")
Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Dendritic spine morphology and dynamics in health and disease
Authors Lee S, Zhang H, Webb D
Received 4 February 2015
Accepted for publication 18 March 2015
Published 3 June 2015 Volume 2015:7 Pages 121—131
DOI https://doi.org/10.2147/CHC.S82214
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Denis Wirtz
Stacey Lee,1 Huaye Zhang,2 Donna J Webb1,3,4
1Department of Biological Sciences, Vanderbilt University, Nashville, TN, 2Department of Neuroscience and Cell Biology, Rutgers Robert Wood Johnson Medical School, Piscataway, NJ, 3Department of Cancer Biology, 4Vanderbilt Kennedy Center for Research on Human Development, Vanderbilt University, Nashville, TN, USA
Abstract: Dendritic spines are actin-rich structures that form the postsynaptic terminals of excitatory synapses in the brain. The development and plasticity of spines are essential for cognitive processes, such as learning and memory, and defects in their density, morphology, and size underlie a number of neurological disorders. In this review, we discuss the contribution and regulation of the actin cytoskeleton in spine formation and plasticity as well as learning and memory. We also highlight the role of key receptors and intracellular signaling pathways in modulating the development and morphology of spines and cognitive function. Moreover, we provide insight into spine/synapse defects associated with several neurological disorders and the molecular mechanisms that underlie these spine defects.
Keywords: dendritic spines, synapses, synaptic plasticity, actin cytoskeleton, glutamate receptors, neurological disorders
Structure and function of dendritic spines
Santiago Ramón y Cajal first described dendritic spines, using Golgi staining, near the end of the 19th century and proposed that these spines were sites of axonal and dendritic contact.1,2 Decades later, with the advent of electron microscopy, these spines were indeed shown to be sites of excitatory synaptic contact between neurons, proving that Cajal’s hypothesis was correct.3,4 These and subsequent studies highlight the importance of dendritic spines and pose interesting questions as to the specific functions of these structures.5,6 Dendritic spines most likely have functions other than to simply connect axons and dendrites. This is supported by the observation that many inhibitory synaptic inputs occur on dendritic shafts in the absence of spines; however, it should be noted that recent data indicate that some inhibitory neurons have functional spines, and inhibitory synaptic inputs can occur on spines of cortical pyramidal neurons.7–10 A widely held theory is that spines serve as biochemical compartments in the cell.5,11 The unique morphology of spines, which consists of an enlarged head and a thin neck, makes them ideal structures to function as postsynaptic biochemical compartments that separate synaptic terminals from dendritic shafts.11,12 In addition, spines could serve as electrical compartments, which can maintain membrane potentials that are distinct from those of the parent dendrites.13–15 The electrical isolation of individual spines might provide a mechanism to allow neurons to integrate and independently regulate the strength of a large number of synaptic inputs.14 Moreover, the compartmentalization of spines most likely contributes significantly to the efficiency of synaptic transmission and plasticity.15–17 Intriguingly, the spine neck width is reported to be an important factor in regulating compartmentalization.16 Other roles for dendritic spines have been proposed,6,17,18 and the specific functions of spines are an active area of interest and debate that warrants continued research.
The functions of dendritic spines are governed, at least in part, by their morphology. They range in morphology from filopodia-like protrusions, which are thought to be spine precursors, to more mature stubby, thin, or mushroom-shaped structures.19 Stubby spines do not have a neck whereas thin and mushroom-shaped spines consist of long necks that are connected to small and large bulbous heads, respectively; filopodia-like protrusions are extensions from the dendrite that lack a bulbous head. Spine morphology is malleable, and their shape can change over time, even on a time scale of minutes or less20–22 (Figure 1). In the case of dendritic filopodia, the dynamic, exploratory nature of these structures could be beneficial in forming connections with axons.23 After an initial interaction between dendritic filopodia and axons, synapses can assemble on a relatively rapid time scale (hours).24,25 For most mammals, spine and synapse formation is widespread during early postnatal development and is followed by a pruning phase during adolescence that eliminates unnecessary or improper synaptic connections.26,27 In adults, dendritic spine formation and elimination are at an equilibrium with a fraction of spines being consistently added or removed.26 Morphological changes, which are usually activity-dependent, also occur in more mature spines and are associated with synaptic plasticity.28 Synaptic plasticity, which entails the strengthening or weakening of synapses over time as well as synapse formation and elimination (structural plasticity), is widely believed to be the cellular basis of learning and memory.28–30 In vivo imaging in the cerebral cortex of mice has shown that spine dynamics/remodeling is associated with different forms of learning.30–33 Synaptic plasticity is also thought to be necessary for the encoding and storage of memory.34 The foundation of this theory dates back to Donald Hebb, who postulated a link between alterations in synaptic activity and memory storage.35
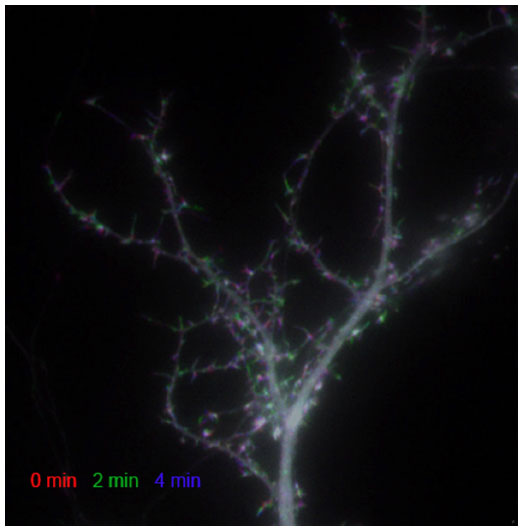 | Figure 1 Dendritic spines are dynamic structures. A three-color temporal overlap of a hippocampal neuron expressing green fluorescent protein-tagged β-actin. |
Experimental attempts to model Hebb’s theory led to the discovery of long-term potentiation (LTP), which typically uses high frequency stimulation to increase synaptic transmission.28 In order to encode information efficiently, an increase in synaptic strength must be counterbalanced by a weakening of synapses by a process termed long-term depression (LTD). LTD can be provoked experimentally with low frequency stimulation, causing a prolonged decrease in synaptic transmission.29,36 These experimental models, LTP and LTD, have been invaluable in generating a wealth of data showing the essential function that synaptic plasticity has in learning and memory. Another line of evidence that supports this link is the well-documented association between abnormalities in dendritic spine/synapse formation and plasticity and numerous neurological disorders, including autism, Alzheimer’s disease (AD), schizophrenia, and intellectual disability.37,38
This review focuses on the function and regulation of dendritic spines and excitatory synapses, and their role in human health and disease. We discuss the molecular mechanisms modulating dendritic spine development, morphology, and function, as well as the defects associated with these structures in certain neurological disorders.
Actin regulation in dendritic spine development and plasticity
Actin is the major cytoskeletal element in dendritic spines where filamentous actin (F-actin), which results from the polymerization of monomeric actin (G-actin), is found at high concentrations. F-actin within spine heads is organized into a branched network that is highly dynamic and regulated by neuronal activity.39–41 Fluorescence recovery after photobleaching experiments with green fluorescent protein-tagged β-actin showed that 85% of the actin turned over in less than 1 minute, and LTD stabilized a significant portion of the dynamic actin.39 Super-resolution imaging of spines also showed fast actin turnover (ie, on a time scale of seconds to minutes) and suggested that the F-actin network comprises mostly short filaments that undergo both retrograde and anterograde actin flow.40 Actin turnover does, however, vary within regions of spines; spine tips have a higher turnover rate than at the base where actin seems to be more stable.42 A dynamic actin cytoskeleton is critical for the morphological malleability of spines, which underlies the formation and plasticity of these structures. Microtubules are also found in at least a subset of spines where they modulate spine morphology and maturation, most likely through their interplay with the actin cytoskeleton.43 However, a detailed discussion of the role of microtubules in regulating dendritic spine function is beyond the scope of this review.
Actin remodeling is regulated by the Rho family of small GTPases that includes Rho, Rac, and Cdc42. These small GTPases are molecular switches that exist in an active (guanosine-5′-triphosphate-bound) and an inactive (guanosine-5′-diphosphate-bound) state. The cycling of the GTPases between the active and inactive states is regulated by three types of proteins, ie, guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), and guanine dissociation inhibitors. GEFs promote the exchange of GDP for GTP, activating the GTPase; GAPs increase intrinsic GTP hydrolysis, returning these proteins to an inactive state; and guanine dissociation inhibitors form soluble complexes with the GTPases and sequester them in an inactive state.44
Rac and Cdc42 induce dendritic spine formation, whereas Rho promotes the retraction and loss of spines.45–47 Rac can promote spine formation through its downstream effector, p21-activated kinase (PAK).48 Cdc42 stimulates spinogenesis and enlargement of spine heads via activation of the actin-binding protein neural Wiskott-Aldrich syndrome protein (N-WASP) and the Arp2/3 complex, which localizes to the postsynaptic density (PSD) and mediates the formation of branched actin filaments.46,49 Moreover, loss of Cdc42 in mice results in deficits in synaptic plasticity and remote memory recall.50 Rho family GEFs and GAPs also have important roles in spine development and function. Mice lacking the Rac GEF karilin-7 exhibit defects in cortical spine density and in working memory.51 The Rac GEF Tiam1 is required for dendritic spine formation, and knockdown of Tiam1 causes a decrease in spine and synapse density.52,53 β-PIX, another GEF, regulates spine formation through activation of Rac and subsequently PAK.48 GEF-H1, a Rho family GEF, inhibits spine formation and negatively regulates spine length through a Rho pathway.54 Rho family GAPs also contribute to the development of dendritic spines and synapses. Expression of the Rac GAP α1-chimaerin leads to a loss of spines by inhibiting new spine formation and by mediating the pruning of existing spines.55,56 Oligophrenin-1, a Rho-GAP, regulates the maturation and plasticity of excitatory synapses by inhibiting Rho activity.57 Furthermore, p190 RhoGAP modulates spine morphogenesis by regulating Rho GTPase activity.58 The function of guanine dissociation inhibitors in regulating spine development and plasticity is currently unknown and represents an exciting avenue for future investigation.
Contribution of ABPs to dendritic spine formation and plasticity
Actin binding proteins (ABPs) also play a large role in modulating actin dynamics. Therefore, a number of ABPs, which can localize to the PSD, are known to regulate spine/synapse formation and plasticity via their ability to modulate actin. As already discussed, N-WASP, which promotes polymerization of branched actin filaments through activation of the Arp2/3 complex, induces spine formation and enlargements of spine heads.46 Knockout of ArpC3, a subunit of the Arp2/3 complex, in forebrain excitatory neurons in mice led to a loss of spines and defects in synaptic plasticity and episodic memory.59 WAVE1, another WASP family member and an effector of Rac, regulates spine morphology and density as well as synaptic plasticity, and loss of this protein results in deficits in learning and memory.60,61 Knockdown of cortactin, which also activates the Arp2/3 complex, similarly led to alterations in spine number and morphology.62 Formins are another class of actin nucleators that are implicated in spine regulation. Formins can be activated by Rho GTPases, but unlike the Arp2/3 complex, formins produce unbranched actin filaments.63,64 One study demonstrated that mammalian diaphanous-related formin 2 promotes filopodia formation;65 however, future studies are needed to further explore the role of formins in spine formation and plasticity. Proteins containing WASP homology 2 actin binding domains are a third class of actin nucleators that were most recently identified.66 Mice lacking Spir-1, the founding member of the WASP homology 2 protein family, have reduced spine density on cortical neurons and exhibit increased fear memory.67
Actin remodeling is mediated by other proteins, such as profilin, cofilin, and gelsolin. Profilin promotes actin polymerization by acting as a nucleotide exchange factor, catalyzing ADP to ATP exchange on G-actin, and by binding G-actin and increasing its incorporation into actin filaments.68,69 Profilin II, a brain-enriched isoform, is associated with stabilization of spine morphology, and blockade of profilin targeting to spines leads to destabilization of spine structure.70 Interestingly, fear conditioning in rats resulted in profilin redistribution into spines in the lateral amygdala, which corresponded with an increase in the size of their postsynaptic densities.71 Mice deficient in profilin II unexpectedly do not have defects in LTP/LTD or learning and memory; however, the number of perforated synapses is increased in the striatum of these mice when compared with wild-type controls.72 Moreover, conditional knockout of profilin I in the mouse forebrain did not result in significant defects in excitatory synaptic transmission or in spine density or morphology.73 Because profilin I and II could have overlapping functions, a double knockout will be necessary to decipher the functions of profilin in regulating spine/synapse development and plasticity. Cofilin, which localizes to the PSD,74 is another key regulator of actin dynamics that binds to and severs actin filaments.75 Cofilin-mediated actin turnover is important for controlling spine length and morphology.65 Furthermore, suppression of cofilin activity is important for the stabilization of mature spines.76 Cofilin localization and activity in spines is modulated by synaptic plasticity.77,78 In addition, cofilin-mediated actin turnover regulates the size of spine heads during LTP and LTD, and loss of cofilin impairs synaptic plasticity and associative learning.77,79–81 The activity of gelsolin, which also severs actin filaments, is important for regulating actin turnover during LTD.39
Neurabins and developmentally regulated brain protein (drebrin) are additional ABPs that contribute to spine development and plasticity. Two neurabins have been identified to date, ie, neurabin I (NrbI) and neurabin II (NrbII), which is also called spinophilin. NrbI is only expressed in the brain whereas NrbII is expressed in a variety of mammalian tissues, including the brain.82,83 NrbI regulates dendritic filopodia length, spine formation and maturation, and synaptic plasticity.84–86 Mice deficient in NrbI have defects in contextual fear memory and LTP, but not in LTD, and display an increase in synaptic transmission.87,88 In contrast, NrbII-deficient mice have defects in LTD, but not LTP.87,89 These mice also exhibit a transient increase in spine density during development, resistance to kainate-induced seizures, and an impairment in conditioned taste aversion learning.89,90 Drebrin is an F-actin binding protein, the expression of which changes from an embryonic (E) isoform to an adult (A) isoform via an alternative splicing mechanism during postnatal development.91 Drebrin A is highly enriched within dendritic spines in the adult brain where it regulates synaptic transmission.92 Drebrin is also an important regulator of spine morphology and development.91,93,94 Drebrin modulates actin dynamics by influencing the interaction of other ABPs, such as cofilin and α-actinin, with actin and by impeding actin-myosin interactions.95–97 Indeed, controlling the balance between drebrin and cofilin is proposed to be essential for regulating actin dynamics in spines.98 Other ABPs and regulators also play important roles in dendritic spine development and plasticity, and we refer the reader to other reviews to gain additional insight into their function in spine formation and plasticity.99–101
Key receptors and intracellular signaling pathways that modulate dendritic spine morphology and development
Glutamate receptors are key regulators of excitatory synaptic transmission and synaptic plasticity in the brain and have critical functions in learning and memory.102 Most excitatory synaptic transmission is mediated by two types of ionotropic glutamate receptors, ie, the N-methyl-D-aspartate receptor (NMDAR) and the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR). Activation of the NMDAR leads to activation of downstream effectors, such as calcium-calmodulin-dependent protein kinase II (CaMKII), reorganization of the actin cytoskeleton, and phosphorylation and insertion of AMPARs into the plasma membrane.103–105 AMPAR membrane insertion and removal as well as AMPAR trafficking are regulated by the actin cytoskeleton via myosin motors and ABPs, such as cofilin and PICK1, indicating the important role that actin plays in modulating AMPAR localization and function.80,106–108 AMPAR insertion into the plasma membrane occurs during synaptic strengthening, such as during LTP, whereas AMPAR removal results in weakening of synapses during LTD.105 Interestingly, LTP promotes the expansion of spine heads whereas LTD leads to spine shrinkage,109 suggesting a link between spine morphology and synaptic efficacy.
A second class of glutamate receptors, metabotropic glutamate receptors (mGluRs), also contribute to spine morphology and development. The mGluRs are G-protein coupled and subdivided in three groups: group 1 includes mGluR1 and 5, group 2 consists of mGluR 2 and 3, and group 3 contains mGluR 4, 6, 7, and 8.110 PSD scaffolding proteins, including Homer and Shank, interact and regulate the activity of group 1 mGluRs.111,112 Moreover, stimulation of group 1 mGluRs results in a significant increase in dendritic spine length, indicating these receptors regulate spine morphology.113 The role of mGluRs in spine formation is currently not clear. In one study, mGluR5 knockout mice exhibited a decrease in spine density in cortical layer IV neurons whereas in another study mGluR5 knockout mice had an increased number of spines.114,115 Future studies are needed to determine the function of mGluRs in modulating spine formation.
Calcium signaling within dendritic spines is an important mechanism for regulating spine morphology as well as synaptic plasticity and function.116 Spines can maintain different concentrations of free intracellular calcium than those found in dendritic shafts, indicating that they can serve as individual calcium compartments.117,118 The calcium concentration in dendritic spines can be regulated by NMDAR channels, by voltage-gated calcium channels, and by release of calcium from internal stores.6 Intracellular calcium can transduce a signal by activating a number of calcium-dependent kinases, including CaMKII. CaMKII is a major component of the PSD within spines and has a central role in synaptic plasticity as well as learning and memory.119 Moreover, a CaMKII isoform, CaMKIIβ, can bind directly to F-actin within spines,120 providing a link between CaMKII signaling and the actin cytoskeleton.
Other membrane receptors are found in spines and influence their morphology and development. Plexin B1 and its ligand semaphorin 4D promote spine formation and changes in spine morphology through a RhoA/Rho-associated kinase pathway.121 Semaphorin 3F, and its receptors neuropilin-2 and Plexin A4, regulate spine morphogenesis and spine size.122 EphB receptor signaling is also critical for spine formation. Mice deficient in EphB1, EphB2, and EphB3 have decreased spine density, abnormal spine morphology (ie, very small or no spine heads), and reduced dendritic filopodia motility.123,124 EphB receptors regulate spine development through a kalirin (GEF)/Rac/PAK pathway and an intersectin (GEF)/Cdc42/N-WASP pathway.125,126 The neuregulin (Nrg)-1 receptor ErbB4 regulates spine size and AMPA-mediated synaptic transmission.127 Loss of ErbB2/B4 in mice causes defects in spine maturation and behavioral abnormalities that include increased aggression and decreased prepulse inhibition, which are observed in schizophrenic patients.128 These phenotypes are rescued by treatment with clozapine, an antipsychotic drug used to treat schizophrenia.128 Nrg-1/ErbB4 signaling also modulates spine formation by a kalirin-dependent mechanism.129
Dendritic spine and synapse defects associated with neurological disorders
A number of neurological disorders, including schizophrenia, AD, autism spectrum disorder, and Down syndrome, are associated with abnormalities in the number, morphology, and plasticity of dendritic spines and synapses. Schizophrenia is a spectrum disorder, which usually develops in late adolescence or early adulthood, and is characterized by impairments in cognition, perception, and motivation.130 In patients with schizophrenia, a reduction in spine density is observed within various brain regions such as the prefrontal cortex, the temporal cortex, the striatum, and pyramidal neurons in the hippocampus.131–135 On the other hand, an increase in spine density is seen in the caudate and putamen patch of patients with schizophrenia.136 Interestingly, alterations in some of the genes/proteins that regulate spine morphology and development, as discussed above, are associated with schizophrenia. NRG1 and ErbB4 are susceptibility genes for schizophrenia, and multiple postmortem studies show aberrant Nrg-1/ErbB4 signaling in the brains of schizophrenic patients.137–139 Disrupted in schizophrenia 1, which regulates Rac activity through its interaction with the GEF kalirin-7, is also a schizophrenia susceptibility gene (Figure 2).140,141 Moreover, patients with schizophrenia have reduced kalirin messenger (m)RNA levels in their dorsolateral prefrontal cortex, and missense mutations in kalirin have been identified in schizophrenic patients.142,143 Cdc42 mRNA levels are lower in patients with schizophrenia, whereas mRNA levels of the Cdc42 effector protein 3 (Cdc42EP3) are increased.142,144 Cdc42EP3 binds septins and regulates septin organization, and Cdc42 inhibits this interaction.145 Therefore, the aberrant expression of Cdc42 and Cdc42EP3 in schizophrenia is proposed to affect opening of the septin barrier in spine necks, in response to glutamate stimulation, which alters molecular diffusion and trafficking between spines and the parent dendrites and leads to impaired synaptic plasticity and spine loss.144 The expression of PSD95, an important scaffolding protein in spines, is also altered in schizophrenic patients.146 Intriguingly, dysbindin, a schizophrenia susceptibility gene, was recently reported to modulate spine dynamics and maturation.147
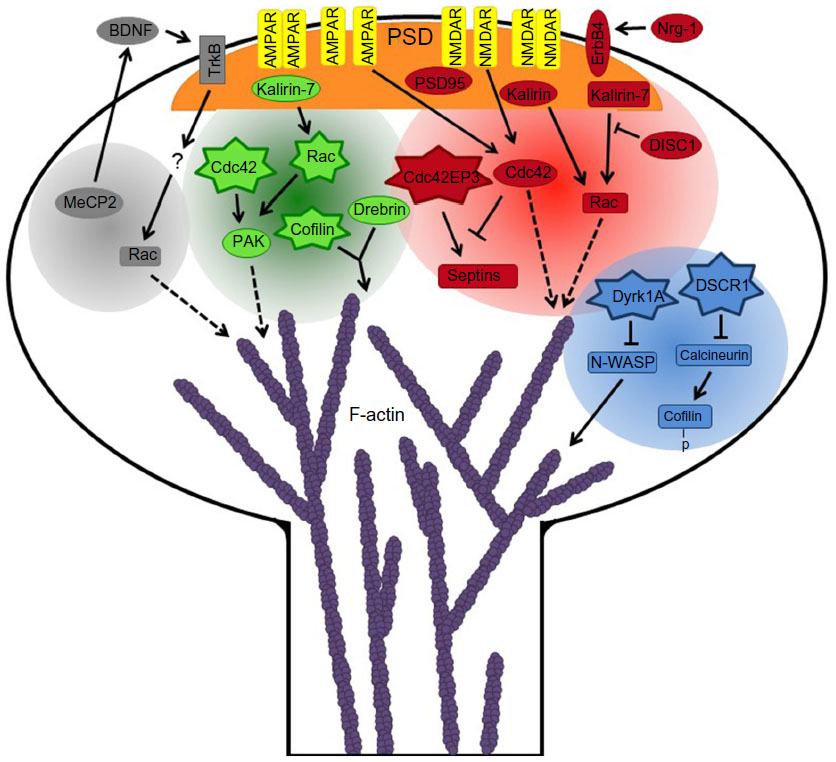 | Figure 2 Schematic of molecular pathways underlying spine defects in several neurological disorders. The neurological disorders include Down syndrome (blue circle), schizophrenia (red circle), Alzheimer’s disease (green circle), and Rett’s syndrome (gray circle). Star shapes indicate upregulation of mRNA and/or protein levels or increased activation of proteins. Ovals represent susceptibility genes (Nrg-1, ErbB4, and DISC1), downregulation of mRNA and/or protein levels, loss-of-function mutations (MeCP2), or mislocalization/dysregulation (PAK). Dashed lines show proteins that regulate actin dynamics through downstream effectors. p-cofilin indicates phosphorylated-cofilin (inactive cofilin). |
AD is a neurodegenerative disease that results in cognitive decline, and usually begins at around 65 years of age. In patients with AD, a significant loss of spines and synapses is seen in the hippocampus and cortex, which are areas of the brain that are significantly affected by AD pathology.38,148 Current data indicate that spine and synapse loss is an early event in AD that precedes neuronal death.38,149 As with schizophrenia, actin regulators and binding proteins are linked to AD. Cofilin aggregates and actin-cofilin rods are found in the brains of AD patients; these abnormal structures could hinder molecular transport and trafficking in neurons, which could contribute to synaptic loss.97,150 Moreover, AD patients have reduced hippocampal levels of drebrin, which could disrupt the balance between drebrin and cofilin and lead to aberrant actin dynamics and synaptic dysfunction.93,151 In AD, Rac and Cdc42 are upregulated, and their effector PAK is mislocalized or dysregulated in AD patients (Figure 2).97,152,153 Furthermore, kalirin-7 expression is altered in the hippocampus of AD patients, which could also contribute to the aberrant spine and synapse phenotype.154
Rett syndrome (RTT) is an X-linked autism spectrum disorder associated with intellectual disability that affects neurodevelopment beginning in early childhood.155 RTT is caused by loss-of-function mutations in MeCP2, which encodes the transcriptional regulator methyl CpG binding protein 2 (MeCP2), as many patients with RTT harbor MeCP2 mutations.156,157 Moreover, deletion of MeCP2 in central nervous system neurons results in an RTT-like phenotype in mice.158 Defects in spines may contribute to RTT, as a decrease in spine number is observed in cortical regions of the brains of RTT patients.159 In addition, a significant decrease in spine density is seen in hippocampal pyramidal neurons in RTT patients.160 The aberrant spine development in RTT may be linked to MeCP2, because mice lacking MeCP2 exhibit a reduction in spine density in the motor cortex and hippocampus.161 One of the transcriptional targets of MeCP2, brain-derived neurotrophic factor (BDNF), and its receptor TrkB, also regulate spine development and plasticity.162,163 Furthermore, RTT patients have decreased levels of BDNF mRNA, and overexpression of BDNF can rescue some of the neuronal and behavioral phenotypes seen in MeCP2 knockout mice, suggesting a function for BDNF signaling in RTT.164,165 Although the specific mechanisms by which BDNF signaling contributes to RTT are currently unknown, it is tempting to speculate that Rho GTPases are involved (Figure 2). BDNF modulates spine formation and plasticity by a Rac-dependent mechanism, and regulation of the activity of the Rho GTPases by bacterial cytotoxic necrotizing factor 1 dramatically improves the behavioral phenotypes, including cognitive defects, in a mouse model of RTT.166,167 However, future investigation will be necessary to determine if Rac or other Rho family GTPases are involved in RTT.
Down syndrome is an intellectual disorder that results from a partial or full additional copy of chromosome 21. A region within chromosome 21, termed the Down syndrome critical region (DSCR), is thought to be responsible for some or possibly all of the features associated with Down syndrome.168 Two genes, DSCR1 and DYRK1A, within this region are of particular interest because their increased expression in mice can recapitulate some Down syndrome phenotypes.169 DYRK1A encodes dual-specificity tyrosine-phosphorylation-regulated kinase 1A (Dyrk1A), which regulates spine formation and actin dynamics (Figure 2). Overexpression of Dyrk1A in hippocampal neurons causes a significant decrease in spine density by inhibiting N-WASP-mediated actin polymerization.170 Furthermore, cortical neurons from Dyrk1A transgenic mice exhibit a reduction in spine density and aberrant spine morphology due to altered actin dynamics.171 This is consistent with the reduction in spine number and altered spine morphology seen in patients with Down syndrome.172,173 DSCR1 encodes DSCR1 protein (also termed RCAN1), which interacts with Fragile X mental retardation protein to regulate spine morphogenesis.174 In addition, DSCR1 inhibits the calmodulin-dependent phosphatase calcineurin, and DSCR1 knockout mice have increased calcineurin activity and defects in spine density, synaptic plasticity, and learning and memory.175 DSCR1 transgenic mice display Down syndrome-like defects in learning and memory, impaired synaptic plasticity, and reduced spine density.176 Alterations in actin dynamics may underlie the spine defects as calcineurin dephosphorylates and activates cofilin, which severs actin filaments.75,177
Remaining knowledge gaps and ongoing research
Although progress has been made over the last few decades in understanding spine development, function, and plasticity, many questions remain. For example, what are the molecular mechanisms that designate a spine to be maintained or pruned? Are the signaling cascades in individual spines that modulate their remodeling and plasticity conserved or unique? What are the key players that regulate spine dynamics as synapses are forming and maturing? Much of our knowledge regarding spine development has been obtained using fixed samples; however, to answer these and other important questions, spine development must be investigated as a real-time process. Moreover, the importance and potential difference in individual spines is increasingly being recognized, making it essential to study spines on an individual basis to understand how they integrate to form circuits. As imaging and other technologies advance, the possibility of investigating individual spines is becoming more feasible. Optogenetic approaches, for instance, may permit selective spine activation to study individual spine dynamics.178 Hopefully, these and other technological developments will pave the way to understanding how individual spines contribute to complex neuronal circuits and ultimately behavior.
Conclusion
In this review, we have summarized some recent insights into spine formation, function, and plasticity. We have highlighted the significance of actin remodeling in these processes and discussed some key actin regulators in dendritic spines. We have also discussed the contribution of crucial synaptic receptors and intracellular signaling pathways to spine development and plasticity. Because of the central role that spines play in cognitive function, defects in their development are associated with a number of neurological disorders, such as schizophrenia, AD, autism spectrum disorder, and Down syndrome. We have outlined the spine/synapse defects associated with these disorders and what is known about the underlying molecular mechanisms that contribute to them. Finally, we have provided insight into some important unanswered questions regarding spines and how addressing these questions will shape the future of dendritic spine research.
Acknowledgment
This work was supported by a National Science Foundation grant (DBI-1450897) to DJW and by a National Institutes of Health grant (NS065183) to HZ.
Disclosure
The authors report no conflicts of interest in this work.
References
Ramón y Cajal S. [Estructura de los centros nerviosos de las aves]. Rev Trim Histol Norm Patol. 1888;1:1–10. Spanish. | |
Ramón y Cajal S. [Significación fisiológica de las expansiones protoplásmicas y nerviosas de la sustancia gris]. Revista de Ciencias Médicas de Barcelona. 1891;22:23. Spanish. | |
Gray EG. Electron microscopy of synaptic contacts on dendrite spines of the cerebral cortex. Nature. 1959;183:1592–1593. | |
Gray EG. Axo-somatic and axo-dendritic synapses of the cerebral cortex: an electron microscope study. J Anat. 1959;93:420–433. | |
Koch C, Zador A. The function of dendritic spines: devices subserving biochemical rather than electrical compartmentalization. J Neurosci. 1993;13:413–422. | |
Yuste R, Majewska A. On the function of dendritic spines. Neuroscientist. 2001;7:387–395. | |
Scheuss V, Bonhoeffer T. Function of dendritic spines on hippocampal inhibitory neurons. Cereb Cortex. 2014;24:3142–3153. | |
Keck T, Scheuss V, Jacobsen RI, et al. Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron. 2011;71:869–882. | |
Kubota Y, Hatada S, Kondo S, Karube F, Kawaguchi Y. Neocortical inhibitory terminals innervate dendritic spines targeted by thalamocortical afferents. J Neurosci. 2007;27:1139–1150. | |
Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis-Davies GC, Higley MJ. Compartmentalization of GABAergic inhibition by dendritic spines. Science. 2013;340:759–762. | |
Muller W, Connor JA. Dendritic spines as individual neuronal compartments for synaptic Ca2+ responses. Nature. 1991;354:73–76. | |
Yuste R, Majewska A, Holthoff K. From form to function: calcium compartmentalization in dendritic spines. Nat Neurosci. 2000;3:653–659. | |
Tsay D, Yuste R. On the electrical function of dendritic spines. Trends Neurosci. 2004;27:77–83. | |
Yuste R. Electrical compartmentalization in dendritic spines. Annu Rev Neurosci. 2013;36:429–449. | |
Harnett MT, Makara JK, Spruston N, Kath WL, Magee JC. Synaptic amplification by dendritic spines enhances input cooperativity. Nature. 2012;491:599–602. | |
Tonnesen J, Katona G, Rozsa B, Nagerl UV. Spine neck plasticity regulates compartmentalization of synapses. Nat Neurosci. 2014;17: 678–685. | |
Grienberger C, Chen X, Konnerth A. Dendritic function in vivo. Trends Neurosci. 2015;38:45–54. | |
Shepherd GM. The dendritic spine: a multifunctional integrative unit. J Neurophysiol. 1996;75:2197–2210. | |
Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat. 1970;127:321–355. | |
Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. | |
Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R. Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci U S A. 1999;96:13438–13443. | |
Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature. 2000;404:876–881. | |
Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. | |
Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. | |
Li Z, Sheng M. Some assembly required: the development of neuronal synapses. Nat Rev Mol Cell Biol. 2003;4:833–841. | |
Chen CC, Lu J, Zuo Y. Spatiotemporal dynamics of dendritic spines in the living brain. Front Neuroanat. 2014;8:28. | |
Petanjek Z, Judas M, Simic G, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2011;108:13281–13286. | |
Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. | |
Ito M, Kano M. Long-lasting depression of parallel fiber-Purkinje cell transmission induced by conjunctive stimulation of parallel fibers and climbing fibers in the cerebellar cortex. Neurosci Lett. 1982;33: 253–258. | |
Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10: 647–658. | |
Xu T, Yu X, Perlik AJ, et al. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462: 915–919. | |
Lai CS, Franke TF, Gan WB. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. | |
Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. | |
Takeuchi T, Duszkiewicz AJ, Morris RG. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130288. | |
Hebb DO. The organization of behavior. New York: John Wiley and Sons; 1949. | |
Ito M, Sakurai M, Tongroach P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J Physiol. 1982;324:113–134. | |
Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. | |
Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. | |
Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5: 239–246. | |
Tatavarty V, Kim EJ, Rodionov V, Yu J. Investigating sub-spine actin dynamics in rat hippocampal neurons with super-resolution optical imaging. PLoS One. 2009;4:e7724. | |
Korobova F, Svitkina T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell. 2010;21:165–176. | |
Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GC, Kasai H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron. 2008;57:719–729. | |
Jaworski J, Kapitein LC, Gouveia SM, et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 2009;61:85–100. | |
Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309. | |
Luo L, Hensch TK, Ackerman L, Barbel S, Jan LY, Jan YN. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 1996;379:837–840. | |
Wegner AM, Nebhan CA, Hu L, et al. N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J Biol Chem. 2008;283:15912–15920. | |
Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10:927–938. | |
Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci. 2005;25:3379–3388. | |
Racz B, Weinberg RJ. Organization of the Arp2/3 complex in hippocampal spines. J Neurosci. 2008;28:5654–5659. | |
Kim IH, Wang H, Soderling SH, Yasuda R. Loss of Cdc42 leads to defects in synaptic plasticity and remote memory recall. Elife. 2014;3. | |
Cahill ME, Xie Z, Day M, et al. Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc Natl Acad Sci U S A. 2009;106:13058–13063. | |
Tolias KF, Bikoff JB, Burette A, et al. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. | |
Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. | |
Kang MG, Guo Y, Huganir RL. AMPA receptor and GEF-H1/Lfc complex regulates dendritic spine development through RhoA signaling cascade. Proc Natl Acad Sci U S A. 2009;106:3549–3554. | |
Buttery P, Beg AA, Chih B, Broder A, Mason CA, Scheiffele P. The diacylglycerol-binding protein alpha1-chimaerin regulates dendritic morphology. Proc Natl Acad Sci U S A. 2006;103:1924–1929. | |
Van de Ven TJ, VanDongen HM, VanDongen AM. The nonkinase phorbol ester receptor alpha 1-chimerin binds the NMDA receptor NR2A subunit and regulates dendritic spine density. J Neurosci. 2005;25: 9488–9496. | |
Nadif Kasri N, Nakano-Kobayashi A, Malinow R, Li B, Van Aelst L. The Rholinked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev. 2009;23:1289–1302. | |
Zhang H, Macara IG. The PAR-6 polarity protein regulates dendritic spine morphogenesis through p190 RhoGAP and the Rho GTPase. Dev Cell. 2008;14:216–226. | |
Kim IH, Racz B, Wang H, et al. Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J Neurosci. 2013;33:6081–6092. | |
Kim Y, Sung JY, Ceglia I, et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442: 814–817. | |
Soderling SH, Langeberg LK, Soderling JA, et al. Loss of WAVE-1 causes sensorimotor retardation and reduced learning and memory in mice. Proc Natl Acad Sci U S A. 2003;100:1723–1728. | |
Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23: 11759–11769. | |
Pruyne D, Evangelista M, Yang C, et al. Role of formins in actin assembly: nucleation and barbed-end association. Science. 2002;297: 612–615. | |
Sagot I, Rodal AA, Moseley J, Goode BL, Pellman D. An actin nucleation mechanism mediated by Bni1 and profilin. Nat Cell Biol. 2002;4: 626–631. | |
Hotulainen P, Llano O, Smirnov S, et al. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 2009;185:323–339. | |
Quinlan ME, Heuser JE, Kerkhoff E, Mullins RD. Drosophila Spire is an actin nucleation factor. Nature. 2005;433:382–388. | |
Pleiser S, Banchaabouchi MA, Samol-Wolf A, et al. Enhanced fear expression in Spir-1 actin organizer mutant mice. Eur J Cell Biol. 2014; 93:225–237. | |
Tilney LG, Bonder EM, Coluccio LM, Mooseker MS. Actin from Thyone sperm assembles on only one end of an actin filament: a behavior regulated by profilin. J Cell Biol. 1983;97:112–124. | |
Pollard TD, Cooper JA. Quantitative analysis of the effect of Acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry. 1984;23:6631–6641. | |
Ackermann M, Matus A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci. 2003;6: 1194–1200. | |
Lamprecht R, Farb CR, Rodrigues SM, LeDoux JE. Fear conditioning drives profilin into amygdala dendritic spines. Nat Neurosci. 2006;9: 481–483. | |
Pilo Boyl P, Di Nardo A, Mulle C, et al. Profilin2 contributes to synaptic vesicle exocytosis, neuronal excitability, and novelty-seeking behavior. EMBO J. 2007;26:2991–3002. | |
Görlich A, Zimmermann AM, Schober D, et al. Preserved morphology and physiology of excitatory synapses in profilin1-deficient mice. PLoS One. 2012;7:e30068. | |
Racz B, Weinberg RJ. Spatial organization of cofilin in dendritic spines. Neuroscience. 2006;138:447–456. | |
Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. | |
Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci. 2009;29: 8129–8142. | |
Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. | |
Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM. Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci U S A. 2012;109:E442–E451. | |
Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. | |
Gu J, Lee CW, Fan Y, et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13:1208–1215. | |
Rust MB, Gurniak CB, Renner M, et al. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010;29:1889–1902. | |
Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci U S A. 1997;94:9956–9961. | |
Nakanishi H, Obaishi H, Satoh A, et al. Neurabin: a novel neural tissue-specific actin filament-binding protein involved in neurite formation. J Cell Biol. 1997;139:951–961. | |
Terry-Lorenzo RT, Roadcap DW, Otsuka T, et al. Neurabin/protein phosphatase-1 complex regulates dendritic spine morphogenesis and maturation. Mol Biol Cell. 2005;16:2349–2362. | |
Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron. 2004;44:321–334. | |
Hu XD, Huang Q, Roadcap DW, Shenolikar SS, Xia H. Actin-associated neurabin-protein phosphatase-1 complex regulates hippocampal plasticity. J Neurochem. 2006;98:1841–1851. | |
Allen PB, Zachariou V, Svenningsson P, et al. Distinct roles for spinophilin and neurabin in dopamine-mediated plasticity. Neuroscience. 2006;140:897–911. | |
Wu LJ, Ren M, Wang H, Kim SS, Cao X, Zhuo M. Neurabin contributes to hippocampal long-term potentiation and contextual fear memory. PLoS One. 2008;3:e1407. | |
Feng J, Yan Z, Ferreira A, et al. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci U S A. 2000;97: 9287–9292. | |
Stafstrom-Davis CA, Ouimet CC, Feng J, Allen PB, Greengard P, Houpt TA. Impaired conditioned taste aversion learning in spinophilin knockout mice. Learn Mem. 2001;8:272–278. | |
Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 2003;23:6586–6595. | |
Ivanov A, Esclapez M, Pellegrino C, Shirao T, Ferhat L. Drebrin A regulates dendritic spine plasticity and synaptic function in mature cultured hippocampal neurons. J Cell Sci. 2009;122:524–534. | |
Hayashi K, Shirao T. Change in the shape of dendritic spines caused by overexpression of drebrin in cultured cortical neurons. J Neurosci. 1999;19:3918–3925. | |
Biou V, Brinkhaus H, Malenka RC, Matus A. Interactions between drebrin and Ras regulate dendritic spine plasticity. Eur J Neurosci. 2008;27:2847–2859. | |
Hayashi K, Ishikawa R, Ye LH, et al. Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. J Neurosci. 1996;16:7161–7170. | |
Ishikawa R, Hayashi K, Shirao T, et al. Drebrin, a development-associated brain protein from rat embryo, causes the dissociation of tropomyosin from actin filaments. J Biol Chem. 1994;269:29928–29933. | |
Zhao L, Ma QL, Calon F, et al. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9:234–242. | |
Shirao T, Gonzalez-Billault C. Actin filaments and microtubules in dendritic spines. J Neurochem. 2013;126:155–164. | |
Tolias KF, Duman JG, Um K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol. 2011;94: 133–148. | |
Lin WH, Webb DJ. Actin and actin-binding proteins: masters of dendritic spine formation, morphology, and function. Open Neurosci J. 2009;3:54–66. | |
Lamprecht R. The actin cytoskeleton in memory formation. Prog Neurobiol. 2014;117:1–19. | |
Peng S, Zhang Y, Zhang J, Wang H, Ren B. Glutamate receptors and signal transduction in learning and memory. Mol Biol Rep. 2011;38: 453–460. | |
Furuyashiki T, Arakawa Y, Takemoto-Kimura S, Bito H, Narumiya S. Multiple spatiotemporal modes of actin reorganization by NMDA receptors and voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A. 2002;99:14458–14463. | |
Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II mediated actin reorganization. J Neurosci. 2006;26:1813–1822. | |
Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. | |
Hanley JG. Actin-dependent mechanisms in AMPA receptor trafficking. Front Cell Neurosci. 2014;8:381. | |
Wang Z, Edwards JG, Riley N, et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. | |
Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol. 2008;10:259–271. | |
Hanley JG. AMPA receptor trafficking pathways and links to dendritic spine morphogenesis. Cell Adh Migr. 2008;2:276–282. | |
Piers TM, Kim DH, Kim BC, Regan P, Whitcomb DJ, Cho K. Translational concepts of mGluR5 in synaptic diseases of the brain. Front Pharmacol. 2012;3:199. | |
Tu JC, Xiao B, Naisbitt S, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. | |
Brakeman PR, Lanahan AA, O’Brien R, et al. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature. 1997;386:284–288. | |
Vanderklish PW, Edelman GM. Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2002;99:1639–1644. | |
Chen CC, Lu HC, Brumberg JC. mGluR5 knockout mice display increased dendritic spine densities. Neurosci Lett. 2012;524:65–68. | |
Wijetunge LS, Till SM, Gillingwater TH, Ingham CA, Kind PC. mGluR5 regulates glutamate-dependent development of the mouse somatosensory cortex. J Neurosci. 2008;28:13028–13037. | |
Bloodgood BL, Sabatini BL. Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol. 2007;17:345–351. | |
Guthrie PB, Segal M, Kater SB. Independent regulation of calcium revealed by imaging dendritic spines. Nature. 1991;354:76–80. | |
Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci. 2005;25:6037–6046. | |
Shonesy BC, Jalan-Sakrikar N, Cavener VS, Colbran RJ. CaMKII: a molecular substrate for synaptic plasticity and memory. Prog Mol Biol Transl Sci. 2014;122:61–87. | |
Shen K, Teruel MN, Subramanian K, Meyer T. CaMKIIbeta functions as an Factin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron. 1998;21:593–606. | |
Lin X, Ogiya M, Takahara M, et al. Sema4D-plexin-B1 implicated in regulation of dendritic spine density through RhoA/ROCK pathway. Neurosci Lett. 2007;428:1–6. | |
Tran TS, Rubio ME, Clem RL, et al. Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature. 2009;462:1065–1069. | |
Henkemeyer M, Itkis OS, Ngo M, Hickmott PW, Ethell IM. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J Cell Biol. 2003;163:1313–1326. | |
Kayser MS, Nolt MJ, Dalva MB. EphB receptors couple dendritic filopodia motility to synapse formation. Neuron. 2008;59:56–69. | |
Penzes P, Beeser A, Chernoff J, et al. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. | |
Irie F, Yamaguchi Y. EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nat Neurosci. 2002;5: 1117–1118. | |
Li B, Woo RS, Mei L, Malinow R. The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron. 2007;54:583–597. | |
Barros CS, Calabrese B, Chamero P, et al. Impaired maturation of dendritic spines without disorganization of cortical cell layers in mice lacking NRG1/ErbB signaling in the central nervous system. Proc Natl Acad Sci U S A. 2009;106:4507–4512. | |
Cahill ME, Remmers C, Jones KA, Xie Z, Sweet RA, Penzes P. Neuregulin 1 signaling promotes dendritic spine growth through kalirin. J Neurochem. 2013;126:625–635. | |
Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience. 2013;251:90–107. | |
Garey LJ, Ong WY, Patel TS, et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–453. | |
Roberts RC, Conley R, Kung L, Peretti FJ, Chute DJ. Reduced striatal spine size in schizophrenia: a postmortem ultrastructural study. Neuroreport. 1996;7:1214–1218. | |
Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. | |
Sweet RA, Henteleff RA, Zhang W, Sampson AR, Lewis DA. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34:374–389. | |
Kolomeets NS, Orlovskaya DD, Rachmanova VI, Uranova NA. Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: a postmortem morphometric study. Synapse. 2005;57:47–55. | |
Roberts RC, Roche JK, Conley RR. Synaptic differences in the patch matrix compartments of subjects with schizophrenia: a postmortem ultrastructural study of the striatum. Neurobiol Dis. 2005;20: 324–335. | |
Stefansson H, Sigurdsson E, Steinthorsdottir V, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. | |
Hashimoto R, Straub RE, Weickert CS, Hyde TM, Kleinman JE, Weinberger DR. Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Mol Psychiatry. 2004;9: 299–307. | |
Pan B, Huang XF, Deng C. Antipsychotic treatment and neuregulin 1-ErbB4 signalling in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:924–930. | |
Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. | |
Hayashi-Takagi A, Takaki M, Graziane N, et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332. | |
Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–566. | |
Kushima I, Nakamura Y, Aleksic B, et al. Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr Bull. 2012;38:552–560. | |
Ide M, Lewis DA. Altered cortical CDC42 signaling pathways in schizophrenia: implications for dendritic spine deficits. Biol Psychiatry. 2010;68:25–32. | |
Joberty G, Perlungher RR, Sheffield PJ, et al. Borg proteins control septin organization and are negatively regulated by Cdc42. Nat Cell Biol. 2001;3:861–866. | |
Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol Psychiatry. 2006;11:737–747. | |
Jia JM, Hu Z, Nordman J, Li Z. The schizophrenia susceptibility gene dysbindin regulates dendritic spine dynamics. J Neurosci. 2014;34: 13725–13736. | |
DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. | |
Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002; 298:789–791. | |
Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000;2:628–636. | |
Kojima N, Shirao T. Synaptic dysfunction and disruption of postsynaptic drebrinactin complex: a study of neurological disorders accompanied by cognitive deficits. Neurosci Res. 2007;58:1–5. | |
Zhu X, Raina AK, Boux H, Simmons ZL, Takeda A, Smith MA. Activation of oncogenic pathways in degenerating neurons in Alzheimer disease. Int J Dev Neurosci. 2000;18:433–437. | |
Ma QL, Yang F, Calon F, et al. p21-activated kinase-aberrant activation and translocation in Alzheimer disease pathogenesis. J Biol Chem. 2008;283:14132–14143. | |
Youn H, Ji I, Ji HP, Markesbery WR, Ji TH. Under-expression of Kalirin-7 Increases iNOS activity in cultured cells and correlates to elevated iNOS activity in Alzheimer’s disease hippocampus. J Alzheimers Dis. 2007;12:271–281. | |
Xu X, Miller EC, Pozzo-Miller L. Dendritic spine dysgenesis in Rett syndrome. Front Neuroanat. 2014;8:97. | |
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG binding protein 2. Nat Genet. 1999;23:185–188. | |
Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. | |
Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. | |
Belichenko PV, Oldfors A, Hagberg B, Dahlstrom A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5:1509–1513. | |
Chapleau CA, Calfa GD, Lane MC, et al. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol Dis. 2009;35:219–233. | |
Belichenko PV, Wright EE, Belichenko NP, et al. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol. 2009;514:240–258. | |
Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. | |
Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21: 4249–4258. | |
Deng V, Matagne V, Banine F, et al. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16:640–650. | |
Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. | |
De Filippis B, Fabbri A, Simone D, et al. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of Rett syndrome. Neuropsychopharmacology. 2012;37:1152–1163. | |
Hale CF, Dietz KC, Varela JA, et al. Essential role for vav guanine nucleotide exchange factors in brain-derived neurotrophic factor-induced dendritic spine growth and synapse plasticity. J Neurosci. 2011;31:12426–12436. | |
Korenberg JR, Kawashima H, Pulst SM, et al. Molecular definition of a region of chromosome 21 that causes features of the Down syndrome phenotype. Am J Hum Genet. 1990;47:236–246. | |
Arron JR, Winslow MM, Polleri A, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. | |
Park J, Sung JY, Park J, Song WJ, Chang S, Chung KC. Dyrk1A negatively regulates the actin cytoskeleton through threonine phosphorylation of N-WASP. J Cell Sci. 2012;125:67–80. | |
Martinez de Lagran M, Benavides-Piccione R, Ballesteros-Yanez I, et al. Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cereb Cortex. 2012;22:2867–2877. | |
Marin-Padilla M. Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res. 1972;44: 625–629. | |
Marin-Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down’s syndrome. A Golgi study. J Comp Neurol. 1976;167:63–81. | |
Wang W, Zhu JZ, Chang KT, Min KT. DSCR1 interacts with FMRP and is required for spine morphogenesis and local protein synthesis. EMBO J. 2012;31:3655–3666. | |
Hoeffer CA, Dey A, Sachan N, et al. The Down syndrome critical region protein RCAN1 regulates long-term potentiation and memory via inhibition of phosphatase signaling. J Neurosci. 2007;27:13161–13172. | |
Martin KR, Corlett A, Dubach D, et al. Over-expression of RCAN1 causes Down syndrome-like hippocampal deficits that alter learning and memory. Hum Mol Genet. 2012;21:3025–3041. | |
Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem. 2005;280:12683–12689. | |
Packer AM, Peterka DS, Hirtz JJ, Prakash R, Deisseroth K, Yuste, R. Two photon optogenetics of dendritic spines and neural circuits. Nat Methods. 2012;9:1202–1205. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.