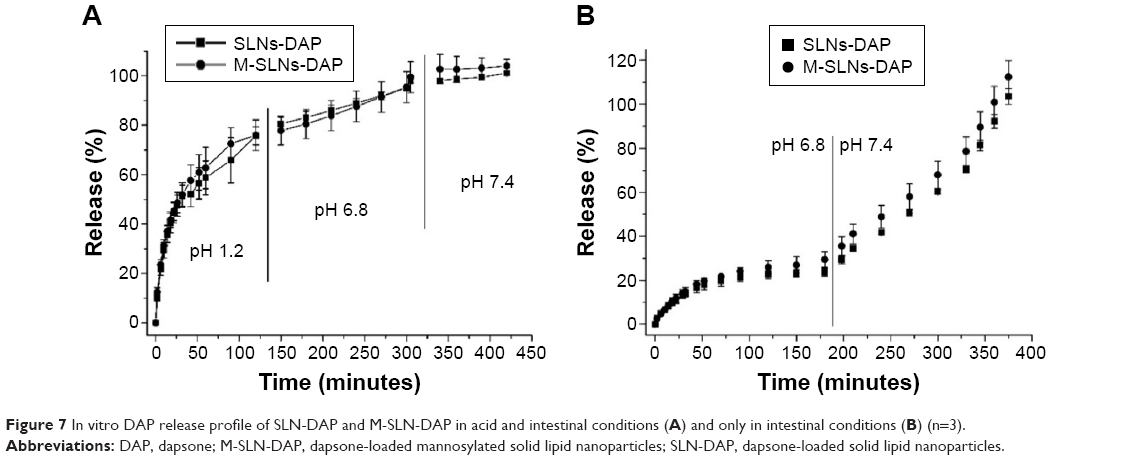 | Figure 7 In vitro DAP release profile of SLN-DAP and M-SLN-DAP in acid and intestinal conditions (A) and only in intestinal conditions (B) (n=3). |
In Figure 7A, the initial burst effect (almost 60% released in the first hour) may be attributed to the disintegration of the lipid matrix in acidic conditions. This issue can be surpassed using gastro-resistant capsules to incorporate the formulations, until they reach the intestine.64
From Figure 7B, the influence of the acidic microenvironment on the release of DAP from nanoparticles can be confirmed. During the first 30 minutes, approximately 30% of the drug was released, which may be attributed to the nonentrapped drug. This percentage was maintained for 3 hours. This period simulates the passage of nanoparticles through the small intestine, showing that the particles remain essentially unchanged and may be available to adhere to the intestinal mucosa for a drug-controlled release favorable for subsequent internalization. Furthermore, the mannose coat may improve the internalization by the M-cell, targeting the drug to the intestinal lymphatic system. When the pH of the medium was changed to pH 7.4, the drug was released faster, reaching 100% of drug release in a total of 6 hours. The sustained release phase of the drug is also not prolonged for longer hours because of the smaller diameter of the particles, which provides a larger surface area for the drug-loaded particles.65,66
Stability of SLN-DAP and M-SLN-DAP
Physical stability of both SLN-DAP and M-SLN-DAP was assessed by analyzing the changes in all the parameters studied before, that is, particle diameter, PDI, ZP, EE, and LC, for 60 days, storing them at room temperature (Figure 8).
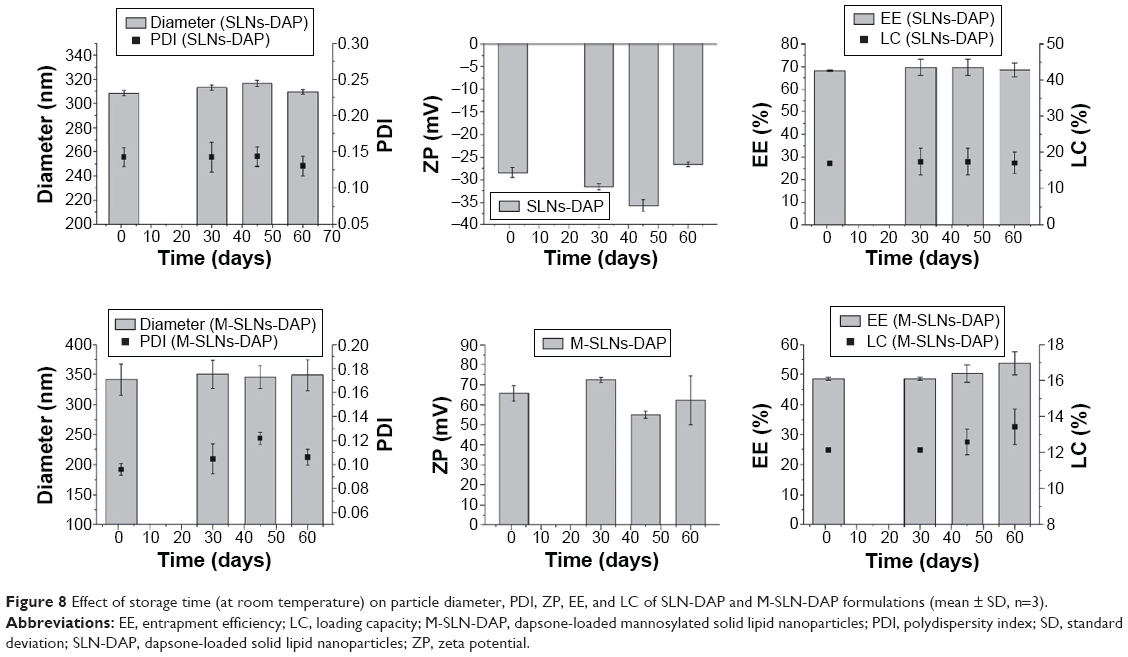 | Figure 8 Effect of storage time (at room temperature) on particle diameter, PDI, ZP, EE, and LC of SLN-DAP and M-SLN-DAP formulations (mean ± SD, n=3). |
Changes in diameter and PDI are usually accepted as indicators of formulation instability. In all samples, no particle aggregation was found through visual observations up to 60 days, which suggests that the nanoparticles were quite stable with no significant changes in the mean particle diameter and PDI.
Overall, all the SLN formulations presented good stability after 60 days, with only slight changes in particle diameter, PDI, and ZP. The EE and LD were also unaffected.
Conclusion
DAP-loaded SLNs and M-SLNs were successfully developed by hot ultrasonication method by employing a three-level, three-factor Box–Behnken Design, after the preformulation study was conducted with different lipids. All the variables studied influenced statistically, at least, one response. Through the response surface methodology, it was possible to identify and obtain particles with optimum diameter distribution, PDI, ZP, satisfactory loading, and high EE. Linearity was found between the observed and the predicted responses, and a very low predicted error was observed in the responses of the optimized formulation, suggesting a very good predictive ability of the design. From the optimized SLNs, it was possible to obtain mannosylated nanoparticles to target the M-cells in the PP of GI tract. All the formulations were characterized. Morphological studies using TEM images showed spherical shape and nonaggregated nanoparticles. We checked if there was any difference between decorated and nondecorated nanoparticles. FTIR analysis showed the characteristic peaks of Schiff’s base, confirming successful mannose coating. A decrease in the enthalpy and broadening of the lipid melting peaks of the DSC thermograms are consistent with the nanostructure of the SLNs. In vitro release profile of DAP revealed that in both pH conditions, the full amount of the drug was released within 6 hours, and the acidic conditions influenced the release rate. The stability tests conducted for 8 weeks at room temperature give a good indication that at least in solution, the formulations maintain their original properties. Our results demonstrated that M-SLN-DAP could be a promising carrier for the specific treatment of leprosy, with an innovative approach to target DAP directly to the immune system for enhanced antileprosy effect and lowered adverse side effects.
Acknowledgments
Alexandre Vieira thanks the CNPq, Ministry of Education of Brazil for the fellowship 246514/2012-4. Luíse Chaves thanks the CAPES Foundation, Ministry of Education of Brazil for the Doctoral fellowship 0831-12-3. Marina Pinheiro is thankful to Fundação para a Ciência e Tecnologia (FCT) for the Post-Doc Grant (SFRH/BPD/99124/2013). Additionally, this work was possible with financial support from FCT/MEC through National Funds and co-financed by FEDER, under the partnership agreement PT2020 – UID/MULTI/04378/2013 -POCI/01/0145/FERDER/007728.
Disclosure
The authors report no conflicts of interest in this work.
References
Pathak K, Raghuvanshi S. Oral bioavailability: issues and solutions via nanoformulations. Clin Pharmacokinet. 2015;54(4):325–357. | ||
Castro PM, Fonte P, Sousa F, Madureira AR, Sarmento B, Pintado ME. Oral films as breakthrough tools for oral delivery of proteins/peptides. J Control Release. 2015;211:63–73. | ||
Harde H, Das M, Jain S. Solid lipid nanoparticles: an oral bioavailability enhancer vehicle. Expert Opin Drug Deliv. 2011;8(11):1407–1424. | ||
Gamazo C, Martin-Arbella N, Brotons A, Camacho AI, Irache JM. Mimicking microbial strategies for the design of mucus-permeating nanoparticles for oral immunization. Eur J Pharm Biopharm. 2015;96:454–463. | ||
Vieira AC, Ferreira Fontes DA, Chaves LL, et al. Multicomponent systems with cyclodextrins and hydrophilic polymers for the delivery of Efavirenz. Carbohydr Polym. 2015;130:133–140. | ||
Mei L, Zhang Z, Zhao L, et al. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv Drug Deliv Rev. 2013;65(6):880–890. | ||
Sigurdsson HH, Kirch J, Lehr C-M. Mucus as a barrier to lipophilic drugs. Int J Pharm. 2013;453(1):56–64. | ||
Chaves LL, Vieira AC, Ferreira D, Sarmento B, Reis S. Rational and precise development of amorphous polymeric systems with dapsone by response surface methodology. Int J Biol Macromol. 2015;81:662–671. | ||
Chen M-C, Sonaje K, Chen K-J, Sung H-W. A review of the prospects for polymeric nanoparticle platforms in oral insulin delivery. Biomaterials. 2011;32(36):9826–9838. | ||
des Rieux A, Pourcelle V, Cani PD, Marchand-Brynaert J, Préat V. Targeted nanoparticles with novel non-peptidic ligands for oral delivery. Adv Drug Deliv Rev. 2013;65(6):833–844. | ||
Araújo F, Pereira C, Costa J, Barrias C, Granja PL, Sarmento B. In vitro M-like cells genesis through a tissue-engineered triple-culture intestinal model. J Biomed Mater Res B: Appl Biomater. 2016;105(4):782–788. | ||
Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. 2013;6(4):666–677. | ||
Witoonsaridsilp W, Paeratakul O, Panyarachun B, Sarisuta N. Development of mannosylated liposomes using synthesized N-octadecyl-d-mannopyranosylamine to enhance gastrointestinal permeability for protein delivery. AAPS PharmSciTech. 2012;13(2):699–706. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Kawakami S, Hashida M. Glycosylation-mediated targeting of carriers. J Control Release. 2014;190:542–555. | ||
Loka RS, McConnell MS, Nguyen HM. Studies of highly-ordered heterodiantennary mannose/glucose-functionalized polymers and concanavalin a protein interactions using isothermal titration calorimetry. Biomacromolecules. 2015;16(12):4013–4021. | ||
Irache JM, Salman HH, Gamazo C, Espuelas S. Mannose-targeted systems for the delivery of therapeutics. Expert Opin Drug Deliv. 2008; 5(6):703–724. | ||
Youngren SR, Mulik R, Jun B, Hoffmann PR, Morris KR, Chougule MB. Freeze-dried targeted mannosylated selenium-loaded nanoliposomes: development and evaluation. AAPS PharmSciTech. 2013;14(3):1012–1024. | ||
Luo Y, Teng Z, Li Y, Wang Q. Solid lipid nanoparticles for oral drug delivery: chitosan coating improves stability, controlled delivery, mucoadhesion and cellular uptake. Carbohydr Polym. 2015;122:221–229. | ||
Kizhakkedathu JN, Creagh AL, Shenoi RA, et al. High molecular weight polyglycerol-based multivalent mannose conjugates. Biomacromolecules. 2010;11(10):2567–2575. | ||
Nakato G, Hase K, Suzuki M, et al. Cutting edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J Immunol. 2012;189(4):1540–1544. | ||
Kim HJ, Brennan PJ, Heaslip D, Udey MC, Modlin RL, Belisle JT. Carbohydrate-dependent binding of langerin to SodC, a cell wall glycoprotein of Mycobacterium leprae. J Bacteriol. 2015;197(3):615–625. | ||
Bhat RM, Prakash C. Leprosy: an overview of pathophysiology. Interdiscip Perspect Infect Dis. 2012;2012:181089. | ||
Kar HK, Gupta R. Treatment of leprosy. Clin Dermatol. 2015;33(1):55–65. | ||
Eichelmann K, González SG, Salas-Alanis J, Ocampo-Candiani J. Leprosy. An update: definition, pathogenesis, classification, diagnosis, and treatment. Actas Dermosifiliogr. 2013;104(7):554–563. | ||
Yamasaki PR, do Nascimento DC, Chelucci RC, et al. Synthesis and evaluation of novel dapsone-thalidomide hybrids for the treatment of type 2 leprosy reactions. Bioorg Med Chem Lett. 2014;24(14):3084–3087. | ||
Anusuya S, Natarajan J. The eradication of leprosy: molecular modeling techniques for novel drug discovery. Expert Opin Drug Discov. 2013;8(10):1239–1251. | ||
Wijagkanalan W, Kawakami S, Takenaga M, Igarashi R, Yamashita F, Hashida M. Efficient targeting to alveolar macrophages by intratracheal administration of mannosylated liposomes in rats. J Control Release. 2008;125(2):121–130. | ||
Wang T, Zhen Y, Ma X, Wei B, Li S, Wang N. Mannosylated and lipid A-incorporating cationic liposomes constituting microneedle arrays as an effective oral mucosal HBV vaccine applicable in the controlled temperature chain. Colloids Surf B Biointerfaces. 2015;126:520–530. | ||
Kumar PV, Asthana A, Dutta T, Jain NK. Intracellular macrophage uptake of rifampicin loaded mannosylated dendrimers. J Drug Target. 2006;14(8):546–556. | ||
Dutta T, Agashe HB, Garg M, Balakrishnan P, Kabra M, Jain NK. Poly (propyleneimine) dendrimer based nanocontainers for targeting of efavirenz to human monocytes/macrophages in vitro. J Drug Target. 2007;15(1):89–98. | ||
Dutta T, Jain NK. Targeting potential and anti-HIV activity of lamivudine loaded mannosylated poly (propyleneimine) dendrimer. Biochim Biophys Acta. 2007;1770(4):681–686. | ||
Guo Y, Liu X, Sun X, Zhang Q, Gong T, Zhang Z. Mannosylated lipid nano-emulsions loaded with lycorine-oleic acid ionic complex for tumor cell-specific delivery. Theranostics. 2012;2(11):1104–1114. | ||
Thomann-Harwood LJ, Kaeuper P, Rossi N, Milona P, Herrmann B, McCullough KC. Nanogel vaccines targeting dendritic cells: contributions of the surface decoration and vaccine cargo on cell targeting and activation. J Control Release. 2013;166(2):95–105. | ||
Chuan J, Li Y, Yang L, et al. Enhanced rifampicin delivery to alveolar macrophages by solid lipid nanoparticles. J Nanopart Res. 2013;15(5):1–9. | ||
Song X, Lin Q, Guo L, et al. Rifampicin loaded mannosylated cationic nanostructured lipid carriers for alveolar macrophage-specific delivery. Pharm Res. 2015;32(5):1741–1751. | ||
Salman HH, Gamazo C, Campanero MA, Irache JM. Bioadhesive mannosylated nanoparticles for oral drug delivery. J Nanosci Nanotechnol. 2006;6(9):3203–3209. | ||
Venishetty VK, Chede R, Komuravelli R, Adepu L, Sistla R, Diwan PV. Design and evaluation of polymer coated carvedilol loaded solid lipid nanoparticles to improve the oral bioavailability: a novel strategy to avoid intraduodenal administration. Colloids Surf B: Biointerfaces. 2012;95:1–9. | ||
Cho HJ, Park JW, Yoon IS, Kim DD. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake. Int J Nanomedicine. 2014;9:495–504. | ||
Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42(1–2):11–18. | ||
Jain A, Agarwal A, Majumder S, et al. Mannosylated solid lipid nanoparticles as vectors for site-specific delivery of an anti-cancer drug. J Control Release. 2010;148(3):359–367. | ||
Pinto MF, Moura CC, Nunes C, Segundo MA, Costa Lima SA, Reis S. A new topical formulation for psoriasis: development of methotrexate-loaded nanostructured lipid carriers. Int J Pharm. 2014;477(1–2):519–526. | ||
Kalam MA, Sultana Y, Ali A, Aqil M, Mishra AK, Chuttani K. Preparation, characterization, and evaluation of gatifloxacin loaded solid lipid nanoparticles as colloidal ocular drug delivery system. J Drug Target. 2010;18(3):191–204. | ||
Das S, Ng WK, Kanaujia P, Kim S, Tan RBH. Formulation design, preparation and physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic drug: effects of process variables. Colloids Surf B Biointerfaces. 2011;88(1):483–489. | ||
Shah B, Khunt D, Bhatt H, Misra M, Padh H. Application of quality by design approach for intranasal delivery of rivastigmine loaded solid lipid nanoparticles: Effect on formulation and characterization parameters. Eur J Pharm Sci. 2015;78:54–66. | ||
Chaves LL, Rolim LA, Gonçalves ML, et al. Study of stability and drug-excipient compatibility of diethylcarbamazine citrate. J Thermal Anal Calorim. 2013;111(3):2179–2186. | ||
Li Y, Taulier N, Rauth A, Wu X. Screening of lipid carriers and characterization of drug-polymer-lipid interactions for the rational design of polymer-lipid hybrid nanoparticles (PLN). Pharm Res. 2006;23(8):1877–1887. | ||
Shah RM, Malherbe F, Eldridge D, Palombo EA, Harding IH. Physicochemical characterization of solid lipid nanoparticles (SLNs) prepared by a novel microemulsion technique. J Colloid Interface Sci. 2014;428:286–294. | ||
Araújo F, Shrestha N, Shahbazi M-A, et al. The impact of nanoparticles on the mucosal translocation and transport of GLP-1 across the intestinal epithelium. Biomaterials. 2014;35(33):9199–9207. | ||
Fattahpour S, Shamanian M, Tavakoli N, Fathi M, Sheykhi SR, Fattahpour S. Design and optimization of alginate – chitosan - pluronic nanoparticles as a novel meloxicam drug delivery system. J Appl Polym Sci. 2015;132(28):42241. | ||
das Neves J, Sarmento B. Precise engineering of dapivirine-loaded nanoparticles for the development of anti-HIV vaginal microbicides. Acta Biomater. 2015;18:77–87. | ||
Rahman Z, Zidan AS, Habib MJ, Khan MA. Understanding the quality of protein loaded PLGA nanoparticles variability by Plackett-Burman design. Int J Pharm. 2010;389(1–2):186–194. | ||
Taurozzi JS, Hackley VA, Wiesner MR. Ultrasonic dispersion of nanoparticles for environmental, health and safety assessment – issues and recommendations. Nanotoxicology. 2011;5(4):711–729. | ||
Bhupinder S, Rishi K, Mousumi N, Naveen A. Developing oral drug delivery systems using formulation by design: vital precepts, retrospect and prospects. Expert Opin Drug Deliv. 2011;8(10):1341–1360. | ||
Neves AR, Lúcio M, Martins S, Lima JLC, Reis S. Novel resveratrol nanodelivery systems based on lipid nanoparticles to enhance its oral bioavailability. Int J Nanomedicine. 2013;8:177–187. | ||
Silva AC, Gonzalez-Mira E, Garcia ML, et al. Preparation, characterization and biocompatibility studies on risperidone-loaded solid lipid nanoparticles (SLN): high pressure homogenization versus ultrasound. Colloids Surf B Biointerfaces. 2011;86(1):158–165. | ||
Chaubey P, Mishra B. Mannose-conjugated chitosan nanoparticles loaded with rifampicin for the treatment of visceral leishmaniasis. Carbohydr Polym. 2014;101:1101–1108. | ||
Costa Jr EdS, Mansur HS. [Preparation and characterization of chitosan/poly(vinyl alcohol)blend chemically crosslinked by glutaraldehyde for tissue engineering application]. Química Nova. 2008;31:1460–1466. Portuguese. | ||
Müller RH, Runge SA, Ravelli V, Thünemann AF, Mehnert W, Souto EB. Cyclosporine-loaded solid lipid nanoparticles (SLN®): Drug–lipid physicochemical interactions and characterization of drug incorporation. Eur J Pharm Biopharm. 2008;68(3):535–544. | ||
Gokce EH, Korkmaz E, Dellera E, Sandri G, Bonferoni MC, Ozer O. Resveratrol-loaded solid lipid nanoparticles versus nanostructured lipid carriers: evaluation of antioxidant potential for dermal applications. Int J Nanomedicine. 2012;7:1841–1850. | ||
Guada M, Sebastián V, Irusta S, Feijoó E, Dios-Viéitez Mdel C, Blanco-Prieto MJ. Lipid nanoparticles for cyclosporine A administration: development, characterization, and in vitro evaluation of their immunosuppression activity. Int J Nanomedicine. 2015;10:6541–6553. | ||
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | ||
He C, Yin L, Tang C, Yin C. Size-dependent absorption mechanism of polymeric nanoparticles for oral delivery of protein drugs. Biomaterials. 2012;33(33):8569–8578. | ||
Üner M, Yener G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspectives. Int J Nanomedicine. 2007;2(3):289–300. | ||
Siram K, Chellan VR, Natarajan T, et al. Solid lipid nanoparticles of diethylcarbamazine citrate for enhanced delivery to the lymphatics: in vitro and in vivo evaluation. Expert Opin Drug Deliv. 2014;11(9):1351–1365. | ||
Mishra A, Vuddanda PR, Singh S. Intestinal lymphatic delivery of praziquantel by solid lipid nanoparticles: formulation design, in vitro and in vivo studies. J Nanotechnol. 2014;2014:1–12. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.