")
Back to Journals » Cancer Management and Research » Volume 10
Delta-6-desaturase inhibitor enhances radiation therapy in glioblastoma in vitro and in vivo
Authors Wang J , Liang H, Sun M , Zhang L , Xu H, Liu W, Li Y, Zhou Y, Li Y , Li M
Received 28 August 2018
Accepted for publication 4 November 2018
Published 7 December 2018 Volume 2018:10 Pages 6779—6790
DOI https://doi.org/10.2147/CMAR.S185601
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Harikrishna Nakshatri
Jie Wang,1 Huaxin Liang,2 Meiyan Sun,3 Lei Zhang,3 Huijing Xu,3 Wei Liu,3 Yan Li,3 Yue Zhou,4 Yingya Li,5 Miao Li2
1Department of Neurology, The China-Japan Union Hospital of Jilin University, Changchun 130033, China; 2Department of Neurosurgery, The China-Japan Union Hospital of Jilin University, Changchun 130033, China; 3College of Laboratory Medicine, Jilin Medical University, Jilin 132013, China; 4Department of Statistics, North Dakota State University, Fargo, ND 58108, USA; 5Department of Cereal Science, North Dakota State University, Fargo, ND 58108, USA
Background: It has been reported that cell inflammation pathways contribute to the development of prostaglandin E2 (PGE2)-inhibitor of DNA-binding protein-1 (ID1)-dependent radioresistance in glioblastoma. Here, we proposed that inhibiting delta-6-desaturase (D6D) could block arachidonic acid synthesis and PGE2 production, thereby reversing PGE2-ID1-dependent radioresistance in glioblastoma cells and xenograft tumor models.
Materials and methods: Two glioblastoma cell lines, namely, U-87 MG and LN-229, were used for the in vitro study. The combination effects of SC-26196 (a D6D inhibitor) and radiation were assessed by the MTS assay, colony formation assay, and cell apoptosis analysis. HPLC/MS analysis was performed to quantify the production of arachidonic acid and PGE2. For the in vivo study, 6-week-old nude mice, each bearing a U-87 MG xenograft tumor, were subjected to 4-week treatments of vehicle, SC-26196, radiation, or the combination of both. Tumor growth was monitored during the treatment, and the tumor tissues were collected at the end for further analysis.
Results: Treatment with SC-26196 significantly improved radiosensitivity in both glioblastoma cell lines in vitro, and radiosensitivity was associated with inhibited synthesis of arachidonic acid and PGE2. The combination of SC-26196 and radiation synergistically inhibited U-87 MG xenograft tumor growth, in association with the induction of tumor apoptosis and suppressed tumor proliferation. SC-26196 also inhibited arachidonic acid and PGE2 production in vivo and limited expression of ID1.
Conclusion: These data suggested that the D6D inhibitor could reverse PGE2-ID1-dependent radioresistance in glioblastoma cells and xenograft tumor models by blocking the synthesis of arachidonic acid and PGE2. Although further investigation is required, the outcomes from this study may guide us in developing a potentially novel combination strategy for current glioblastoma therapy.
Keywords: delta-6-desaturase, glioblastoma, radiation therapy, inflammation pathway
Introduction
Glioblastoma is the most common cancer that begins within the brain and the most aggressive malignancy in the central nervous system.1,2 Common treatments for glioblastoma include surgery, chemotherapy, and radiation therapy. However, resistance to chemotherapy and radiation is the major obstacle for glioblastoma treatment, and the median survival rate after treatment remains very poor.1–4
Various mechanisms have been proposed that may account for radioresistance in glioblastoma, and interference with these mechanistic pathways may help to sensitize glioblastoma to radiation therapy.5–17 For example, it has been reported that cellular inflammatory pathways play important roles in radioresistance in glioblastoma, and inhibiting inflammatory pathways could sensitize glioblastoma cells/tumors to radiation therapy.18–20 In a recent study, Cook et al21 reported that prostaglandin E2 (PGE2), a major pro-inflammatory metabolite derived from arachidonic acid, could induce inhibitor of DNA-binding protein-1 (ID1)-dependent radioresistance in glioblastoma cells. Therefore, a strategy that targets the cell inflammatory pathways seems promising and could aid in radiation therapy for glioblastoma treatment.
Delta-6-desaturase (D6D) is the key enzyme that converts upstream ω-6 fatty acids (primarily linoleic acid) to arachidonic acid. Research has shown that the levels of D6D mRNA are higher in tumors than in normal tissue from patients with brain tumors, and inhibiting D6D led to decreased levels of arachidonic acid and its derived pro-cancer/pro-inflammation metabolites, which eventually resulted in anti-inflammation and anti-cancer effects.22 However, to the best of our knowledge, D6D has not been investigated as a therapy target in glioblastoma. Therefore, for the first time, we proposed that inhibition of D6D by SC-26196, a D6D inhibitor, can suppress the inflammation pathways of cancer cells, thus sensitizing glioblastoma cells to radiation therapy. In this study, by using in vitro and in vivo glioblastoma models, we demonstrated that inhibiting D6D, by blocking the synthesis of arachidonic acid and PGE2, could reverse PGE2-ID1-dependent radioresistance in glioblastoma cells and xenograft tumor models (Scheme 1). Although further investigation is required, the outcomes from this study may guide us in developing a potentially novel combination strategy for current glioblastoma therapy.
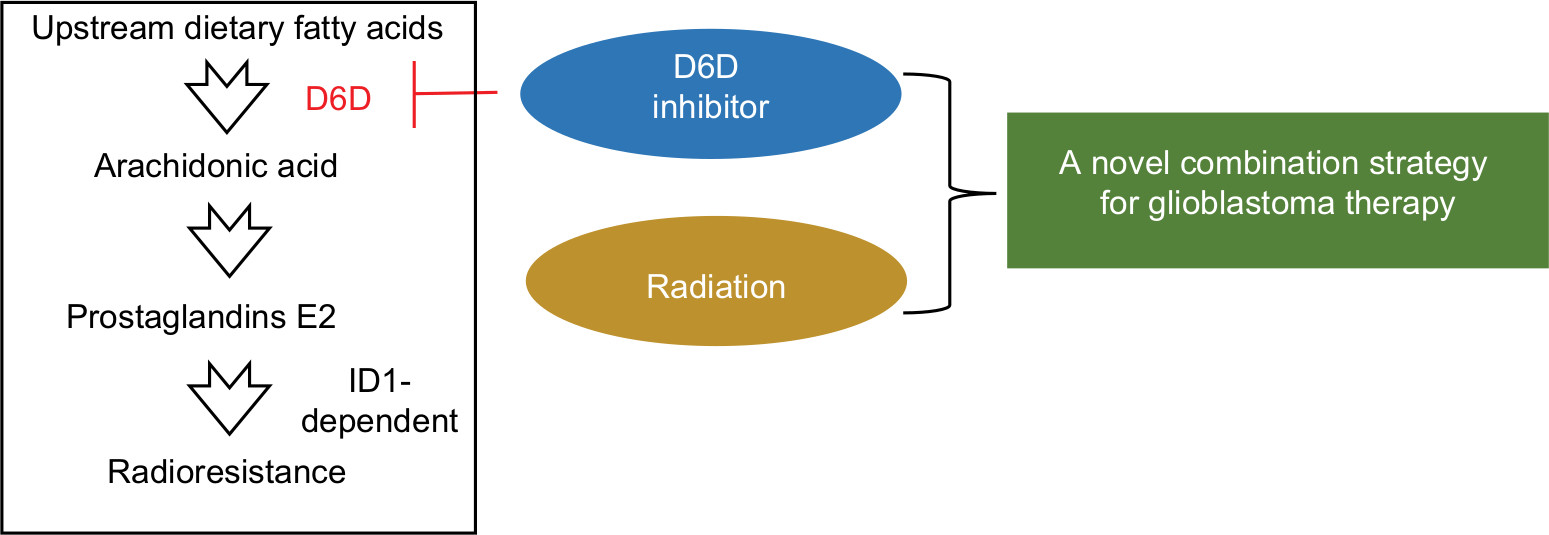 | Scheme 1 Proposed mechanisms by which D6D inhibition could reverse the PGE2-ID1-dependent radioresistance in glioblastoma. Abbreviations: D6D, delta-6-desaturase; PGE2, prostaglandin E2; ID1, inhibitor of DNA-binding protein-1. |
Materials and methods
Cell lines
U-87 MG and LN-229 cell lines were originally purchased from American Type Culture Collection. U-87 MG cells were grown in Eagle’s minimum essential medium supplemented with 10% FBS. LN-229 cells were grown in DMEM supplemented with 10% FBS. Cells were cultured in an incubator at 37°C containing 95% humidified atmosphere with 5% CO2.
MTS assay
Cell proliferation after treatments of SC-26196 (Sigma-Aldrich Co., St Louis, MO, USA), radiation, or the combination of both was assessed using an MTS Cell Proliferation Colorimetric Assay Kit (Abcam, Cambridge, MA, USA). Briefly, U-87 MG and LN-229 cells were seeded at 5,000 cells per well into 96-well plates and incubated overnight. Then, the cells were exposed to different treatments including SC-26196 alone, X-ray irradiation alone, and the combination of both at various doses. After a 48-hour incubation, 20 µL/well of MTS reagent was added, and the absorbance was measured using a plate reader at 490 nm within 4 hours of incubation. All experiments were repeated at least three times.
Colony formation assay
The colony formation assay was performed to assess cell survival upon SC-26196, radiation, and their combination treatment. Briefly, cells were seeded at 500 cells per well into six-well plates, incubated overnight, and exposed to SC-26196 alone, radiation alone, or a combination of both. After a 10-day incubation, the cells were fixed with 10% neutral buffered formalin and stained with 0.05% (w/v) crystal violet solution. Cell colonies were then counted using an inverted microscope. All experiments were repeated at least three times.
Cell apoptosis analysis
Cell apoptosis upon various treatments was analyzed in vitro using the annexin V-FITC Apoptosis Detection Kit (Sigma-Aldrich Co.). Briefly, 2.0×105 cells were seeded overnight in each well of six-well plates followed by treatment with SC-26196 alone, radiation alone, or a combination of both. After a 48-hour incubation, the cells were harvested and suspended in binding buffer at a concentration of 1.0×106 cells/mL. Then, annexin V-FITC and PI staining solution were added to the cell suspension, followed by a 10-minute incubation in the dark. The samples were then subjected to apoptosis analysis using a flow cytometer. All experiments were repeated at least three times.
In vitro detection of arachidonic acid and PGE2
U-87 MG and LN-229 cells were seeded at 2.0×105 cells per well in six-well plates and incubated overnight, followed by treatment with SC-26196 alone, radiation alone, or a combination of both. After a 48-hour incubation, the cells and culture medium were collected. Arachidonic acid and PGE2 were extracted by mixing with chloroform:methanol (2:1). The organic phase was collected and evaporated to dryness. The samples were then reconstituted with 100 µL of ethanol for HPLC/MS analysis using an internal standard method. All experiments were repeated at least three times.
Animal model
All animal studies were carried out by following the Animal Experiment Standard Operating Procedure issued by the Experimental Animal Committee of Jilin University with an approved permit No. SCXK 2007-0011. A total of 32 female nude mice (6 weeks old) were housed four per cage in the pathogen-free animal facility of the Animal Service Center of Jilin University with water and food ad libitum. The mice were kept in a temperature/humidity-controlled room with a 12-hour light/dark cycle.
Tumor xenografts were established by subcutaneously injecting 2×106 U-87 MG cells (in 100 µL of PBS) into the hind flank of mice. After 7 days of cell injection, the mice were then divided into four different treatment groups (eight mice per group), and each received a 4-week treatment of 1) vehicle control, 2) SC-26196 (intraperitoneal injection, 30 mg/kg, every third day), 3) local X-ray irradiation (5 Gy, every third day), or 4) a combination of SC-26196 and radiation (radiation was performed 1 hour after SC-26196 injection). Tumor growth was monitored twice a week, and tumor volume was calculated as: V = tumor length × width2/2. After a 4-week treatment, the mice were euthanized with an overdose of pentobarbital, and the tumor tissues were collected for further analysis.
Immunofluorescence study
Fresh collected tumor tissues were fixed with 10% neutral buffered formalin overnight and dehydrated with ethanol and xylene. Then, the tissues were embedded in paraffin for sectioning. Tissues sections were mounted onto histological slides, rehydrated,, and incubated with primary antibodies for cleaved-PARP, Ki-67, and ID1. After washing, the slides were incubated with the corresponding secondary antibodies for fluorescence study. Cell nuclei were counter stained with DAPI. The images were acquired with a Zeiss Axio Imager M2 microscope with a magnification of 200×. Quantification data were acquired from eight tumor tissues per treatment group.
In vivo detection of arachidonic acid and PGE2
Tumor tissues from each mouse were weighed and homogenized. The arachidonic acid and PGE2 from tumor tissues were then extracted using chloroform/methanol (2/1). The sample solution was vacuumed to dryness and reconstituted with 100 µL of ethanol for HPLC/MS analysis. Quantification data were acquired from eight tumor tissues per treatment group.
Statistics
For the in vitro study, every experiment was repeated at least three times. For in vivo experiments, data were acquired from eight mice per treatment group. Statistical differences between the mean values for different groups were evaluated by ANOVA, and significant differences were considered with a P-value <0.05. Data are represented as the mean ± SD.
Results
The D6D inhibitor sensitized glioblastoma cells to radiation in vitro
To test whether inhibiting D6D could sensitize glioblastoma to radiation therapy, two cell lines, namely U-87 MG and LN-229, were exposed to the D6D inhibitor SC-26196, radiation, or a combination of the two at various doses. The results from the MTS assay showed that treatment with SC-26196 alone or with radiation alone only resulted in a limited anti-proliferation effect in both cell lines (<35% inhibition at the highest tested concentrations; Figure 1A and B). In comparison, the concurrent treatment of SC-26196 and radiation led to significantly improved anti-proliferation activities in both cell lines in a synergistic manner (~80% inhibition at the highest tested concentrations; Figure 1A and B).
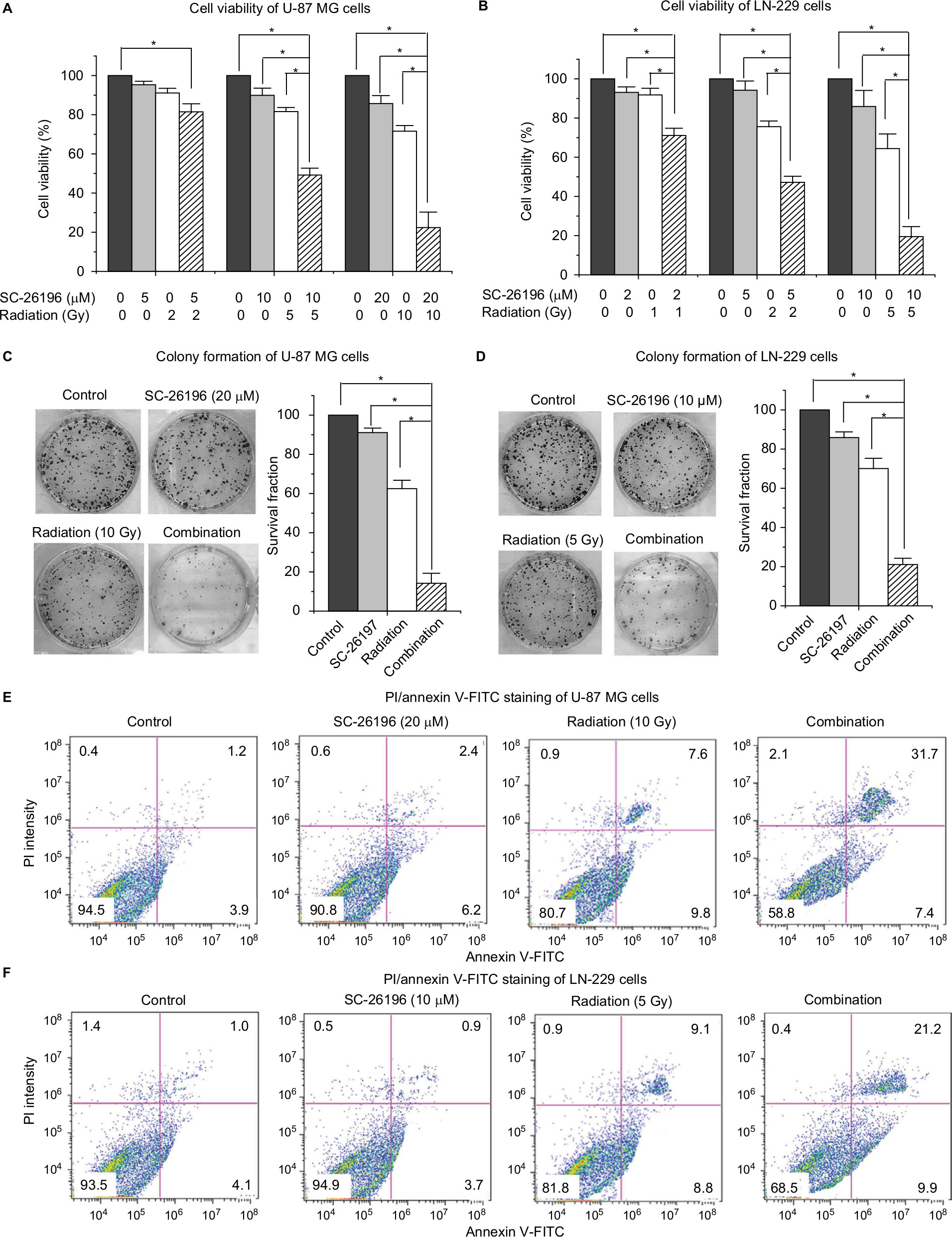 | Figure 1 D6D inhibitor sensitized glioblastoma cells to radiation in vitro. Notes: (A and B) MTS assay of U-87 MG and LN-229 cells treated with vehicle control, SC-26196, radiation, or their combination at indicated dose. (C and D) Colony formation assay of U-87 MG and LN-229 cells treated with vehicle control, SC-26196, radiation, or their combination at the indicated doses. (E and F) Apoptosis analysis (annexin V-FITC/PI double staining) of U-87 MG and LN-229 cells treated with vehicle control, SC-26196, radiation, or their combination at the indicated doses. All quantification data are represented as mean ± SD with at least three separate experiments. The data in control treatment group were normalized to 100%. *Significant difference with P<0.05. Abbreviation: D6D, delta-6-desaturase. |
A colony formation assay was performed to confirm the synergistic effect from combination treatment. Consistent with the MTS assay, treatment with SC-26196 alone or radiation alone at the indicated doses only led to a moderate anti-proliferation effect in U-87 MG and LN-229 cells (<40% inhibition; Figure 1C and D). However, the combination treatment resulted in significantly less colony formation compared to the single treatment groups and vehicle control in both cell lines (>80% inhibition; Figure 1C and D).
Radiation is known to kill cancer cells by damaging DNA and leading to cell death. Therefore, we further performed PI/annexin V-FITC staining analysis to investigate the combination effect of the D6D inhibitor and radiation in U-87 MG and LN-229 cells. The results showed that D6D inhibitor alone did not trigger cell apoptosis in both cell lines, while radiation alone led to limited cell apoptosis compared to vehicle controls (<20% from annexin V positive-PI positive and annexin V positive-PI negative populations; Figure 1E and F). In comparison, the combination treatment in both cell lines resulted in a significant induction of cell apoptosis compared to the radiation alone and control groups (~30%–40% apoptotic population; Figure 1E and F). The activation of apoptosis after the combination treatment in both cell lines was also confirmed by Western blot analysis, which showed alterations in a series of pro- and anti-apoptotic proteins (Figure S1).
In addition to U-87 MG and LN-229 cells, we also tested the combinational effect of SC-26196 and radiation on other human glioblastoma cell lines including A172 and U-138 MG cells. The results showed that SC-26196 was also able to sensitize these cell lines to radiation treatment at various tested doses (Figure S2).
The D6D inhibitor improved radiosensitivity in glioblastoma cells by inhibiting PGE2 production
It has been reported that PGE2 could induce ID1-dependent radioresistance in glioblastoma cells, and therefore, we proposed that the observed D6D inhibitor-induced radiosensitivity was due to limited PGE2 production.21 To prove our hypothesis, by using HPLC/MS analysis, we assessed the production of the pro-inflammatory metabolites PGE2 and its precursor arachidonic acid in U-87 MG and LN-229 cells after various treatments. Data showed that treatment with the D6D inhibitor (including the combination of D6D inhibitor and radiation) led to a significant decrease in arachidonic acid and PGE2 in both cell lines (Figure 2A and B). More importantly, the results from the MTS assay showed that when the cells were treated with the combination of SC-26196, radiation, along with arachidonic acid or PGE2, they were able to develop resistance to radiation again (Figure 2C and D), indicating that the suppressed arachidonic acid and/or PGE2 production was responsible for the improved radiosensitivity from D6D inhibitor treatment. Further investigation showed that the concurrent application of arachidonic acid and celecoxib (a selective cyclooxygenase-2 [COX-2] inhibitor that blocks the conversion of arachidonic acid to PGE2) failed to restore radioresistance in the cells (Figure 2C and D), confirming that it was PGE2, rather than arachidonic acid, that played important role in the development of radioresistance in glioblastoma cells.
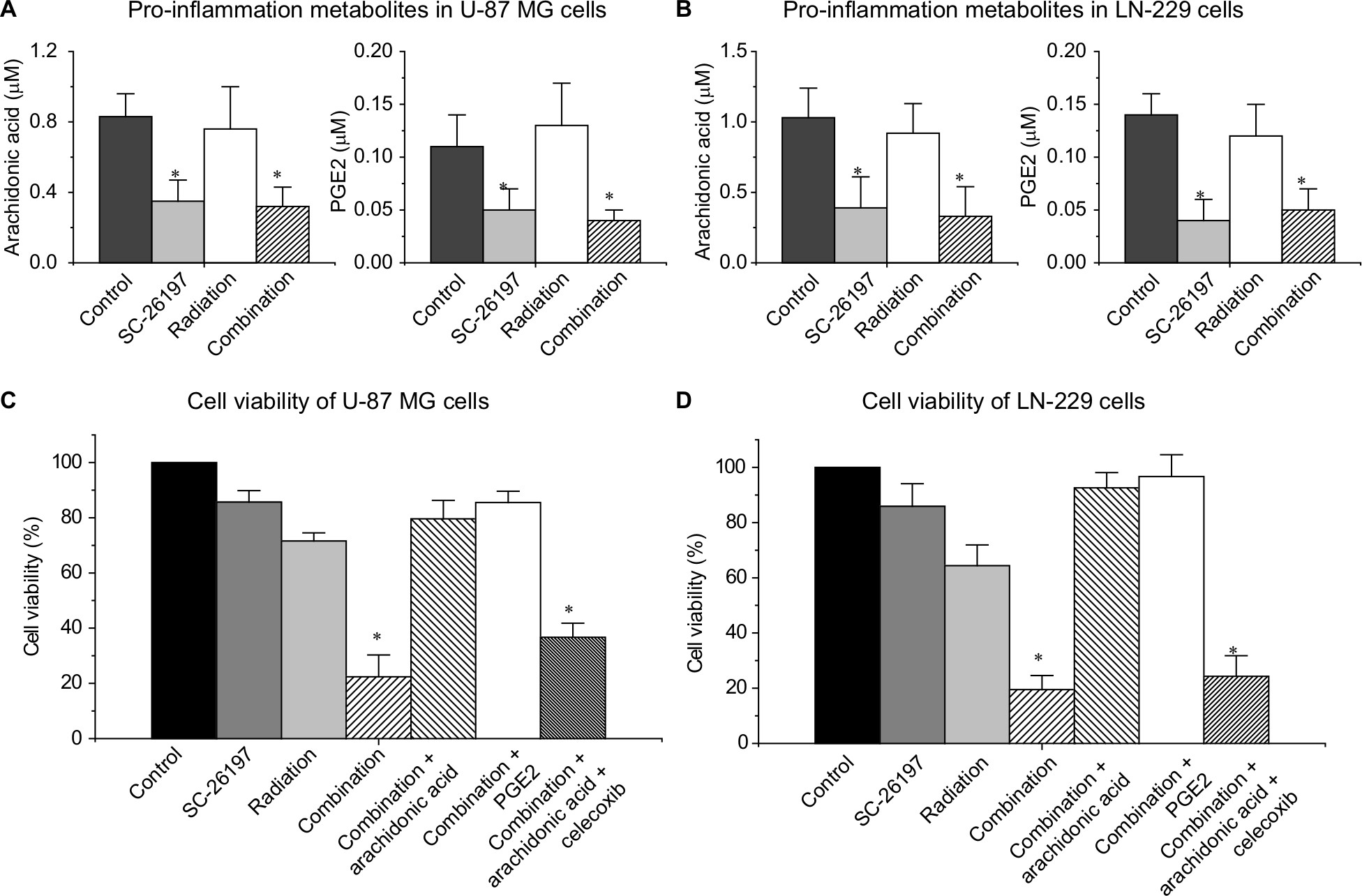 | Figure 2 The D6D inhibitor improved radiosensitivity in glioblastoma cells by inhibiting PGE2 production. Notes: (A) HPLC/MS quantification of arachidonic acid and PGE2 in 1.5×106 U-87 MG cells treated with vehicle control, SC-26196 (20 µM), radiation (10 Gy), or their combination. (B) HPLC/MS quantification of arachidonic acid and PGE2 in 1.5×106 LN-229 cells treated with vehicle control, SC-26196 (10 µM), radiation (5 Gy), or their combination. (C) MTS assay of U-87 MG cells treated with vehicle control, SC-26196 (20 µM), radiation (10 Gy), SC-26196 + radiation, SC-26196 + radiation + arachidonic acid (20 µM), SC-26196 + radiation + PGE2 (20 µM), or SC-26196 + radiation + arachidonic acid + celecoxib (10 µM). (D) MTS assay of LN-229 cells treated with vehicle control, SC-26196 (10 µM), radiation (5 Gy), SC-26196 + radiation, SC-26196 + radiation + arachidonic acid (20 µM), SC-26196 + radiation + PGE2 (20 µM), or SC-26196 + radiation + arachidonic acid + celecoxib (10 µM). All quantification data are represented as the mean ± SD of at least three separate experiments. *Significant difference vs the control group with P<0.05. Abbreviations: D6D, delta-6-desaturase; PGE2, prostaglandin E2. |
The D6D inhibitor sensitized xenograft glioblastoma tumors to radiation therapy
To confirm the in vitro results, we established a xenograft tumor model by injecting U-87 MG cells subcutaneously into the hind flank of nude mice. The mice were exposed to a 4-week treatment of SC-26196 alone, radiation alone, or a combination of both. Tumor growth was monitored twice a week. The results showed that SC-26196 alone did not significantly inhibit tumor growth (with an average tumor size of 183.9±14.3 mm3 vs 208.7±14.5 mm3 that in the control group), while radiation alone moderately suppressed tumor growth (with an average tumor size of 151.6±15.1 mm3; Figure 3A–C). In comparison, the combination of SC-26196 and radiation synergistically inhibited tumor growth (average tumor size of 66.8±28.3 mm3).
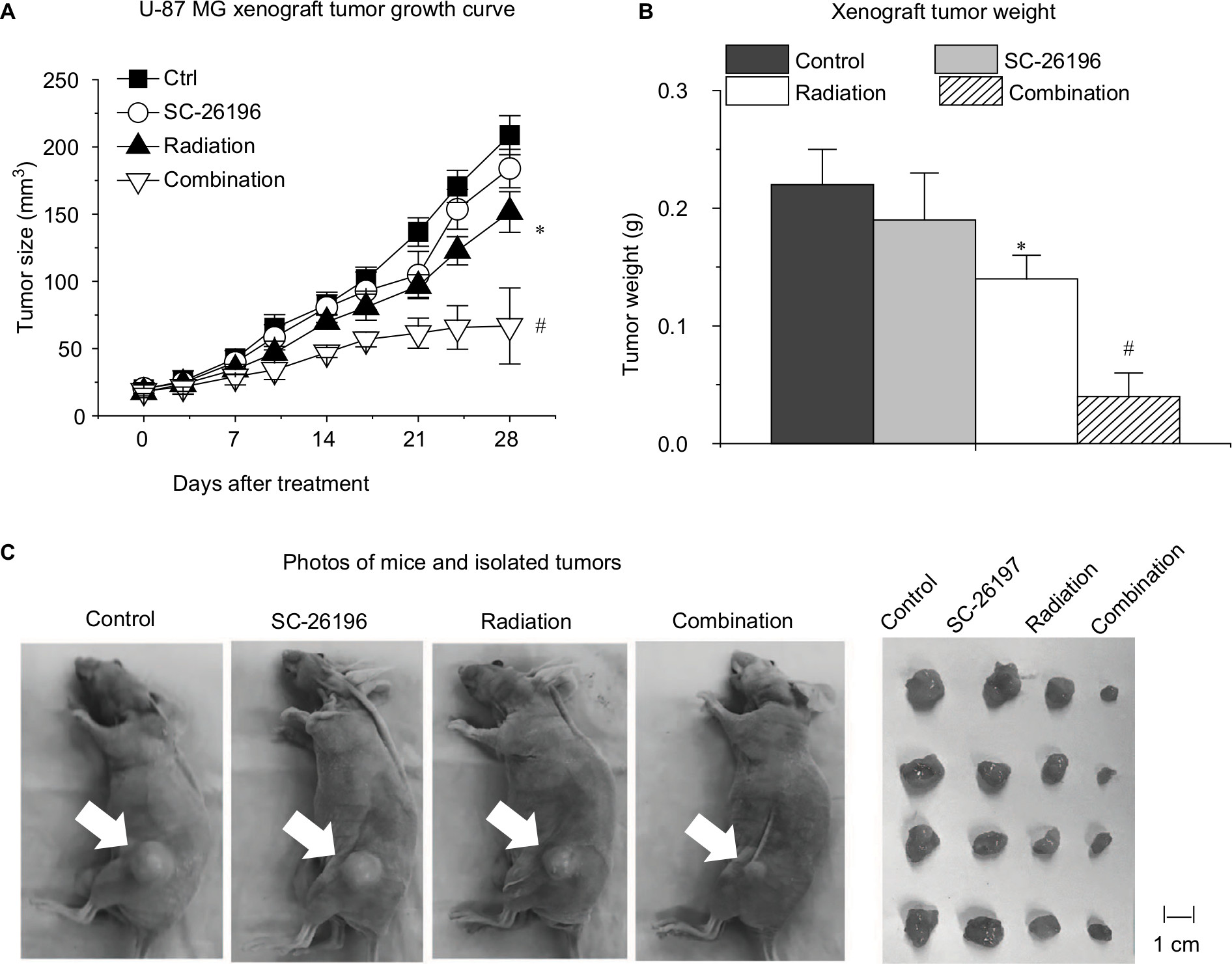 | Figure 3 The D6D inhibitor sensitized a xenograft glioblastoma tumor model to radiation therapy. Notes: (A) Measured tumor size during a 4-week treatment of vehicle control, SC-26196, radiation, or their combination. (B) Measured tumor weight at the endpoint. (C) Representative photos of mice and isolated tumor tissues at the end of the treatment. White arrows indicate tumor site. All quantification data are represented as the mean ± SD with eight mice per treatment group. *A significant difference vs control with P<0.05. #A significant difference vs control and the single treatment group with P<0.05. Abbreviations: D6D, delta-6-desaturase; PGE2, prostaglandin E2. |
Immunofluorescence assay showed that the combination treatment synergistically induced tumor apoptosis, as evidenced by cleaved-PARP positive staining (Figure 4A). Meanwhile, the expression of Ki-67, a cell proliferation marker, was significantly inhibited by the combination treatment in a synergistic manner compared to the control group and radiation treatment group (Figure 4B).
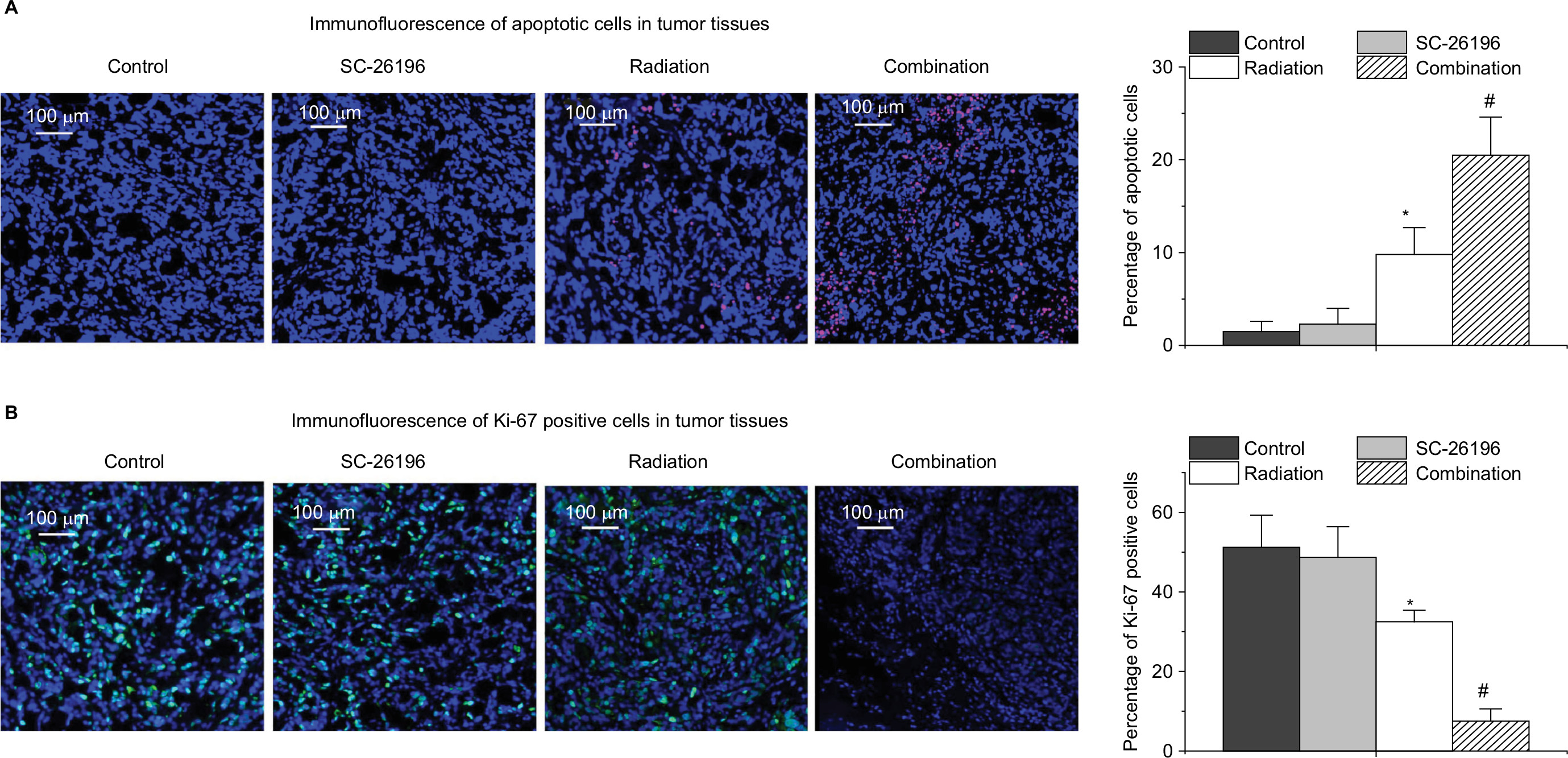 | Figure 4 Immunofluorescence analysis of tumor apoptosis and proliferation. Notes: (A) Representative images for cleaved PARP expression (stained in red) in tumor tissues. Cell nuclei were counter stained with DAPI. For quantification, the percentage of cleaved PARP-positive cells vs the total number of cells in each sample was calculated. (B) Representative images for Ki-67 expression (stained in green) in tumor tissues. Cell nuclei were counter stained with DAPI. For quantification, the percentage of Ki-67-positive cells vs the total number of cells in each sample was calculated. The images were acquired with a Zeiss Axio Imager M2 microscope with a magnification of 200×. All the quantification data are represent as the mean ± SD with eight mice per treatment group. *A significant difference vs control with P<0.05 and #a significant difference vs control and the single treatment group with P<0.05. |
The D6D inhibitor reversed radioresistance in vivo by suppressing the PGE2-ID1 pathway
Inhibitor of DNA-binding proteins are transcriptional regulators that play important roles in tumor progression.23,24 Interestingly, it has been established that PGE2 contributes to radioresistance by inducing the ID1-dependent pathway.21 Hence, here, we proposed that the D6D inhibitor could overcome tumor radioresistance by inhibiting PGE2 production, thereby consequently limiting the PGE2-ID1 signaling pathway.
By using HPLC/MS analysis, we observed that D6D inhibitor treatment (alone or in combination with radiation) could significantly inhibit the production of arachidonic acid (PGE2 precursor), thereby limiting PGE2 levels (Figure 5A). Consequently, an immunofluorescence assay showed that the expression level of ID1 in tumor tissues after D6D inhibitor treatment was significantly decreased compared to that of samples without D6D inhibitor treatment (Figure 5B). These results suggested that the observed increase in radiosensitivity in xenograft glioblastoma tumor may be derived from the ability of the D6D inhibitor to suppress the PGE2-ID1 pathway (Scheme 1).
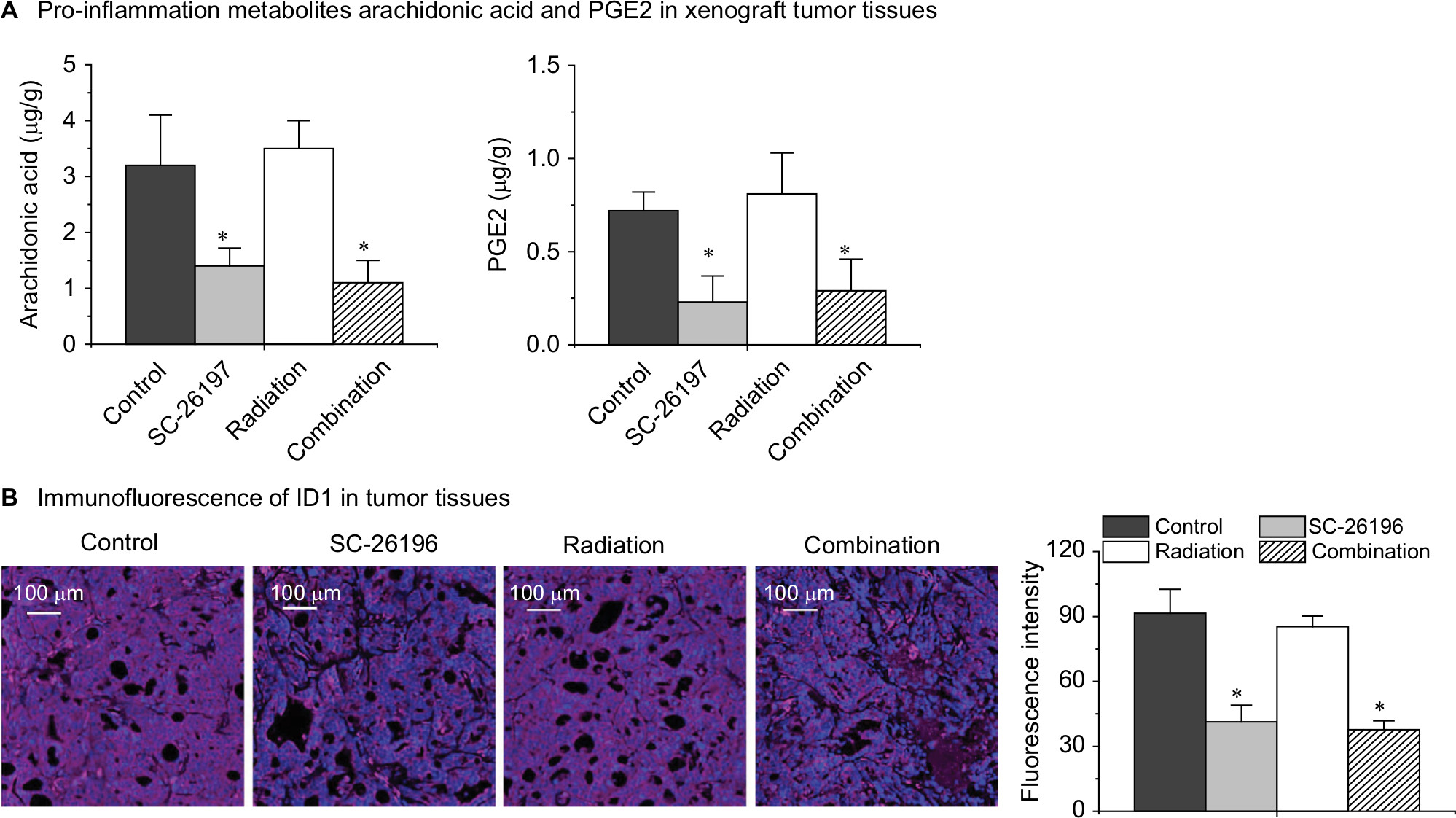 | Figure 5 The D6D inhibitor reversed tumor radioresistance by suppressing the PGE2-ID1 pathway. Notes: (A) HPLC/MS quantification of the pro-inflammation metabolites arachidonic acid and PGE2 in tumor tissues. (B) Representative images for ID1 expression (stained in red) in tumor tissues. Cell nuclei were counter stained with DAPI. For quantification, the fluorescence intensities of ID1 staining were calculated. The images were acquired with a Zeiss Axio Imager M2 microscope with a magnification of 200×. All the quantification data represent the mean ± SD with eight mice per treatment group. *A significant difference vs control with P<0.05. Abbreviations: D6D, delta-6-desaturase; PGE2, prostaglandin E2. |
Discussion
In this study, we have demonstrated that treatment with a D6D inhibitor, SC-26196, could significantly improve radiosensitivity in both glioblastoma cell lines U-87 MG and LN-229. It was also observed that the combination of a D6D inhibitor and radiation synergistically inhibited U-87 MG xenograft tumor growth in association with the induction of tumor apoptosis and suppression of tumor proliferation. Our data suggested that this synergistic effect may be derived from the ability of the D6D inhibitor to inhibit PGE2 production, which eventually limited ID1-dependent radioresistance (Scheme 1).
He et al22 previously reported that treatment with D6D inhibitor alone could inhibit skin cancer and lung cancer growth in a xenograft tumor model, which was probably due to interference with inflammatory pathways (evident by the decreased content of arachidonic acid and its tumor-promoting metabolites). However, in the present animal study, although we observed decreased levels of arachidonic acid and PGE2 in tumor tissues after D6D inhibitor treatment, no significant anti-cancer effect was observed in animals that received the D6D inhibitor treatment alone. A possible reason for that may be that a much lower dosage and frequency of D6D inhibitor treatment was used in our study (30 mg/kg body weight, every third day) compared to the previous report (100 mg/kg body weight/day).
In addition to the PGE2-ID1-dependent mechanism, there are also many other proposed mechanisms that may be responsible for radioresistance in glioblastoma where the inflammatory pathways are involved. For example, Wagemakers et al19 reported that a COX-2 inhibitor could enhance the tumor’s response to radiation by affecting the tumor vasculature; Suzuki et al20 reported that a COX-2 inhibitor enhanced the radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress; and Stanzani et al18 suggested that pro-inflammatory pathways, especially STAT3 and IL6, are major players in the radioresistance of glioblastoma-initiating cells. Therefore, in our future studies, we will focus on a mechanistic investigation to elucidate how the D6D inhibitor and/or blockade of inflammatory pathways could contribute to reversing the radioresistance in glioblastoma. Research outcomes in this area will shed light on how to target inflammatory pathways to overcome radioresistance in glioblastoma therapy.
Conclusion
Here, we demonstrate that by inhibiting D6D, by blocking the synthesis of arachidonic acid and PGE2, could reverse ID1-dependent radioresistance in glioblastoma cells and a xenograft tumor model. However, in the present work, we chose to use readily available commercial cell lines and an easily established subcutaneous xenograft model to initiate our research on this topic, which may not entirely reflect the genotypes and phenotypes of primary tumors. Therefore, in our follow-up studies, we are looking into obtaining primary cell samples and establishing an orthotopic xenograft model, to continue our investigation, which may lead to the development of a potentially novel combination strategy for current glioblastoma therapy.
Acknowledgments
This work was supported by the National Natural Science Fund of the People’s Republic of China (grant 81771304), the National Natural Science Youth Fund of the People’s Republic of China (grants 81601234 and 81601073), and the Health Special Fund of Jilin Province, China (SCZSY201616) and the Science and Technology Innovation Development Fund of Jilin (grant 201750246).
Disclosure
The authors report no conflicts of interest in this work.
References
Kleihues P, Ohgaki H. Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro Oncol. 1999;1(1):44–51. | ||
Ray S, Bonafede MM, Mohile NA. Treatment patterns, survival, and healthcare costs of patients with malignant gliomas in a large US commercially insured population. Am Health Drug Benefits. 2014;7(3):140–149. | ||
Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829–848. | ||
Ozawa T, Holland EC. Rethinking glioma treatment strategy. Oncotarget. 2014;5(20):9532–9533. | ||
Kelley K, Knisely J, Symons M, Ruggieri R. Radioresistance of brain tumors. Cancers (Basel). 2016;8(4):42. | ||
Han X, Xue X, Zhou H, Zhang G. A molecular view of the radioresistance of gliomas. Oncotarget. 2017;8(59):100931–100941. | ||
Mukherjee B, McEllin B, Camacho CV, et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009;69(10):4252–4259. | ||
Kim Y, Kim KH, Lee J, et al. Wnt activation is implicated in glioblastoma radioresistance. Lab Invest. 2012;92(3):466–473. | ||
Narayan RS, Fedrigo CA, Stalpers LJ, Baumert BG, Sminia P. Targeting the Akt-pathway to improve radiosensitivity in glioblastoma. Curr Pharm Des. 2013;19(5):951–957. | ||
Mehta M, Khan A, Danish S, Haffty BG, Sabaawy HE. Radiosensitization of primary human glioblastoma stem-like cells with low-dose AKT inhibition. Mol Cancer Ther. 2015;14(5):1171–1180. | ||
Palumbo S, Tini P, Toscano M, et al. Combined EGFR and autophagy modulation impairs cell migration and enhances radiosensitivity in human glioblastoma cells. J Cell Physiol. 2014;229(11):1863–1873. | ||
Taylor IC, Hütt-Cabezas M, Brandt WD, et al. Disrupting NOTCH slows diffuse intrinsic pontine glioma growth, enhances radiation sensitivity, and shows combinatorial efficacy with bromodomain inhibition. J Neuropathol Exp Neurol. 2015;74(8):778–790. | ||
Carruthers R, Ahmed SU, Strathdee K, et al. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol Oncol. 2015;9(1):192–203. | ||
Gao L, Li F, Dong B, et al. Inhibition of STAT3 and ErbB2 suppresses tumor growth, enhances radiosensitivity, and induces mitochondria-dependent apoptosis in glioma cells. Int J Radiat Oncol Biol Phys. 2010;77(4):1223–1231. | ||
Yuan X, Du J, Hua S, et al. Suppression of autophagy augments the radiosensitizing effects of STAT3 inhibition on human glioma cells. Exp Cell Res. 2015;330(2):267–276. | ||
Chiang MF, Chen HH, Chi CW, et al. Modulation of Sonic hedgehog signaling and WW domain containing oxidoreductase WOX1 expression enhances radiosensitivity of human glioblastoma cells. Exp Biol Med (Maywood). 2015;240(3):392–399. | ||
Besse A, Sana J, Fadrus P, Slaby O. MicroRNAs involved in chemo- and radioresistance of high-grade gliomas. Tumour Biol. 2013;34(4):1969–1978. | ||
Stanzani E, Martínez-Soler F, Mateos TM, et al. Radioresistance of mesenchymal glioblastoma initiating cells correlates with patient outcome and is associated with activation of inflammatory program. Oncotarget. 2017;8(43):73640–73653. | ||
Wagemakers M, van der Wal GE, Cuberes R, et al. COX-2 inhibition combined with radiation reduces orthotopic glioma outgrowth by targeting the tumor vasculature. Transl Oncol. 2009;2(1):1–7. | ||
Suzuki K, Gerelchuluun A, Hong Z, et al. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro Oncol. 2013;15(9):1186–1199. | ||
Cook PJ, Thomas R, Kingsley PJ, et al. Cox-2-derived PGE2 induces Id1-dependent radiation resistance and self-renewal in experimental glioblastoma. Neuro Oncol. 2016;18(10):1379–1389. | ||
He C, Qu X, Wan J, et al. Inhibiting delta-6 desaturase activity suppresses tumor growth in mice. PLoS One. 2012;7(10):e47567. | ||
Lasorella A, Benezra R, Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer. 2014;14(2):77–91. | ||
Ling F, Kang B, Sun XH. ID proteins: small molecules, mighty regulators. Curr Top Dev Biol. 2014;110:189–216. |
Supplementary materials
Materials and methods for supplemental figures
Western blot analysis
Western blot analysis was performed to assess the changes in apoptotic proteins in U-87 MG and LN-229 cells after treatments with SC-26196, radiation, or a combination of both. After exposed to various treatments, the cells were incubated with CelLytic solution (Sigma-Aldrich Co., St Louis, MO, USA) for protein extraction. The same amount of protein from each sample was then loaded into 10% SDS-PAGE gels followed by standard Western blotting procedures.
MTS assay of A172 and U-138 MG cells
The A172 and U-138 MG cell lines were originally purchased from American Type Culture Collection. A172 cells were grown in DMEM supplemented with 10% FBS. U-138 MG cells were grown in Eagle’s minimum essential medium supplemented with 10% FBS. Cells were cultured in an incubator at 37°C containing 95% humidified atmosphere with 5% CO2.
Cell proliferation after treatment with SC-26196, radiation, or a combination of both was assessed using an MTS Cell Proliferation Colorimetric Assay Kit (Abcam). Briefly, A172 and U-138 MG cells were seeded at 5,000 cells per well into 96-well plates and incubated overnight. Then, the cells were exposed to different treatments including SC-26196 alone, X-ray irradiation alone, or a combination of both at various doses. After a 48-hour incubation, 20 µL/well of MTS reagent was added, and the absorbance was measured using a plate reader at 490 nm within 4 hours of incubation. All experiments were repeated at least three times.
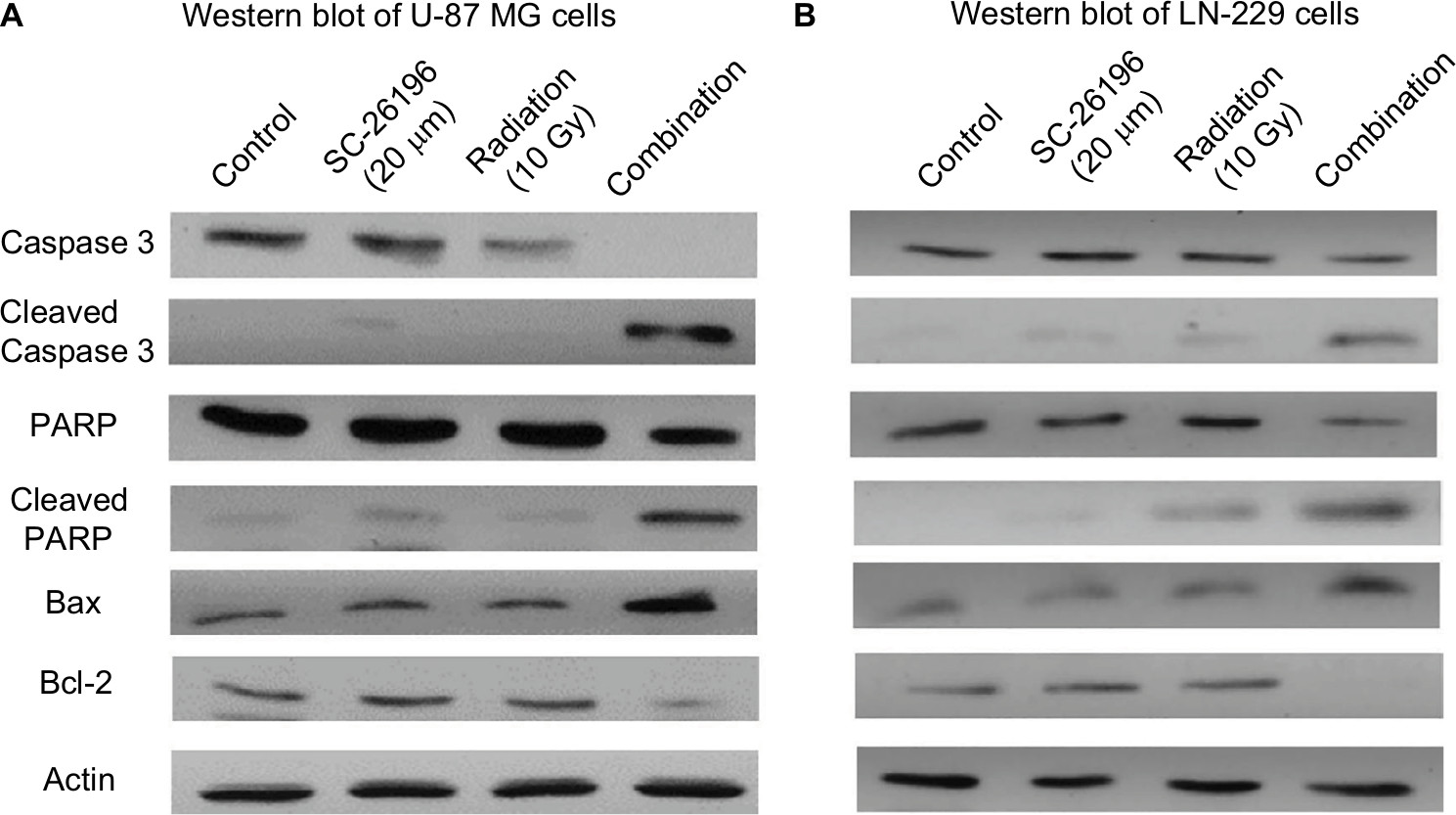 | Figure S1 The combination treatment of D6D inhibitor and radiation induced apoptosis in U-87 MG and LN-229 cells. Notes: (A) Western blot analysis of caspase 3 (pro and cleaved), PARP (pro and cleaved), and Bax and Bcl-2 in U-87 MG cells treated with vehicle control, SC-26196, radiation, or their combination at the indicated doses. β-Actin served as a loading control. (B) Western blot analysis of caspase 3 (pro and cleaved), PARP (pro and cleaved), and Bax and Bcl-2 in LN-229 cells treated with vehicle control, SC-26196, radiation, or their combination at the indicated doses. β-Actin served as a loading control. Abbreviation: D6D, delta-6-desaturase. |
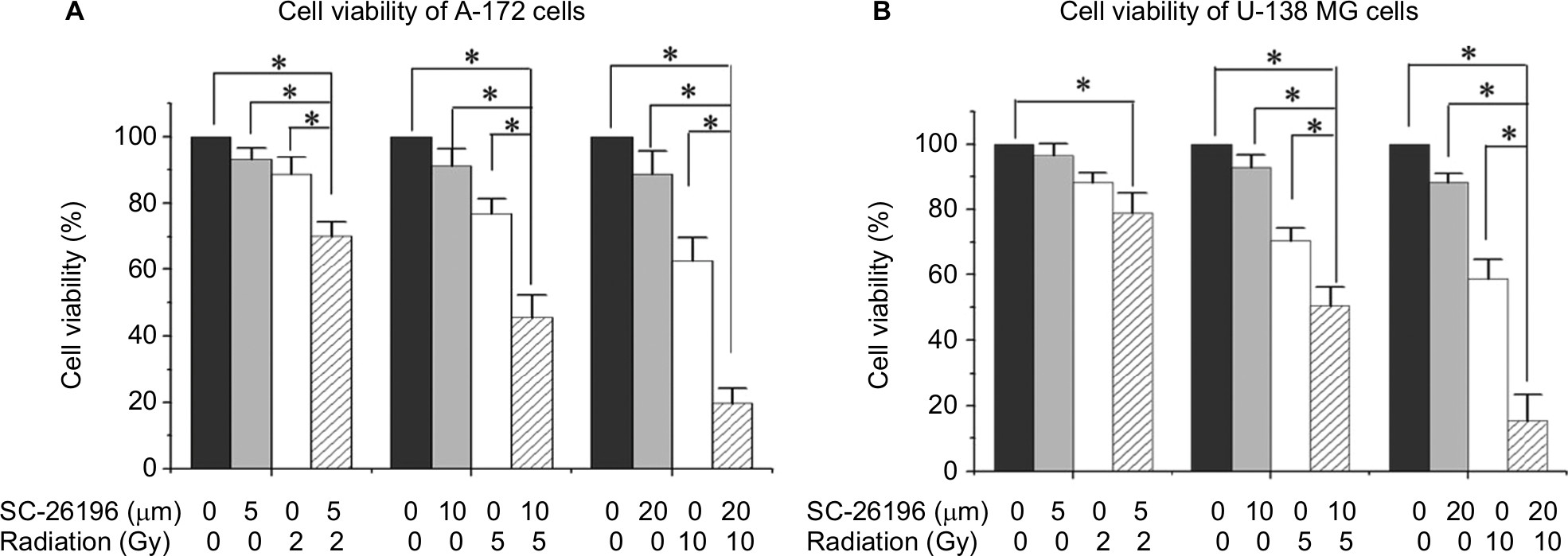 | Figure S2 The D6D inhibitor sensitized A172 and U-138 MG cells to radiation. Notes: (A) MTS assay of A172 cells treated with vehicle control, SC-26196, radiation, or their combination at the indicated doses. (B) MTS assay of U-138 MG cells treated with vehicle control, SC-26196, radiation, or their combination at the indicated doses. All quantification data are represented as the mean ± SD of at least three separate experiments. The data in the control treatment group were normalized to 100%. *A significant difference with P<0.05. Abbreviation: D6D, delta-6-desaturase. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.