")
Back to Journals » Clinical Ophthalmology » Volume 15
Descriptive Study of a Cohort of 488 Patients with Inherited Retinal Dystrophies
Authors Coco-Martin RM , Diego-Alonso M , Orduz-Montaña WA, Sanabria MR, Sanchez-Tocino H
Received 22 November 2020
Accepted for publication 4 February 2021
Published 9 March 2021 Volume 2021:15 Pages 1075—1084
DOI https://doi.org/10.2147/OPTH.S293381
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Rosa M Coco-Martin,1,2 Miguel Diego-Alonso,1,3 W Andres Orduz-Montaña,1 M Rosa Sanabria,1,4 Hortensia Sanchez-Tocino1,3
1Instituto Universitario de Oftalmobiología Aplicada (IOBA), University of Valladolid, Valladolid, Spain; 2Red Temática de Investigación Cooperativa en Salud de Oftalmologia (Oftared), Instituto de Salud Carlos III, Madrid, Spain; 3Department of Ophthalmology, Hospital Universitario Río Hortega, Valladolid, Spain; 4Department of Ophthalmology, Complejo Hospitalario De Palencia, Palencia, Spain
Correspondence: Rosa M Coco-Martin
Instituto de Oftalmobiologia Aplicada (IOBA), University of Valladolid, Campus Miguel Delibes, Pº de Belén Nº 17, Valladolid, 47011, Spain
Tel +34 983423559, ext. 4738
Fax +34 983423274
Email [email protected]
Purpose: To analyze the distribution of inherited retinal diseases (IRDs), describe the clinical characteristics of patients, and determine the percentages of patients with genetic diagnosis in the Castilla y Leon region of Spain.
Methods: All patients with an IRD seen in the two major referral units of Castilla y Leon during a 20-year period were included. The ages at symptom onset, diagnosis, and the last visit; sex; family history; history of consanguinity; type of inheritance; status of the fundus and electroretinogram findings; lens and macular status, visual acuity; and visual field data were recorded. Patients were divided into those with retinitis pigmentosa (RP) and all others. Gene mutations were gathered when available.
Results: Four hundred eighty-eight patients with IRDs were studied: 216 (44.26%) with RP of which 34 (15.74%) had syndromic diseases, and 272 had other conditions being 161 (59,19%) macular dystrophies. The mean delay in diagnosis was 6– 16.2 years respectively. For the RP group the mean age at the last visit was 47.96± 17,26; mean age of cataract surgery was 48.30 ± 12.01 years; and the foveal area was preserved in 74 (35.07%) patients, atrophic in 101 (47.87%), and edematous in 36 (17.06%). A genetic study had been performed in 58 (26.85%) of patients with RP and 71 (26,1%) of the rest, being indeterminate in 17 (29.31%) out of RP group and 20 (28.16%) out of the others.
Conclusion: Clinical characteristics are comparable to other published series. There is a significant delay in diagnosis. The number of patients with IRDs and available genetic diagnosis, thus being possible candidates for undergoing personalized treatments including gene therapy in our region is low and must be improved.
Keywords: inherited retinal dystrophies, retinitis pigmentosa, genetic diagnosis, visual acuity, visual field
Introduction
Inherited retinal diseases (IRD) are a group of rare Mendelian neurodegenerative conditions caused by mutations in genes that encode for proteins essential for the functioning and maintenance of photoreceptors, retinal pigment epithelium and other retinal neurons, thus leading to retinal dystrophies or optic neuropathies.1 The prevalence rates vary between 1/750 and 1/5000, depending on the region and level of consanguinity or ethnicity.2–4 IRDs are one of the main causes of irreversible blindness in young patients and have a highly significant impact on quality of life and health economics.5,6
IRD can be classified in panretinal pigmentary retinopathies affecting primarily rods or cones in which pigmentary clumping occurs secondary to photoreceptor death; macular dystrophies with only central retinal involvement; stationary conditions in which the photoreceptors do not function but do not die; optic nerve disease primarily due to involvement of ganglion cells; and other less frequent diseases such as vitreoretinopathies.7,8 The most frequent IRD is retinitis pigmentosa (RP) that usually is an isolated ocular condition, but 20% to 30% of cases may be syndromic.2,9,10
The inheritance patterns of these pathologies are usually monogenic, autosomal dominant (AD), autosomal recessive (AR), or X-linked recessive (XL) but on rare occasions can be digenic, mitochondrial, or have another pattern.2 More than 3,000 mutations have been identified in more than 300 genes in syndromic and non-syndromic diseases, and that number is constantly increasing thanks to greater access to next-generation sequencing (NGS).5,11
Establishment of organized and shared networks to create registries and biobanks would facilitate the implementation of epidemiologic studies and would help to identify the causal mutations using exome and whole genome sequencing provided by NGS techniques, which has become an endeavor of the highest priority.5 This advancement is crucial to elucidate the molecular pathologies of disease, diagnose unresolved cases, improve genetic counseling, and develop new advanced therapies including gene therapy, all of which will change the paradigm of IRDs, as new treatments may improve vision and prevent blindness.12 Thus, identifying the genes involved and the clinical status of our patients will help identify patients who are candidates for treatments based on personalized medicine or involvement in one of the more than 300 ongoing clinical trials for this group of diseases.12
The objective of the current study was to describe the relative frequencies of different diseases with the clinical patterns of various subtypes of monogenic retinal degenerations and estimate the genetic diagnostic rates, describing the causative genes in a Spanish region to know the starting point and set goals for improvement. Thus, this study will increase the knowledge about these dystrophies in our population and provide useful information to plan research and management by the public health system.
Patients and Methods
This retrospective, cross-sectional, descriptive study was performed at the Instituto Universitario de Oftalmobiologia Aplicada (IOBA) Retina Unit, University of Valladolid, Spain, and the RP Unit of the Hospital Universitario Río Hortega (HURH), Valladolid, Spain, both referral centers in the Castilla y Leon area, a sparsely populated region with only 2,500,000 inhabitants.
This study followed the tenets of the Helsinki Declaration of 1964 (last amendment, 2013). The Clinical Research Ethics Committee of the Valladolid East Health Area approved the study with appropriate participants’ informed consent.
Patients
The HURH database included 82 patients with RP seen between 1995 and 2019. The IOBA database included 518 consecutive patients with a diagnosis of an IRD seen between 1998 and 2019. All double entries in both units, cases with a doubtful diagnosis (those without clear clinical findings nor genetically confirmed diagnosis) or an incomplete dataset were excluded.
Data Collection
For the RP group, the variables studied included sex; ages at symptom onset, diagnosis, and the last visit; family history; lens status; inheritance pattern, and genetic testing data. The electrophysiology status was recorded as normal, diminished, or abolished for the scotopic and photopic responses. The ocular fundus was classified as having a classic pattern in the spicules, scarce pigment, granular pattern, atrophy, perivenous pattern, and sector RP. The macular status was classified as preserved, atrophic, or edematous based on ocular fundus, autofluorescence and Spectral Domain Optical Coherence Tomography (OCT). The best-corrected visual acuity (BCVA) was recorded using a Snellen chart and converted to the logarithm of the minimum angle of resolution (LogMAR) using a validated procedure.13 The BCVA was collected for each decade of life for each eye; for analysis, the patients were divided into five groups according to the low vision scale of the World Health Organization based on the BCVA in the eye with the better vision, ie, no low vision (LV) (LogMAR≤0 = ≥1 Snellen), mild LV (LogMAR>-0 - <0.5 = <1 - >0,5 Snellen), moderate LV (LogMAR> 0.5 - <1.00 = <0.5 - >0.1 Snellen), severe LV (LogMAR>1.00 - <1.3 = <0.1 - >0.05 Snellen), and blindness (LogMAR>1.30 = <0.05 Snellen). The visual field (VF) data were divided into five groups: central VF <5º; central VF between 5º and 10º; central VF >10º; total abolition of the VF; and central rather than peripheral involvement.
For the remaining IRD patients, we recorded the age; sex; baseline BCVA; ages at symptom onset and diagnosis; inheritance pattern, and mutated gene if available. The inheritance pattern was based on ophthalmic examination, clinical progression, and pedigree data.
A positive genetic test result means that found variants were identified as pathogenic: frameshift, premature stop codon, splice site variants affecting canonical sites, and reported variants known to cause retinal diseases in ClinVar [https://www.ncbi.nlm.nih.gov/clinvar/] or the Human Gene Mutation Database [https://go.qiagen.com/HGMD] or whose frequency in the control population were less than 0.5% and comply with the pathogenicity predictions following established bioinformatic algorithms. A negative genetic test result means that no clear pathogenic mutation was found in any of the tested genes.
Statistical Analysis
For the quantitative variables, the descriptive statistics used were the mean, standard deviation (SD), and the range (maximal and minimal values). These values were calculated in Excel tables and with the SPSS program version 24 for Windows (IBM Corp., Armonk, NY, USA) for 1989–2017. For the qualitative variables, the numbers and percentages of each category were used.
The Student’s t-test was used to compare the means between two groups for quantitative variables, and the chi2 test for qualitative variables through a contingency table or Fisher’s exact test if the expected frequencies were small. P < 0.05 was considered significant. The analyses were performed with the statistical package SPSS, version 24.
Results
The clinical records of 600 patients were reviewed with suspected IRD, and high clinical suspicion of a hereditary cause could be confirmed in 488 patients. Of them, 216 (44.26%) patients had RP and 272 (55.74%) patients presented other different IRDs (Table 1).
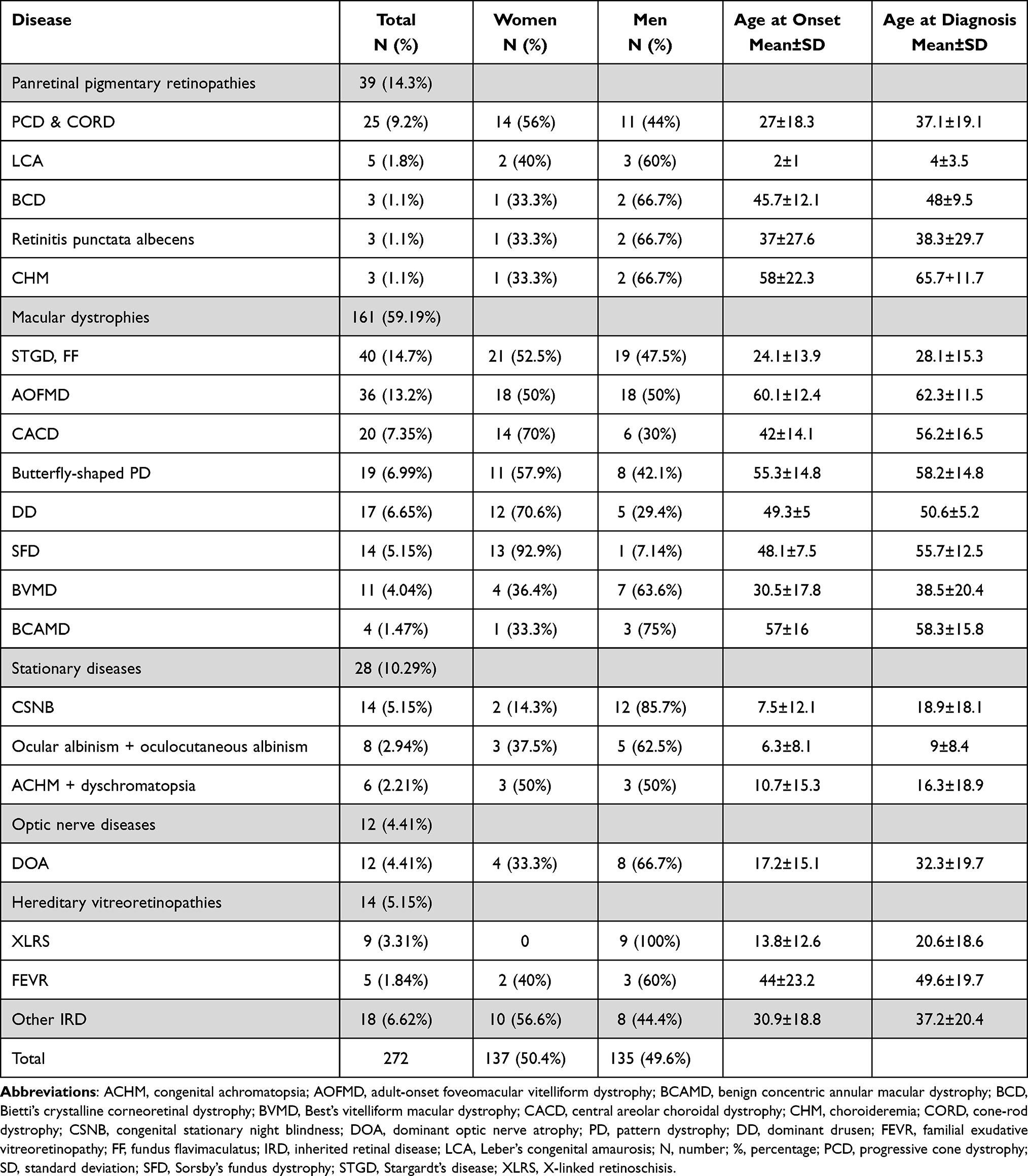 |
Table 1 Inherited Retinal Diseases Other Than Retinitis Pigmentosa: Distribution by Sex and Age at Baseline and Diagnosis |
Of the RP subgroup, 112 (51.85%) were men and 104 (48.15%) were women. Thirty patients (13.9%) had Usher syndrome (7 type I, 21 type II, and 2 type III). Two patients had Bardet-Biedl syndrome, one retinal degeneration related to Sjögren-Larsson syndrome and one to Crouzon syndrome.
The inheritance pattern could not be established in 22 RP patients due to lack of information. Of the remaining 194 RP patients, 47 (24.23%) were classified as sporadic cases, 104 (53.61%) with AR, 29 (14.95%) as AD, 13 (6.7%) as XL, and one (0.52%) with dominant XL that may also be a female carrier with severe involvement. In the syndromic RP group, 19 patients (67.86%) were AR, 8 (28.57%) were sporadic and one patient was AD (3.57%).
One hundred and five (48.61%) patients of the total RP sample had at least one family member affected by the disease. Twenty-one (11.11%) of 189 RP patients were children of consanguineous parents and 33 (17.46%) of “relative” consanguineous parents (both born in a village with less than 500 inhabitants). In fact, 11 out of 30 patients with AR RP and available diagnosis testing had a homozygous mutation.
Fifty-eight of 216 (26.85%) RP patients had undergone genetic testing, and 41 (70.68%) out of them were positive (Table 3), although 11 of the remaining 17 patients who were negative had been tested before 2013 before implementation of the NGS techniques. Interestingly, in the group without a genetic diagnosis, 70.6% of subjects had useful remaining vision.
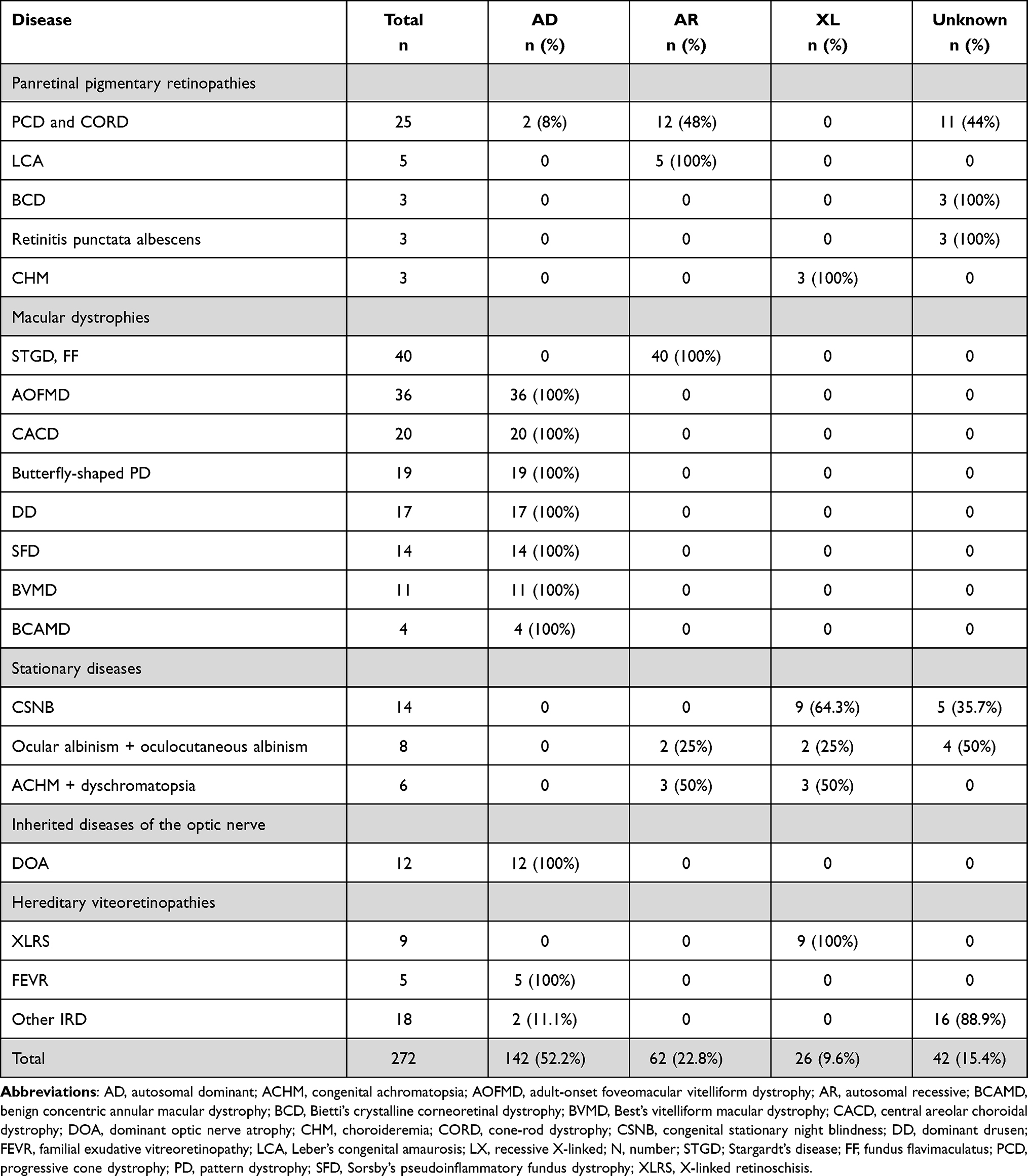 |
Table 2 Pattern of Inheritance of Each Disease |
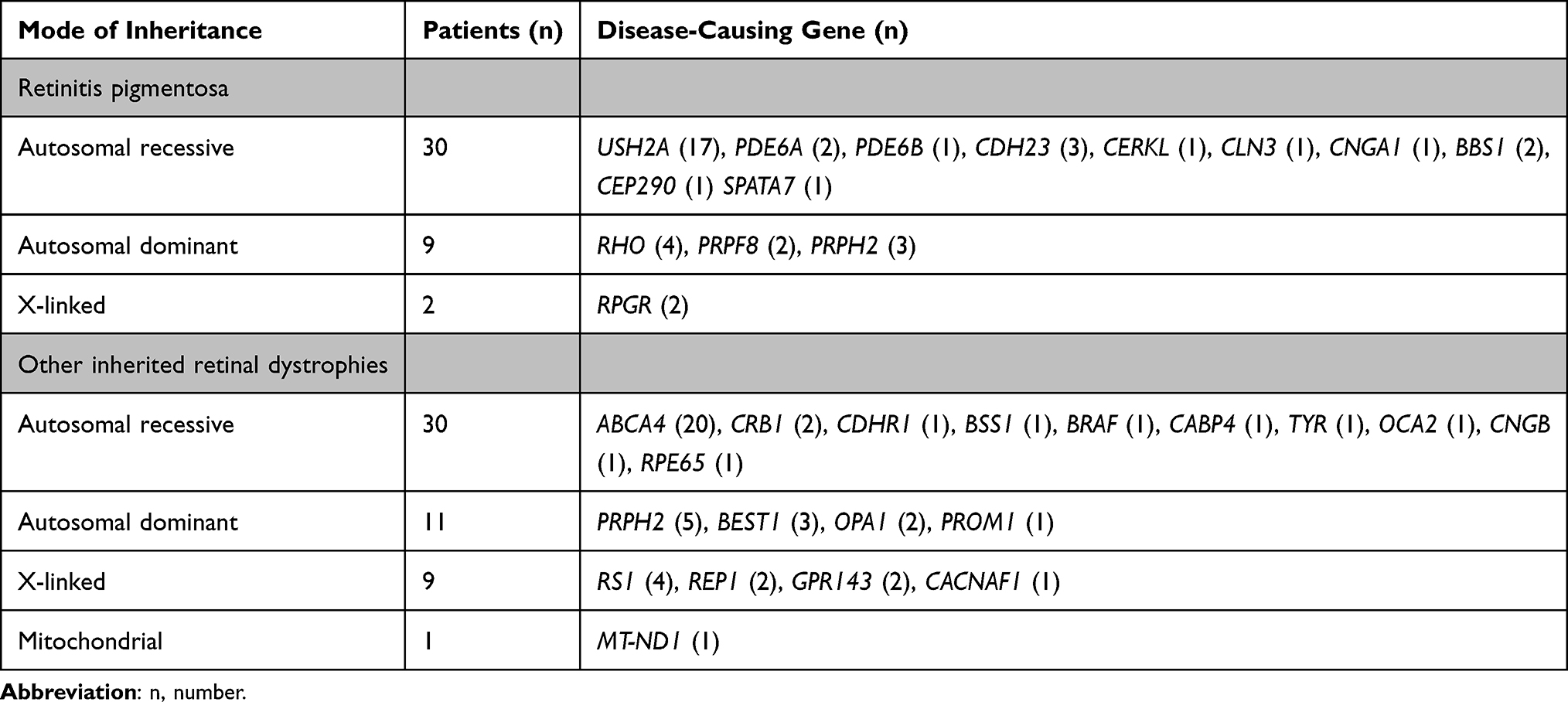 |
Table 3 Results of Genetic Testing |
Mean age at symptom onset in patients with RP was 17.30±15.27 years (range, 1–73) and mean age at diagnosis was 31.48±17.68 years (range, 2–82), indicating an average delay in diagnosis of 16.21 years. The mean age of patients with RP at the last visit was 47.96 ± 17.26 years (range; 3–89) and no significant differences were found between syndromic (T of Student for mean age at symptom onset: p value = 0.064, T of Student for mean age at diagnosis: p value = 0.167) or between the different patterns of inheritance (Student’s T test for mean age at symptom onset: p value = 0.216, Student’s T test for mean age at diagnosis: p value = 0.843).
Electroretinography (ERG) data were collected from 69 individuals with RP. The scotopic ERGs were not available in 2 patients (2.94%), it was decreased in 15 (21.74%), and abolished in 51 (73.91%). The photopic ERGs were normal in six (8.69%) patients, 18 (26.09%) had a diminished recording, and 45 (65.22%) had an abolished registry.
The ocular fundus could be classified in 211 patients with RP; of those, 166 (78.67%) had a classical pattern of pigmented spicules, 28 (13.27%) had not a pigmentary fundus, eight (3.79%) had extensive atrophy, six (2.84%) had sector RP, two (2.84%) perivenous RP, and one (1,42%) a granular pattern.
Information about the lens was obtained in 208 patients on their last visit. Cataract developed at a mean age of 42.44±15.84 years (range, 2–86), and 65 patients (30.09%) were pseudophakic. The mean age at which cataract surgery was performed in the overall sample was 48.30 ± 12.01 years (range, 14–82). No significant differences were found between syndromic and non-syndromic RP (Student’s T test for mean age at cataract surgery: p value = 0.501).
The macular status was recorded in 211 patients with RP at their last visit and was preserved in 74 (35.07%), atrophic in 101 (47.87%), and edematous in 36 (17.06%). The BCVA could be analyzed in 195 patients and data grouped according to low vision parameters and throughout life are presented in Figure 1. Visual field data were available in 190 patients with RP and data in different groups of age are presented in Figure 2.
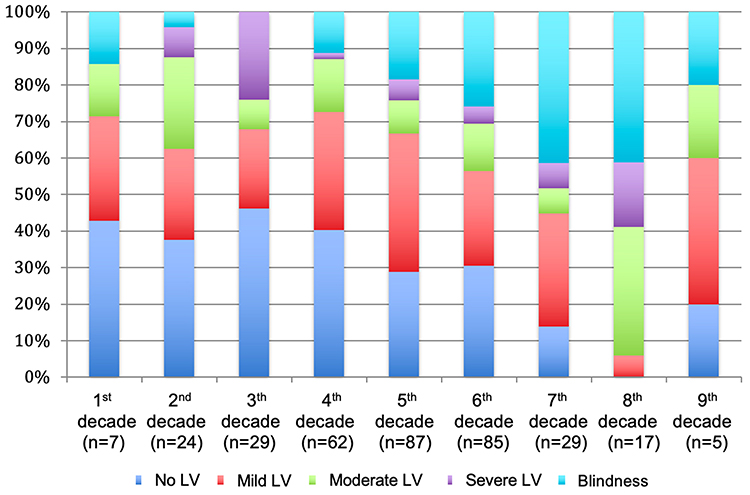 |
Figure 1 Percentages of patients based on low-vision categories. The figure shows the percentages of patients according to the decade of life for the total retinitis pigmentosa sample A higher number of patients without Low Vision (LV) or with mild LV is observed in the first decades, which decreases throughout life while blind patients increase. |
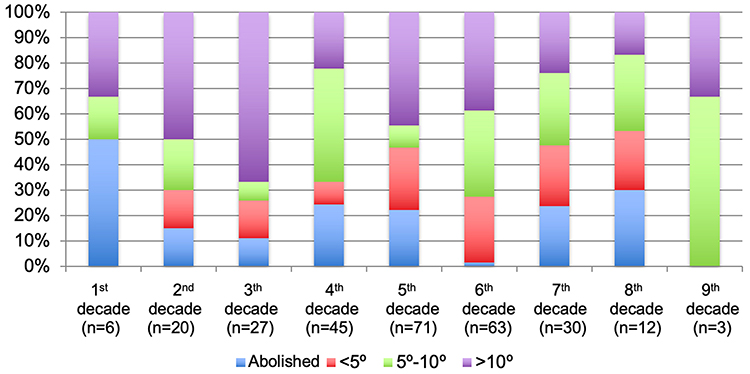 |
Figure 2 Percentages of patients based on the status of the visual field according to the decade of life for the total retinitis pigmentosa sample. The figure shows the percentages of patients according to the decade of life for the total retinitis pigmentosa sample. The number of analysed patients is very small in the first and last decades of life, so these percentages should be taken with caution. Unrecordable CVs or lower than 5 degrees preserved increase from the 2nd to the 7th decades of life, and those greater than 5 degrees decrease as expected. |
Concerning IRD other than RP the frequency distribution of each disease, distribution by sex, mean age at symptom onset, and mean age at diagnosis are presented in Table 1. The delay in diagnosis for progressive cone dystrophy and cone-rod dystrophy was 10 years; in central areolar choroidal dystrophy (CACD) 14 years; in Sorsby’s fundus dystrophy 7 years; in Best’s vitelliform macular dystrophy 8 years; in congenital stationary night blindness 11 years; in congenital achromatopsia 6 years, and in dominant optic atrophy 15 years. The inheritance patterns are presented in Table 2.
Only 71 (26.1%) of the 272 patients diagnosed with other IRD had a genetic diagnosis, but it was negative or undetermined in 20 (28.1%). Nine patients with Stargardt’s, 4 albinisms, 1 achromatopsia and 2 CORD had a homozygous mutation. We also observed a founder mutation in PRPH2 in a family with CACD. The results are presented in Table 3. A comparison between our data and results obtained by other research groups is presented in Table 4.
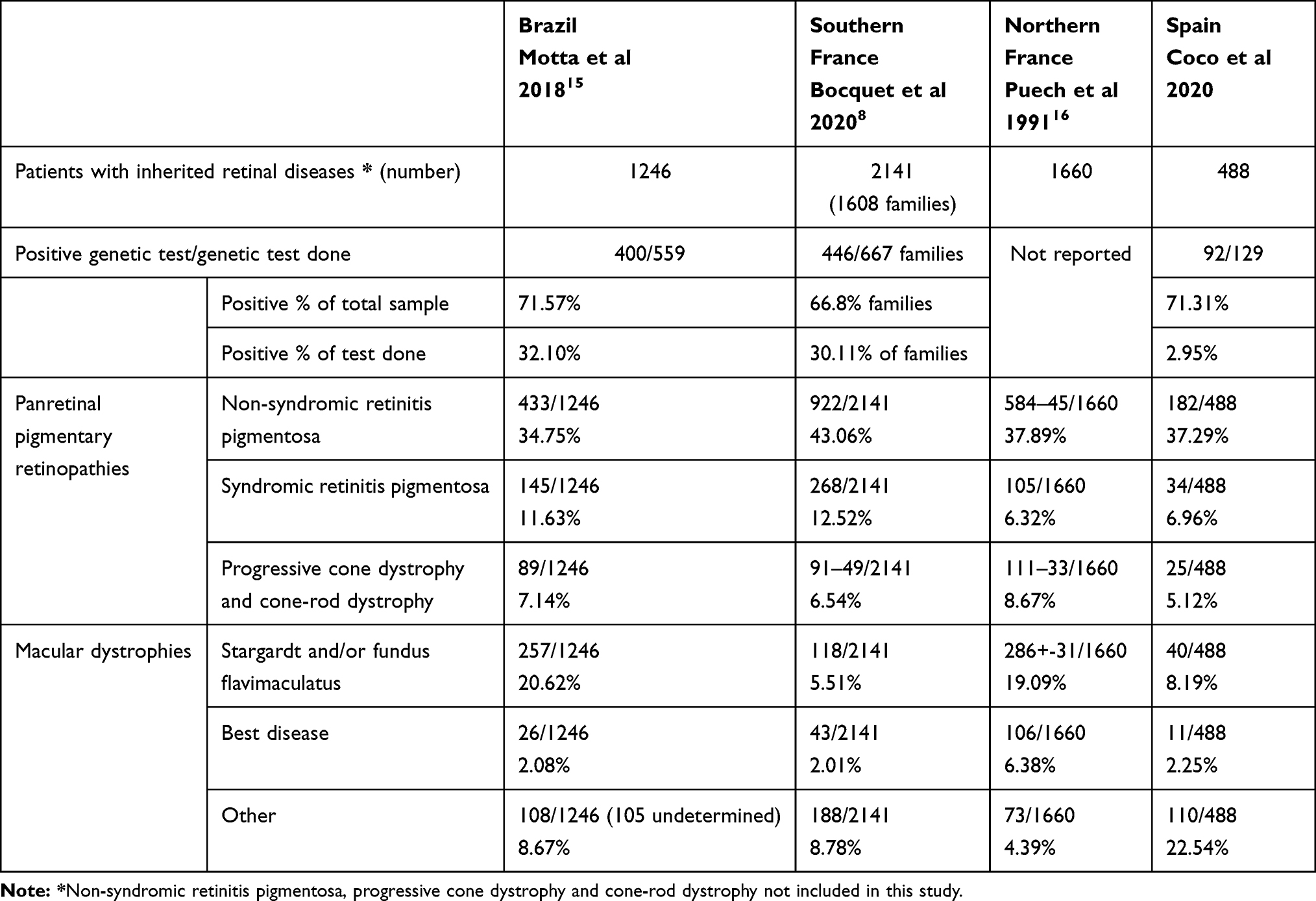 |
Table 4 Comparison of Inherited Retinal Diseases in the Current Series with Other Series |
Discussion
The current study presents interesting data about the relative frequencies of inherited retinal diseases in a specific IRD unit and their clinical appearance, inheritance patterns, age at diagnosis, and results of genetic testing in a Spanish region with 2,500,000 inhabitants. As expected, RP was the most frequently IRD found in the current study. The RP frequencies vary little between published studies and range from 34.75% to 43.06%.8,15,16 Usher syndrome was the most frequent form of syndromic pigmentary retinopathy as reported previously.8,14–16 Regarding macular dystrophies, Stargardt’s disease was the most frequently seen entity in the current study and in most studies but with important variations ranging from 5.51% reported by Bocquet et al in Southern France to 20.62% reported by Motta et al in Brazil.8,15 The reported frequencies of other macular dystrophies vary widely among studies of different populations, and the current study has the highest percentage.8,15,16 Such wide variations can be due to several factors. First it is not clear if some macular dystrophies included in the current study also were included in other studies; second the definite diagnosis of a particular macular dystrophy can be challenging; moreover, geographic variations in the frequency of some mutations can promote geographic disparities in the frequency of some entities as happens with the higher rate of CACD in the Spanish population compared with others.17,18 Although consanguinity can play an important role in the prevalence of IRD in some populations,19 the current study had consanguinity data, including relative consanguinity data (11.11% and 17.46%, respectively), similar to those published in other populations.15,20 Besides, one third of homozygous mutations are found in AR diseases as expected.
Concerning the inheritance patterns in the RP group, the rate of AR inheritance in the current sample was high (53.61% vs 16.0%-39% in other studies), while sporadic cases occurred less frequently than in other studies (24.23% vs 40%-51% in other studies).4,21–24 However, 28 patients in our sample did not provide family history data and thus they were not included in this analysis; if they had been considered as sporadic cases, this figure would have approached 50%.
Within the RP group, the ERG was abolished or diminished in most patients as expected and analogously to data reported by Berson et al.20,25
With small variations, the clinical presentations in the current sample were similar to those already described. First of all, most had the classic pattern of pigmentation in the spicules on the ocular fundus. The percentage of cataract patients (39.42%) was similar to the data provided by Testa et al24 but slightly lower than in other studies (45%-52%),26 which can be explained in part because the percentage of pseudophakic patients (31.25%) is higher than that found in other studies (15.4%-20%).10,27–29 Besides, the mean age at the time of cataract surgery in young adulthood was similar to that reported previously.27–29 Lastly, the prevalence of macular edema in our RP sample (17.06%) was lower than in other studies (23%-50%),10,30,31 although it was similar to that reported by Hirakawa et al.32 This can be explained because in our series, almost half of the patients had macular atrophy, and only patients with a well-preserved macular area can have macular edema.
Genetic diagnosis of IRDs is desirable for many reasons as it can provide a definitive diagnosis which can deliver great relief to patients and families, it also helps physicians to better define the inheritance pattern and the risk for the remaining family members, and it is crucial to identify patients for potential enrollment in clinical trials or new advanced therapies. But patients should have good cell tissue remnants to have any possibility for gene therapy functioning, and the fact that 71.6% still have a good remaining central vision, makes us hope that they may be susceptible to receiving these treatments when the genetic diagnosis is made. The first important difficulty for genetic diagnosis of IRDs is that the clinical diagnosis of different phenotypes may be due to mutations in the same gene and vice versa.33 Therefore, the wide variety of IRDs and their relative low frequency makes exact diagnosis difficult for the general ophthalmologist and even for retinal specialists, and historically this has been crucial to focus gene testing. However, with the advent of high-throughput genomic sequencing techniques this has been made easier and the success rates have improved significantly. Thus, we think that our negative cases need to be reassessed using these new tools that are increasingly able to find not known mutations or mutations in second alleles (mainly deep intronic mutations in large genes). Besides, functional studies must also be carried out to help clarify some variants of unknown significance. Another important factor that must be taken into account to evaluate our findings on genetic diagnosis data is the difficulty of access to genetic diagnosis in Castilla y Leon, which explains a diagnosis rate lower in our study (22.95%), than that from other studies (30.11% and 32.10%),8,15 which needs to be improved.
Another key point is diagnostic delay. Delayed diagnosis are known common problems in IRDs and may result from deferral of patients with referring symptoms and/or the inexperience of doctors who first treated them as we have already said. Data from the Survey of the Delay in Diagnosis for Rare Diseases in Europe confirm that it occurs in the diagnosis of any rare disease.34 Moreover, late diagnosis of non-progressive retinal dystrophies may be due in part to their moderate visual impairment causing only unappreciated disability and minimal fundus changes in children who do not complain.34 Entities with a more characteristic phenotype can be recognized early, but in many cases a conclusive diagnosis of an IRD cannot be made until several visits and electrophysiologic and imaging evaluations are performed and interpreted correctly. Better diagnosis rates are obtained if patients are seen in referral units, but it can take years to reach them and access to them must also be improved.
But the current study has important limitations. First, the sample size of each disease was small except for that of RP. However, these are rare diseases and in this context our study enables relevant clinical findings of these diseases to be highlighted. Another constraint was the heterogeneity in the follow-up of the patients (some, up to 20 years of follow-up and others who came for isolated visits). In addition, the presence of macular edema may have been underestimated, especially in patients with few visits during the first years in which OCT was unavailable, although this happened only in a small number of patients. A major problem was that the vast majority of patients do not have a molecular diagnosis and the state-of-the-art in IRD is based on both clinical and molecular diagnosis. In this context comments about inheritance patterns must be taken with caution although genealogical trees were informative in most of the patients catalogued. Finally, many of the genetic studies with negative results were very old and patients need to be reassured using NGS techniques. Nevertheless, this is something we wanted to point out to be able to ask regional government for a change in their policy of denying test to this group of patients and families.
The major strength of the current study was that the patients were part of specialized practices and the sample size was adequate considering that the diseases are rare. It also emphasizes the need to improve access to referral units and to genetic testing in our population. Lastly, knowing the rate of genetic testing in actual clinical practice can be a good starting point to plan future actions such as genetic studies with new diagnostic techniques for as many subjects as possible, which would allow classification of the pathology from the genotypic point of view in our population, to move forward in the search for new mutations and properly diagnose patients with no definitive diagnoses.
Conclusions
The clinical data of our sample are quite similar to other published series of IRDs. Only 26.85% of the patients with RP and 26.1% of the rest of the IRDs had undergone genetic testing, but in more than two thirds (70.68%) of those with RP the test results were positive and had elucidated the mutated gene. Most RP patients (71.6%) had residual vision, which allows them to be candidates for treatments aimed at preserving their vision, and we need to promote their prompt genetic diagnosis. The current study confirmed the substantial delays in diagnosing most of these diseases and the need to perform more genetic studies in the current region. By highlighting our weaknesses, we can lobby our regional managers to improve these data.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval
This Study was declared in accordance with ethical standards Hospital Universitario Rio Hortega and the Clinical Research Ethics Committee – Health Area East – of Valladolid (CEIC-VA-EAST-HCUV) with the reference number PI 18-1171TFMNOHCUV. Administrative permissions were acquired by our team to access the data used in our research.
Consent for Publication
Not applicable.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study received no funding.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Traboulsi EI. Hope and major strides for genetic diseases of the eye. J Genet. 2009;88(4):395–397. doi:10.1007/s12041-009-0060-8
2. Chizzolini M, Galan A, Milan E, et al. Good epidemiologic practice in retinitis pigmentosa: from phenotyping to biobanking. Curr Genomics. 2011;12(4):260–266.
3. Na KH, Kim HJ, Kim KH, et al. Prevalence, age at diagnosis, mortality, and cause of death in retinitis pigmentosa in Korea-a nationwide population-based study. Am J Ophthalmol. 2017;176(4):157–165.
4. Bertelsen M, Jensen H, Bregnhoj JF, Rosenberg T. Prevalence of generalized retinal dystrophy in Denmark. Ophthalmic Epidemiol. 2014;21(4):217–223.
5. Bakkar MM, Alzghoul EA, Haddad MF. Clinical characteristics and causes of visual impairment in a low vision clinic in northern Jordan. Clin Ophthalmol. 2018;12:631–637.
6. Muñoz-Cuellar JC, RM. Clinical characterization and frequency of observation of hereditary retinal diseases: multicentric study in Panama in 2012-2013. J Pharm Pharmacol. 2016;4:618–630.
7. Bocquet B, Lacroux A, Surget MO, et al. Relative frequencies of inherited retinal dystrophies and optic neuropathies in Southern France: assessment of 21-year data management. Ophthalmic Epidemiol. 2013;20(1):13–25.
8. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40.
9. Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–186.
10. Costa KA, Salles MV, Whitebirch C, Chiang J, Sallum JMF. Gene panel sequencing in Brazilian patients with retinitis pigmentosa. Int J Retina Vitreous. 2017;3(1):33. doi:10.1186/s40942-017-0087-6
11. Gill JS, Georgiou M, Kalitzeos A, Moore AT, Michaelides M. Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br J Ophthalmol. 2019.
12. Clinical Trials Database. U.S National Library, Bethesda. 2000. Available from: https://clinicaltrials.gov.
13. Holladay JT. Visual acuity measurements. J Cataract Refract Surg. 2004;30(2):287–290.
14. Motta FL, Martin RP, Filippelli-Silva R, Salles MV, Sallum JMF. Relative frequency of inherited retinal dystrophies in Brazil. Sci Rep. 2018;8(1):15939.
15. Puech B, Kostrubiec B, Hache JC, Francois P. Epidemiology and prevalence of hereditary retinal dystrophies in the Northern France. J Fr Ophtalmol. 1991;14(3):153–164.
16. Gamundi MJ, Hernan I, Muntanyola M, et al. High prevalence of mutations in peripherin/RDS in autosomal dominant macular dystrophies in a Spanish population. Mol Vis. 2007;13:1031–1037.
17. Tishkoff SA, Kidd KK. Implications of biogeography of human populations for ‘race’ and medicine. Nat Genet. 2004;36(11 Suppl):S21–7.
18. Nirmalan PK, Krishnaiah S, Nutheti R, Shamanna BR, Rao GN, Thomas R. Consanguinity and eye diseases with a potential genetic etiology. Data from a prevalence study in Andhra Pradesh, India. Ophthalmic Epidemiol. 2006;13(1):7–13.
19. Berson EL, Rosner B, Simonoff E. Risk factors for genetic typing and detection in retinitis pigmentosa. Am J Ophthalmol. 1980;89(6):763–775.
20. Ayuso C, Garcia-Sandoval B, Najera C, Valverde D, Carballo M, Antinolo G. Retinitis pigmentosa in Spain. The Spanish multicentric and multidisciplinary group for research into retinitis pigmentosa. Clin Genet. 1995;48(3):120–122.
21. Najera C, Millan JM, Beneyto M, Prieto F. Epidemiology of retinitis pigmentosa in the Valencian community (Spain). Genet Epidemiol. 1995;12(1):37–46.
22. Boughman JA, Fishman GA. A genetic analysis of retinitis pigmentosa. Br J Ophthalmol. 1983;67(7):449–454.
23. Testa F, Rossi S, Colucci R, et al. Macular abnormalities in Italian patients with retinitis pigmentosa. Br J Ophthalmol. 2014;98(7):946–950.
24. Berson EL. Light deprivation and retinitis pigmentosa. Vision Res. 1980;20(12):1179–1184.
25. Fujiwara K, Ikeda Y, Murakami Y, et al. Risk factors for posterior subcapsular cataract in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2017;58(5):2534–2537.
26. Jackson H, Garway-Heath D, Rosen P, Bird AC, Tuft SJ. Outcome of cataract surgery in patients with retinitis pigmentosa. Br J Ophthalmol. 2001;85(8):936–938.
27. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809.
28. Liew G, Moore AT, Bradley PD, Webster AR, Michaelides M. Factors associated with visual acuity in patients with cystoid macular oedema and Retinitis Pigmentosa. Ophthalmic Epidemiol. 2018;25(3):183–186.
29. Hagiwara A, Yamamoto S, Ogata K, et al. Macular abnormalities in patients with retinitis pigmentosa: prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol. 2011;89(2):e122–5.
30. Hajali M, Fishman GA, Anderson RJ. The prevalence of cystoid macular oedema in retinitis pigmentosa patients determined by optical coherence tomography. Br J Ophthalmol. 2008;92(8):1065–1068.
31. Battu R, Mallipatna A, Elackatt NJ, Schouten J, Webers CAB. Challenges of managing retinal dystrophies: an experience from south India. Ophthalmic Genet. 2018;39(1):1–3.
32. Hirakawa H, Iijima H, Gohdo T, Tsukahara S. Optical coherence tomography of cystoid macular edema associated with retinitis pigmentosa. Am J Ophthalmol. 1999;128(2):185–191.
33. EURORDIS: European Organization for Rare Diseases. Eurordis policy fact sheet. 2013. https://www.eurordis.org/sites/default/files/publications/Fact_Sheet_Eurordiscare2.pdf.
34. Zeitz C, Robson AG, Audo I. Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res. 2015;45:58–110.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.