")
Back to Journals » Local and Regional Anesthesia » Volume 13
Delayed Emergence from Anesthesia: What We Know and How We Act
Authors Cascella M , Bimonte S , Di Napoli R
Received 9 June 2020
Accepted for publication 1 September 2020
Published 5 November 2020 Volume 2020:13 Pages 195—206
DOI https://doi.org/10.2147/LRA.S230728
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Stefan Wirz
Marco Cascella,1 Sabrina Bimonte,1 Raffaela Di Napoli2
1Division of Anesthesia and Pain Medicine, Istituto Nazionale Tumori – IRCCS – “Fondazione G. Pascale, Naples, Italy; 2Department of Anesthesiology, Institut Jules Bordet, Université Libre De Bruxelles, Bruxelles 1000, Belgium
Correspondence: Marco Cascella
Division of Anesthesia and Pain Medicine, Istituto Nazionale Tumori – IRCCS – “Fondazione G. Pascale, Via Semmola 1. 80100, Naples, Italy
Tel +39 0815903221
Fax +39 0815903778
Email [email protected]
Abstract: The emergence from anesthesia is the stage of general anesthesia featuring the patient’s progression from the unconsciousness status to wakefulness and restoration of consciousness. This complex process has precise neurobiology which differs from that of induction. Despite the medications commonly used in anesthesia allow recovery in a few minutes, a delay in waking up from anesthesia, called delayed emergence, may occur. This phenomenon is associated with delays in the operating room, and an overall increase in costs. Together with the emergence delirium, the phenomenon represents a manifestation of inadequate emergence. Nevertheless, in delayed emergence, the transition from unconsciousness to complete wakefulness usually occurs along a normal trajectory, although slowed down. On the other hand, this awakening trajectory could proceed abnormally, possibly culminating in the manifestation of emergence delirium. Clinically, delayed emergence often represents a challenge for clinicians who must make an accurate diagnosis of the underlying cause to quickly establish appropriate therapy. This paper aimed at presenting an update on the phenomenon, analyzing its causes. Diagnostic and therapeutic strategies are addressed. Finally, therapeutic perspectives on the “active awakening” are reported.
Keywords: general anesthesia, anesthesia emergence, delayed emergence, emergence delirium
Introduction
According to the World Health Organization (WHO) Global Health Estimate, tens of millions of patients undergo general anesthesia (GA) worldwide, each year.1 Interestingly, despite a wealth of studies on the subject, we still do not have a clear picture of the neurobiological mechanisms basis of GA. This fascinating puzzle, however, is gradually being completed. We begin to understand, for example, that induction and emergence (anesthesia emergence, AE) are non-mirror processes.2–4. The ending stage of GA characterized by the progression from the patient’s unconsciousness status to complete wakefulness and restoration of consciousness (RoC) has precise neurobiology which largely differs from that of the induction phase.5–7 In practical terms, knowledge of the impact of pharmacological factors such as general anesthetics, neuromuscular blocking agents (NMBAs), opioids, or non-pharmacological conditions such as hydroelectrolytic alterations, hypothermia, on AE processes could help in understanding, preventing, and managing the multiple complications that can occur at this stage.
On these premises, this paper is aimed at presenting a brief update on the neurobiological mechanisms underlying the AE phase. Among the potential complications that can occur during this phase, particular attention is paid to the phenomenon of the delayed awakening, analyzing its causes, and addressing diagnostic and therapeutic strategies. Moreover, therapeutic perspectives, and the ongoing research designed for establishing the possibility of accelerating awakening from anesthesia, are discussed.
Mechanisms of Anesthesia Emergence
On the molecular level, the effect of general anesthetics is achieved through their action on different types of receptors including gamma-aminobutyric acid type A (GABAA) receptors, N-methyl D-aspartate (NMDA) receptors, α2 receptors, and other receptors such as opioid receptors as well as ion channel anesthetic targets such as members of the neuronal hyperpolarization-activated cyclic nucleotide-gated (HCN) family channels and two-pore domain potassium (K2P) channels. Functionally, the drug-receptor interaction leads to several changes in cortical and subcortical signals, inducing alterations in the connectivity across brain regions. Complex mechanisms underlie alterations of cortico-cortical and cortical-subcortical functional connectivity. The different general anesthetics activate different molecular patterns, expressed as different functional alterations in brain connectivity and different electrophysiological correlates. Volatile agents, for instance, interfere with frontal-posterior connectivity and this effect reverberates on the gamma (20–60 Hz) oscillations which have a pivotal role in arousal and maintenance of consciousness.8 Again, propofol provokes a quick anteriorization of alpha rhythms (8–12 Hz) and promotes the propagations of slow-delta oscillations across the cortex, inducing a functional disruption of the connectivity between distinct cortical areas. Yet, dexmedetomidine impairs the thalamo-cortical functional connectivity mostly expressed as spindle waves (12–16 Hz) in the frontal area.9 The matter is extremely complex, as alterations in connectivity within distinct brain regions lead to different depths of anesthesia. Thus, changes in thalamic-cortical connectivity lead to the induction of the loss of consciousness (LoC) whereas changes in the cortico-cortical functional connectivity and a further impact on cortico-subcortical functioning induce the completion of the induction mechanism and the maintaining of the surgical anesthesia status.
During the AE phase, mechanisms responsible for LoC and anesthesia maintenance are gradually reversed. Nevertheless, these “passive” processes are associated with specific awakening mechanisms. Of note, these active processes include several ascending arousal brain pathways where the thalamus plays a key role.10 Apart from the thalamus, other arousal-promoting brain regions such as the substantia nigra, the ventral and laterodorsal tegmental areas of the midbrain, the dorsal raphe, and the locus ceruleus (LC) as well as the basal forebrain (BF), and lateral hypothalamus are involved. Thus, it has been postulated the existence of a mesencephalic arousal pathway.
The hypothalamus is also implicated in these wake-promoting processes. Orexin also known as hypocretin, is an endogenous wakefulness-promoting substance. Hypothalamic orexinergic neurons are involved in both the sleep-to-wake transition and maintenance of wakefulness.11 This orexinergic system is functionally connected with other structures such as basal ganglia that regulate the awakening processes during the AE. Interestingly, serotonergic neurons in the dorsal raphe nucleus receive projections from orexinergic neurons, concurring in sleep-wakefulness modulation.12
Among these complex arousal networks, there are the LC norepinephrine (LCNE) system and posterior hypothalamic histaminergic tuberomammillary nuclei (TMN). In particular, the LCNE is the main structure of the so-called LCNE arousal system which includes the posterior cingulate cortex, thalamus, and basal ganglia. Another arousal pathway is the brainstem ascending reticular arousal system (ventral and dorsal pathways) which originates from the pontine/midbrain regions and develops cholinergic cortical projections through interactions with the thalamus, hypothalamus, and the BF region. In summary, i) AE is not just a passive process due to a cessation of the action of general anesthetics; ii) the arousal mechanisms at the end of GA are produced by structures deeper in the brain, rather than being activated within the neocortex; iii) rather than a single awakening system, it is more correct to refer to an arousal network. It is composed by the cholinergic basal forebrain, dopaminergic ventral tegmental area, anterior cingulate cortex, orexinergic hypothalamic, serotonergic raphe, LCNE, and histaminergic TMN neurons.
Emergence Complications and Inadequate Emergence
The emergence complications are potentially manifold. They are mostly due to a rising sympathetic tone during this anesthesia stage or by issues concerning airway tone and reflexes. These complications may encompass coughing and potential intracranial and/or intraocular pressures raise, respiratory accidents such as laryngospasm and subsequent oxygenation issues, as well as hemodynamic instability with changes in blood pressure and heart rhythm. Hypertension can lead to bleeding complications that may require urgent new surgery (eg, in neurosurgical setting). Furthermore, up to 20% of GA awareness with recall episodes are attributable to the awakening phase of GA.13
Moreover, emergence complications can manifest as inadequate emergence. This term refers to an alteration of consciousness activity in the first postoperative period. There are two subtypes of inadequate emergence:
- Emergence delirium
- Delayed emergence
The emergence delirium (ED), also termed as emergence agitation or emergence excitement, is characterized by agitation, restlessness, and hyperactivity during the transition from unconsciousness to complete wakefulness. It is a well-known phenomenon in pediatric anesthesia with an estimated incidence of 50–80% in children14 but can occur also in adults, in approximately 6% of GA.15 Although it is often confused with the hyperactive postoperative delirium, this latter occurs after a “lucid interval” from an emergence without cognitive alterations. On the contrary, in the ED the agitation is already present in the RoC phase. The potential association between ED and hyperactive postoperative delirium must be better investigated.16
The other form of inadequate emergence is the delayed emergence (DE) from anesthesia, also termed as hypoactive emergence; the term emergence somnolence is less used. Despite the majority of patients exhaustively respond to stimulation within 60 minutes from the last administration of the anesthetic agents, numerous factors can delay the awakening processes. On these bases, the DE is defined as the inability to regain an adequate level of consciousness, remaining the patient unresponsive or deeply sedated, 30–60 minutes after the end of GA.17,18 Concerning the magnitude of the phenomenon, despite previous investigations indicated an incidence of DE of about 9%,18 to date it is not possible to refer to definitive data on its real incidence.
The two subtypes of inadequate emergence must necessarily be distinct. In the case of delayed awakening, indeed, the transition from unconsciousness to complete wakefulness usually occurs along a normal trajectory, although slowed down. On the other hand, this awakening trajectory could proceed abnormally, possibly culminating in ED.
Clinical Features and Assessment of Delayed Emergence
This manifestation of inadequate emergence is not an uncommon complication of anesthesia and often represents a challenge for clinicians who must make an accurate diagnosis of the underlying cause to quickly establish appropriate therapy. Moreover, although this condition is benign in most cases, rarely delayed awakening may be due to more serious causes such as stroke or anoxic-ischemic brain injury or other neurologic or no-neurologic diseases.
Clinically, DE presents with an altered mental state featuring sedation, lack of initiative, and lack of adequate response to stimuli. It is often associated with respiratory complications, and a lack of protective airway reflexes with an increased risk of aspiration.19 Depression of consciousness can be associated with hypoventilation that, in turn, can lead to progressive acidemia and hypoxia. Moreover, the reduced tone of the pharyngeal and laryngeal structures can lead to an increase in the resistance of the airways with the triggering of obstructive phenomena.
A clinical approach is particularly useful for identifying possible causes. Since there is no dedicated tool for monitoring recovery after anesthesia, the clinical assessment is carried out by referring to the scales commonly adopted for evaluating the state of consciousness in various contexts. To obtain a rapid and reproducible measure of the depth of consciousness, the Glasgow Coma Scale (GCS) is largely used. Although GCS has been developed for the evaluation and prediction of outcomes in traumatic brain injuries, it represents a practical method for assessing the state of consciousness in other settings as well. The GCS calculates the verbal, movement, and visual responses to stimulation. By translating the information provided by the tool into the context of the AE, a GCS < 8 defines a very slow awakening phase and an unconscious patient; on the other hand, a score > 12 is suggestive of awakening. Other scales used for this purpose are the Richmond Agitation-Sedation Scale (RASS) and the Nursing Delirium Screening Scale (Nu-DESC); the latter is suitable for exploring both forms of inadequate emergence, ED and DE.20
Causes of Delayed Emergence
In most cases, a delayed awakening from anesthesia can be attributed to the residual action of one or more anesthetic agents and adjuvants used in the peri-operative period. The list of potentially implicated drugs includes benzodiazepines (BDZs), propofol, opioids, NMBAs, and adjuvants. The prolonged effect of these drugs can be linked to dosage errors or metabolic deficits or other causes. Particular conditions may alter the pharmacological action, even in the absence of a posological error. These conditions must be necessarily considered in the diagnostic process because most of them are progressive and life-threatening if not promptly treated. Hypothermia, hypoglycemia, severe hyperglycemia, electrolyte imbalances such as hypernatremia, and hyponatremia in the peri-operative period can influence the AE lasting. Patient-related factors, such as the use of CNS drugs, severe hypothyroidism, liver disease, hypoalbuminemia, kidney diseases such as uremia are also involved. An inappropriate ventilation technique may involve hyperventilation/hypoventilation with an impact on recovery times through hypocapnia and cerebral vasoconstriction (hyperventilation) or CO2 narcosis (hypoventilation). Finally, surgical or neurological complications such as intraoperative cerebral hypoxia, hemorrhage, embolism or thrombosis have been described.21
Following an attempt to catalog the multiple causes of the complication, pharmacokinetic (PK) conditions (drug disposition) can be distinguished from pharmacodynamic (PD) causes (drug sensitivity). Other causes of DE are linked to metabolic alterations or to rare neurological and psychiatric conditions (Table 1). This proposed nosographic approach has only an exemplary purpose, since, several factors may be simultaneously present, such as an excessive dosage of drugs associated with a metabolic deficit. Further, specific conditions such as age can influence both the pharmacological activity (PD) and the PK processes. Again, the duration of surgery can prolong the awakening from anesthesia by impacting on several factors such as drug accumulation and hypothermia. Finally, because anesthesia is achieved through a combination of drugs, and these drugs, in addition to interacting with each other, can interfere with other drugs taken by the patient, drug interference is an important cause of ED. Of note, these drug interactions can concern both PK, in terms of interferences in one of the different stages (absorption, transport, metabolism, excretion), and PD through synergism, summation, and potentiation phenomena.
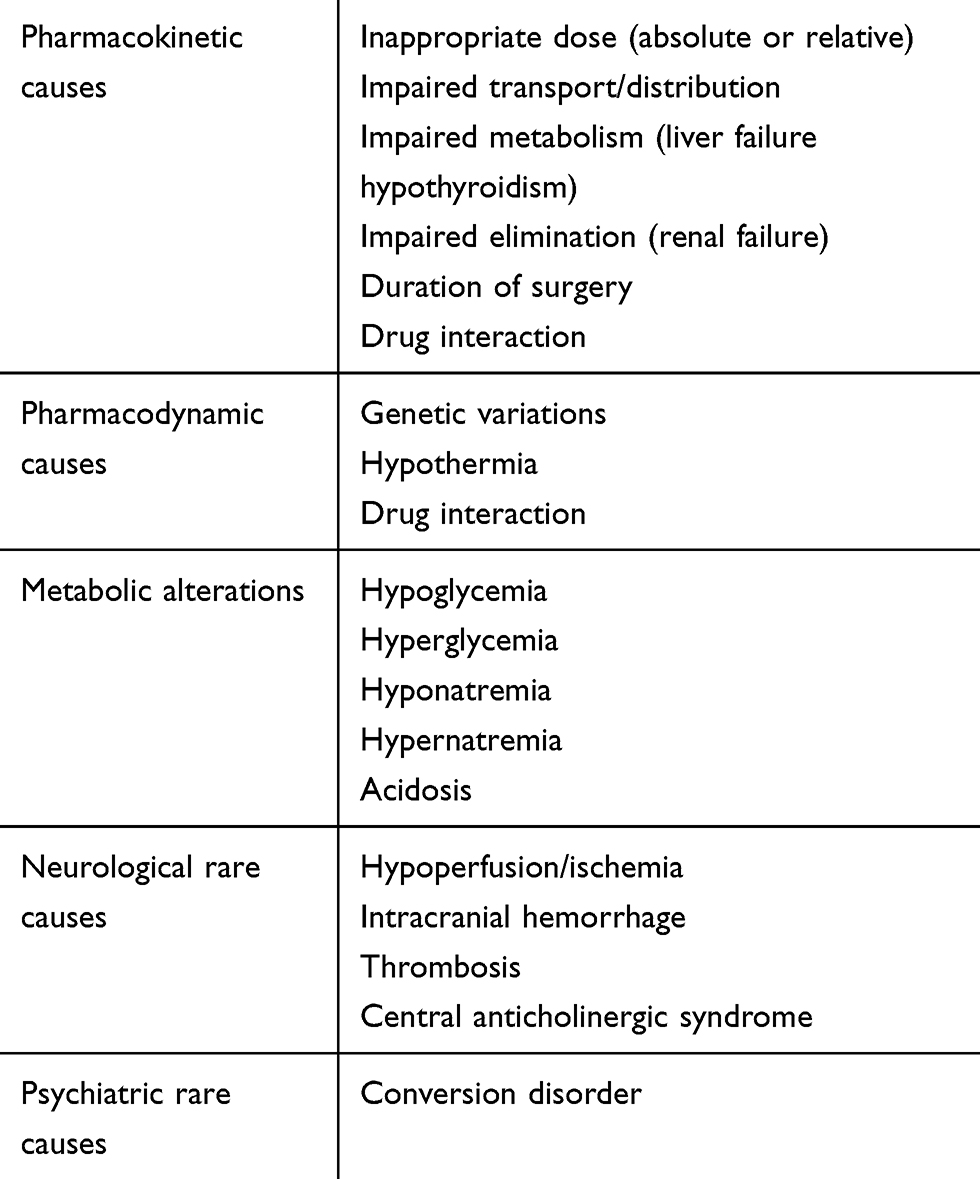 |
Table 1 Causes of Delayed Emergence from Anesthesia |
Pharmacokinetic Causes
In accordance with the principles of PK, once the administration of a drug has ceased, its residual effects are influenced by a series of factors relating to dose, metabolism, blood transport, distribution, and excretion.
The inappropriate dose of drugs represents one of the main causes of delayed awakening from AG. An improper dosage may be due to a malfunction of devices that deliver drugs or to a calculation error. A special issue concerns the body weight. Because a relative drug overdose is common in obese patients who receive a dosage of anesthetic agents based on real weight, the optimal dose for most medications used under anesthesia should be calculated on the lean body weight.
Different conditions can alter the metabolism and elimination of drugs and contribute to DE such as liver or kidney failure. In these patients, the reduced drug metabolism or a reduced clearance leads to an increased duration of action. Moreover, renal failure can impair the protein binding through alteration of the acid-base status. Severe hypothyroidism or can also reduce drug metabolism. The drug interactions between different anesthetic agents used or between anesthesia drugs and medicaments used for chronic pathologies, should be better known. For example, because midazolam is metabolized by the same P450 iso-enzyme as alfentanil, the simultaneous administration of the two medications prolongs the duration of the pharmacological effects of both.22 A case of lengthened recovery from GA due to gabapentin/ketamine interaction was also reported.23
A reduced volume of distribution and plasma protein binding leads to an increased drug-free plasma concentration with an enhancement of the drug effect. This aspect is very important in fragile patients with hypoalbuminemia.
Hypothermia is frequently encountered under anesthesia. It reduces the metabolism and elimination of drugs and enhances the effects of anesthetics. The affinity of hemoglobin for oxygen increases because hypothermia affects both the dissociation of pH and oxygen, contributing to poor tissue oxygenation.
The lowering of body temperatures in the awakening phase increases oxygen consumption leading to an increase in lactate levels. In addition, the reduced perfusion and metabolism of the drugs increase the duration of the action of sedatives, hypnotics, and some neuromuscular relaxants. In particular, an internal temperature of <33 °C also reduces the minimum alveolar concentration (MAC) value of the inhalation agents. The MAC decreases from 5% to 7% for each °C decrease in internal temperature, enhancing sedation, and lengthening the recovery time.24 Hypothermia can also induce direct CNS activity depression.
Pharmacodynamic Causes
The pharmacodynamic effects of general anesthetics and drugs used in anesthesia are generally influenced by the patient’s age. Geriatric patients show increased sensitivity to general anesthetics, opioids, and BDZs due to the progressive decline in brain functioning. Knowledge of this concept must necessarily entail a dosage adjustment of the drugs used for anesthesia. For example, the opioid dosage should be reduced by almost 50% in geriatric patients.25
The effects on recovery time from anesthesia in the elderly are not always related to the dose. For instance, Fredman et al26 evaluated the effect of several doses of midazolam in elderly patients undergoing short urological interventions and demonstrated that midazolam significantly depressed mental function and delayed awakening and discharge from post-anesthesia care unit (PACU) regardless of dose. Regarding other factors potentially involved, a debated argument concerns the gender difference in sensitivity to anesthetics. Buchanan et al27 showed that women have less sensitivity to the hypnotic action of anesthetics with, in turn, faster recovery than men. Again, emergence times were faster in premenopausal women than in postmenopausal ones. Other studies illustrated that women have an increased sensitivity to the non-depolarizing NMBAs whereas males seem to be more sensitive than females to propofol.28 Yet, compared to males, females seem to be more sensitive to opioid receptor agonists. Nevertheless, the question is still clearly to be explained and requires further investigation.
Another issue regards the factors related to the anesthetic drugs and adjuvants used. The pharmacological effect is linked to the type of drug used and the combination of agents in different clinical contexts. Concerning BDZs, while these drugs are commonly used both in premedication and for the induction of AG, the CNS depressing effect, also in terms of duration, is often underestimated. By using BDZ, the effects of propofol are enhanced because both classes of medications act on the gamma-aminobutyric acid (GABA) pathway. Particular attention must be paid in the use of high and repeated doses of BDZs, especially in relation to the duration of the intervention. For instance, although the maximum effect of midazolam occurs in approximately 5 to 10 minutes, it has an elimination half-life of 1.5 to 2.5 hours, with an increase for subsequent doses. Elderly patients are very sensitive and are at a higher risk of overdose problems. Moreover, these drugs, especially when administered with high-dose opioids, can manifest a conspicuous action on respiratory depression, inducing hypercapnia, and delayed awakening. Finally, an overlooked aspect concerns patients who take BDZs on chronic therapy. In this regard, Maeda et al19 proved that chronic BDZ therapy represents an independent predictor of DE.
Propofol has a rapid distribution half-life which leads to rapid awakening from a bolus dose of approximately 8–10 minutes. However, the sensitive half-life — the time needed to reduce the concentration of 50% of the medication at the site of the effect — is dependent on the duration of the infusion. Therefore, the duration of the effect is influenced by the context-sensitive half-life, the amount of drug administered, and the co-administration of other drugs. Target controlled infusion (TCI) systems allow calculating the recovery times of anesthesia when this anesthetic is given by continuous infusion. However, many variables such as hypothermia can alter calculations and there seems to be no difference between TCI and conventional continuous infusion rates on emergence times.
In the case of propofol administration, less predictable variables are patient-related factors. Genetic variations of the enzymes that metabolize propofol, although rare, can be a causative factor in DE. Yonekura et al29 have explored the influence of the genetic polymorphisms of the main enzymes that metabolize propofol — cytochrome P450 2B6 (CYP2B6) and uridine 5′-diphospho-glucuronosyl transferase 1A9 (UGT1A9) — in a patient who had delayed awakening from propofol (from 70 minutes to 3 hours) twice over 2 years. The authors excluded other potential causes and, finally, the genetic analysis confirmed the presence of the variant CYP2B6 516 G/T and UGT1A9 I399 C/C, of the enzymes CYP2B6 (responsible for the hydroxylation of propofol) and UGT1A9 (which catalyzes the glucuronidation of propofol) both involved in propofol metabolism. In particular, CYP2B6 and UGT1A9 are highly polymorphic genes and specific gene variants can reduce the enzymatic activity and, consequently, the degradation of propofol, prolonging the plasma concentrations of propofol.30 Genetic variations in propofol metabolism should be addressed in patients with incomprehensible DE.
In the case of volatile anesthetic agents, AE depends on the pulmonary elimination of the drug and the MAC-awake. This parameter represents the anesthetic concentration associated with suppression of the response to verbal command in 50% of the patients when volatile concentration decreases during the AE. Interestingly, for each volatile agent, the ratio MAC‐awake/MAC can be used for describing the AE: with a ratio approaching 1, the patient will have faster recovery times.31 During the administration of an inhaled agent, after a short period of equilibrium, the MAC directly represents the partial pressure of the anesthetics in the CNS and is independent of the uptake and distribution of the agent in other tissues. According to the pharmacokinetics of halogenates, the awakening time is precisely correlated to alveolar ventilation and inversely associated with the solubility of gases in the blood. Consequently, alveolar hypoventilation lengthens the time taken to exhale the anesthetic and delays the emergency. Moreover, other factors can influence the potency of inhalation anesthetics. For instance, the MAC value decreases with the increasing age of the subject.
Not all halogenated have the same profile as the use of an agent with low blood solubility causes a faster AE. For example, an old comparative study showed that mean times (with 95% confidence interval) from discontinuation of anesthesia administration to the opening of eyes were 8.2 min for sevoflurane (blood solubility of 0.69) and 11.5 min for isoflurane (blood solubility of 1.46).32
Furthermore, the time required for the emergence increases as the duration of the anesthesia increases (ie it increases the context-sensitive half-life, due to the absorption of the tissues). In the case of desflurane, the AE timing seems to be independent of the anesthesia duration if it lasts less than four hours.33 Nevertheless, MAC-awake does not change. Theoretically, indeed, once a patient has emerged from anesthesia performed by volatile agents, it is unlikely that he/she may present a loss of consciousness again due to anesthetic action solely. On the other hand, from a practical point of view, loss of responsiveness after emergence from a volatile anesthetic due to hypoventilation-induced hypercapnia can occur, although rarely.
Opioids produce analgesia, sedation but also respiratory depression, reducing the sensitivity of the brain stem chemoreceptors to carbon dioxide. The effect may result in hypercapnia and delayed awakening, although it varies from subject to subject and is influenced by the concomitant administration of other anesthetic drugs. Respiratory depression responds quickly to opioid antagonists. However, when a short-acting antagonist such as naloxone is administered, the patient should be carefully monitored for the possible recurrence of respiratory depression.
The unrecognized residual neuromuscular block at the end of anesthesia, due to the failure or incorrect use of neuromuscular monitoring, in addition to lengthening the recovery times from anesthesia, represents the main cause of awareness in the awakening phase.13 Residual neuromuscular block can give the appearance of DE in a completely conscious patient. Nevertheless, residual effects of NMBAs can cause hypoventilation that leads to hypoxemia and/or hypercapnia, which can further exacerbate sedation. Again, non-PD conditions can interfere with drug activity. Electrolyte disturbances, for example, cause cell wall hyperpolarization and prolonged blockage while acidosis gives protons to the tertiary amines, increasing the affinity of the receptors. Other factors can interfere with the PK of these drugs. Hypothermia, for example, reduces their metabolism. Again, butyrylcholinesterase (BChE) gene polymers prolong the blockade produced by succinylcholine and mivacurium, reducing their metabolism.34 There are acquired enzyme deficiency forms due to substances and drugs such as methylxanthines (eg, caffeine, theophylline), quinidine, barbiturates, opioids (eg, morphine, and codeine), atropine, epinephrine, phenothiazines, vitamins (eg, folic acid, and vitamin K). Finally, increasing age, pregnancy, severe liver disease, and burn injuries can reduce BChE activity.
Other drugs used during anesthesia, such as clonidine, could lead to ED. However, the evidence is scarce, as only care reports are described in the literature.35 Again, dexmedetomidine could lengthen recovery times from GA especially when given in addition to propofol.36 Concerning the chronic pharmacotherapy, apart from BDZs, other CNS drugs such as barbiturates, anticholinergics, antidepressants, antipsychotics, can be associated with delayed recovery of consciousness after anesthesia. Finally, Makarem et al proved that preoperative drug or alcohol abuse can be associated with a higher risk of DE.37
Metabolic Alterations
Hypoglycemia is one of the most feared complications found in diabetic patients undergoing surgery.38 Because GA masks cognitive dysfunction and the clinical signs of hypoglycemia such as diaphoresis and tachycardia, severe and prolonged hypoglycemia can have serious brain effects. Although hypoglycemia can provoke DE mostly in individuals with a history of poorly controlled diabetes, starvation, or alcohol consumption, the occurrence of hypoglycemia-related DE has been also reported in non-diabetic patients.39
Even severe hyperglycemia can prolong the time of awakening from anesthesia. It causes osmotic diuresis and dehydration. The consequences of dehydration vary from drowsiness to acidosis. Moreover, blood hyperosmolality and hyperviscosity predispose to thrombosis and cerebral edema.
Although mild hyponatremia is generally asymptomatic, a serum sodium concentration <120 mEq/L causes confusion and irritability. Preoperative hyponatremia (sodium level <135 mEq/L) is associated with a higher risk of perioperative major coronary events and 30-day mortality.40 Serum sodium concentration <110 mEq/L leads to seizures, coma, and a rise in mortality. The causes of intraoperative hyponatremia are manifold. Inappropriate antidiuretic hormone secretion (SIADH) can result from brain trauma, subarachnoid hemorrhage, and drug administration (eg, opioids, haloperidol, vasopressin). Hyponatremia may be secondary in the patient with brain injury and prolonged use of mannitol. Fluid overload and hyponatremia with hypo-osmolarity can develop when extensive volumes of irrigation fluid such as glycine solution are absorbed by open venous sinuses during transurethral resection of the prostate with the potential occurrence of pulmonary edema, and cerebral edema (ie, TURP syndrome). Under anesthesia, hyponatremia delays emergence through the development of cerebral edema and uncompensated plasma osmotic changes.
Hypernatremia (sodium levels > 145 mEq/L) can be caused by a fluid volume deficit, disproportionate sodium intake, kidney failure, or other diseases. Severe hypernatremia (sodium levels > 152 mEq/L) is less likely to develop in the postoperative period. Clinically, excess sodium causes cellular dehydration with brain dehydration. Symptoms include thirst, drowsiness, confusion, and coma.
Hypocalcemia, severe hypercalcemia, and hypermagnesemia are further potential metabolic alterations capable of inducing CNS depression.
Neurological and Psychiatric Rare Causes
Prolonged hypoxemia periods can lead to hypoxic damage of varying magnitude. Ischemic episodes are often the result of failing brain perfusion secondary to low mean arterial pressure (MAP) which escapes brain self-regulation mechanisms (between 60 and 160 mmHg MAP). Of note, carotid surgery and seated operations present a high risk of hypoperfusion. In addition to damage from hypoperfusion/ischemia, intracranial hemorrhage, thrombosis or cerebral infarction can occur in association with intraoperative arrhythmias (eg, cerebral thromboembolism due to atrial fibrillation or flutter), or with severe changes in blood pressure in terms of hypo or hypertension.
Central anticholinergic syndrome is a rare syndrome precipitated by drugs or substances that increase the release of serotonin (levodopa, and carbidopa-levodopa association, amphetamines and derivatives, cocaine), impair its reuptake (selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, tricyclic antidepressants, valproate, carbamazepine, methadone, meperidine, tramadol, pentazocine, metoclopramide, St. John’s wort), increased sensitivity of postsynaptic receptors (lithium), or act as direct serotonin agonists (triptans, lysergic acid diethylamide). Atropine, scopolamine, and antihistamines (promethazine) can also be implicated. Furthermore, Cohen et al41 described a case of a 32-year-old woman sedated (2 mg of midazolam and 30 mg of propofol) for a minor surgery who developed the syndrome and DE and was successfully treated with 2 mg of physostigmine. In brief, the syndrome is believed to be due to a decrease in inhibitory anticholinergic activity in the brain and usually manifests when 2 or more serotonergic drugs are used simultaneously. Symptoms and signs are confusion, agitation, hallucinations, dilated pupils, and peripheral anticholinergic effects such as tachycardia, blurred vision, dry mouth, diaphoresis, and urinary retention. Severe forms are characterized by muscle rigidity, hyperthermia and multi-organ failure. Delayed awakening from anesthesia is possible. Of note, the symptoms are quickly reversed by physostigmine but can reappear when its effect stops; thus, multiple doses may be required.
Local anesthetics used for subarachnoid anesthesia, or through the epidural route, can be distributed intracranially, causing a series of complications ranging from seizures to cardiovascular depression to cardiac arrest. A delay in recovery from anesthesia with drowsiness and numbness, up to a manifest state of coma, can occur.42
In very rare cases the delay in awakening is not attributable to organic conditions, being an expression of psychiatric manifestations such as conversion disorder which manifests with neurological symptoms (eg, paralysis), despite the absence of an obvious organic cause.43
Operational Strategies
Given the difficulty of estimating the true incidence and the multiple etiologies of the DE, prevention by means of accurate intra-operative monitoring systems, combined with an appropriate operational strategy such as prevention of hypothermia, is fundamental. Regarding intraoperative monitoring, neuromuscular monitoring (quantitative > qualitative) is of paramount importance whereas brain monitoring of anesthesia through processed EEG-based devices can be particularly useful in the awakening phase, although a difficult to quantify disturbance presumably produced by sub-cortical sleep-wake cycle regulating systems can affect their reliability during AE.44
The therapeutic interventions include, first of all, the control of mechanical ventilation (to evaluate the end-tidal CO2 levels) if the patient is still intubated, oxygenation, and hemodynamic stability (ABC check) (Figure 1). Verification of the patient’s body temperature must necessarily be included in this first assessment.
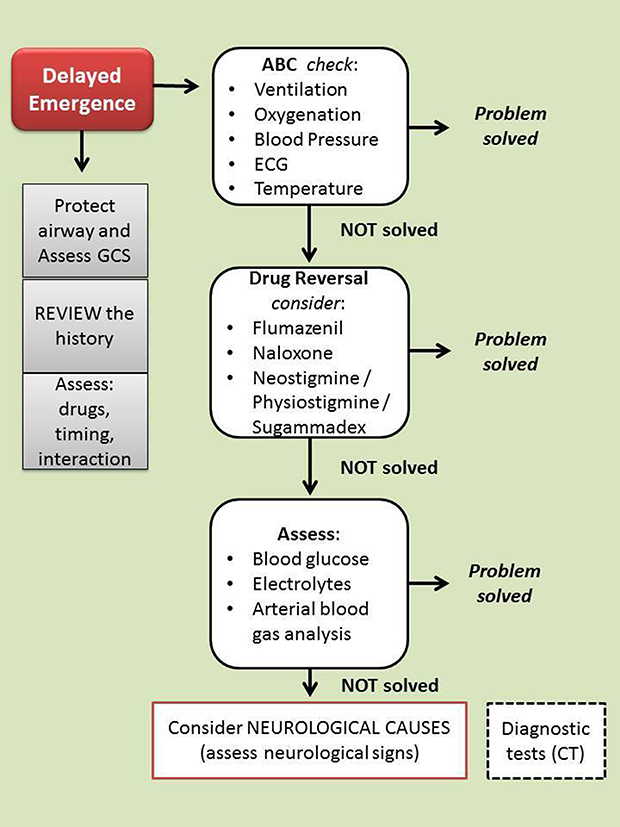 |
Figure 1 Algorithm for delayed emergence management. In the case of delayed awakening, it is advisable to protect the airways and evaluate the level of consciousness (with any associated neurological signs). At the same time, it is necessary to begin to evaluate (and correct) the possible causes. In succession the most frequent causes must be checked (eg, dosage error, or residual effect of drugs). When no cause is clearly identifiable, it is appropriate to decide to carry out further diagnostic tests (eg CT scan). Legend: GCS, Glasgow Coma Scale; CT, Computed Tomography. |
A physical exam is also mandatory. It is aimed at evaluating the features of spontaneous ventilation, patient’s pupil size (although several medications may affect pupillary reactions), spontaneous or induced movements, presence or absence of reflexes (cough and gag reflexes), and response to external stimuli such as verbal input, and tactile stimulation. The neurological examination may show decerebration or decortication, or focal signs due to regional ischemia.
At the same time, it is necessary to reassess the medical history, examining the medical record and all the information that may be useful such as drugs, timing, and interaction. In this context, potential risk factors such as older age, body habitus (obese and underweight patients), history of hypertension, preexisting psychological disorders,37 or neurologic conditions such as seizures or cerebral vascular disorders or rare neurologic disorders (eg, idiopathic hypersomnia) must be evaluated. This rapid check must include preexisting cardiac and pulmonary disease, renal or hepatic disease (eg, cirrhosis-induced hepatic encephalopathy), subclinical or clinical hypothyroidism, daily medication with BDZs and/or barbiturates, other CNS drugs as well as substance dependence, and metabolic alterations. Perioperative use of herbal medications — not discontinued 2 to 3 weeks prior to surgery — such as St. John’s wort, valerian root, and kava kava should be investigated.
The anesthesia record must be reviewed. In particular, the intraoperative administration of drugs (eg, opioids), general anesthetics administration, and any unexpected intraoperative event must be evaluated. It should be considered that males are probably more likely to have prolonged awakening as women have faster recovery times than men.27 (Table 2).
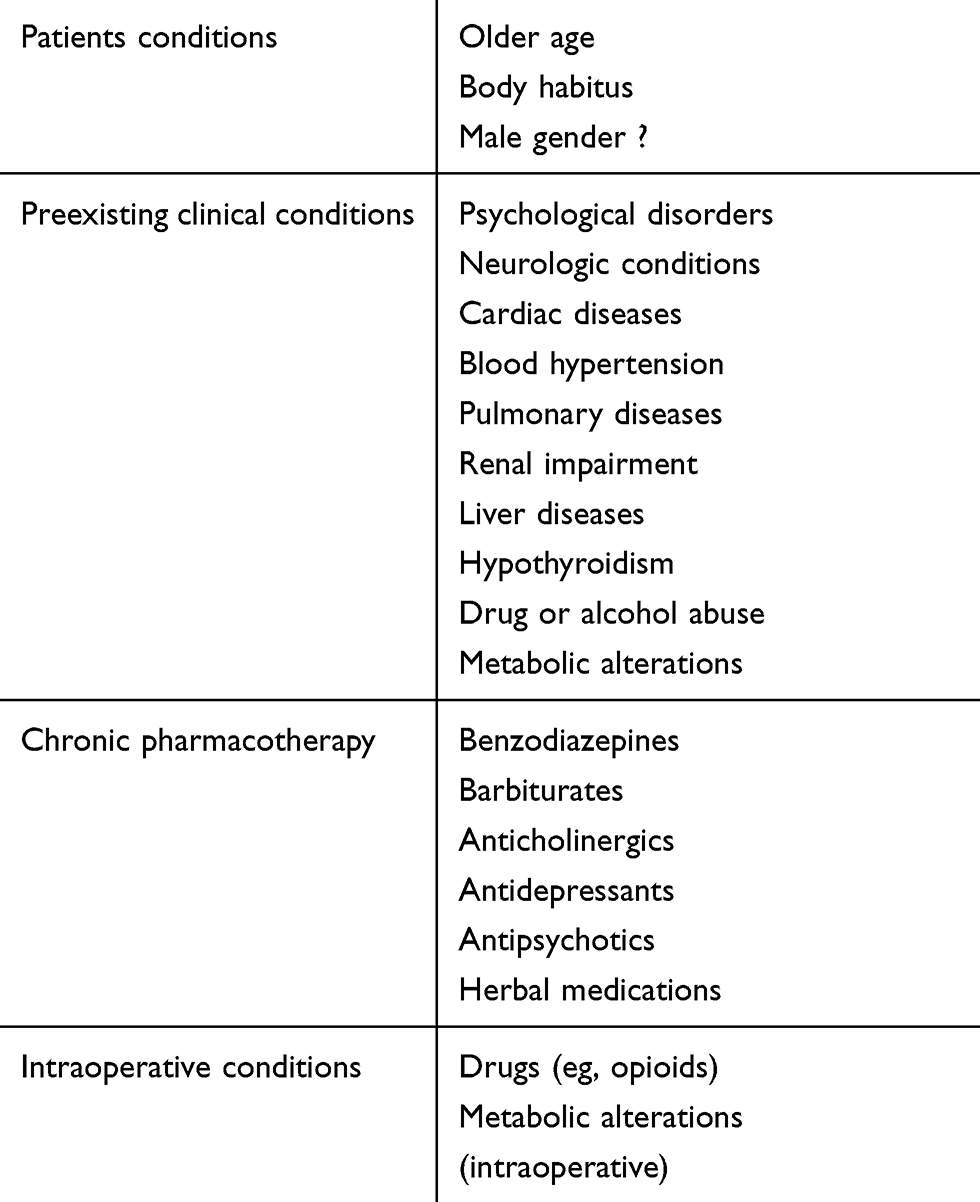 |
Table 2 Risk Factors for Delayed Emergence from Anesthesia |
The airway protection is mandatory and the assessment of the state of consciousness can be carried out with tools such as the GCS.
If the problem has not been resolved, the use of antagonist drugs such as naloxone (eg,0.04 mg IV q 2 mins, up to 2mg), flumazenil (eg, 0.2–1 mg IV q 1 min, up to 1mg), neostigmine (or physostigmine 0.5 to 1 mg IV) or sugammadex, this latter for the reversal of the residual neuromuscular block from rocuronium, should be considered. In the case of residual blockage, it is advisable to continue sedation and supportive therapy for the period of time necessary to eliminate the blockage. When anesthesia was performed through a continuous infusion of propofol, it is appropriate to check the correctness of the infusion plan. Thiamine (100 mg IV) can be used for the management of alcohol withdrawal.
Although in most cases the complication is solved in this step, the problem can still persist. Other potential causes, referring to metabolic issues, must be searched. In this regard, a sample for acid-base balance can offer important information, guiding the most appropriate treatment. In very rare cases the problem can persist and the rarest possible causes such as neurologic conditions must be examined. At this stage, it is appropriate to decide to carry out further diagnostic tests and the patient should be transported for urgent computed tomography.
Perspectives
The emergence from the GA is commonly considered as a slow and passive process in which, at the end of the surgery, the administration of intravenous and/or inhalation anesthetic drugs is suspended and this determines the RoC. Recent studies suggest that induction and awakening are asymmetric processes.4–7 In fact, the neural circuits that mediate induction do not completely overlap those that mediate the AE. The better understanding of these two processes has allowed researchers to start new studies focused on the actively induced transition from unconsciousness to awakening. Although data are not conclusive, the prospects seem to be interesting.
Methylphenidate is a dopamine uptake inhibitor tested for this aim. Preclinical studies in rats have shown that it can induce awakening from GA during a continuous TCI of propofol.45 A randomized clinical trial (RCT) was conducted at Ohio State University (NCT02327195). Methylphenidate (20 mg orally, 2 hours prior to surgery) was compared to placebo in adults undergoing isoflurane anesthesia. Although the recruitment status of this investigation is indicated as completed (n = 54), to date the results have not been published. Another RCT was conducted at Massachusetts General Hospital (NCT02051452). Patients were randomized to receive methylphenidate (n = 16) or placebo (n = 16). The study is listed as finished and we are waiting for published data. Another clinical study was scheduled to evaluate the drug in healthy volunteers, but it was closed prematurely (NCT02429076).
Another medication is modafinil. It is a long-acting wakefulness-promoting agent used in the treatment of somnolence from obstructive sleep apnea (OSA). Although its precise mechanism of action is still to be characterized, it is probably linked to an increase in monoaminergic transmission combined with an enhanced GABAergic inhibition. Studies conducted in OSA patients evaluated the PACU length of stay rather than the emergence time and, however, showed no advantages in the intervention group (200 mg of modafinil before anesthesia) compared to placebo.46
In preclinical investigations, caffeine also offered important prospects for accelerating awakening from anesthesia.47 In a recent RCT, Fong et al demonstrated that in healthy individuals (n = 8) anesthetized twice with 1.2% isoflurane for 1 hour, the intravenous administration of caffeine citrate (15 mg/kg) during the final 10 minutes of each session, induced a significant (p =0.002) reduction in awakening times (42%) compared to placebo.48 Although other studies on the subject are ongoing (NCT03577730), the results have not yet been posted.
Other compounds that could be used are dextroamphetamine and atomoxetine, physostigmine, and nicotine. However, only preclinical evidence is currently available.49–51
The research aimed at identifying strategies for active awakening must necessarily overcome a fundamental gap represented by the evidence that mechanisms that regulate the emergence phase are manifold. Probably, acting through different agents strengthening different arousal networks could lead to more satisfying results.
Conclusions
The delayed recovery of consciousness during anesthesia constitutes an important challenge for anesthesiologists who must quickly evaluate the possible causes and implement therapeutic strategies. Pending drugs that can promote “active” awakening, prevention is the best strategy, using intraoperative monitoring and designing safe anesthesia. When the complication has occurred, a careful evaluation of the possible causes can allow a rapid resolution of the problem. In particularly rare cases, because the problem can be due to a serious neurological injury, a diagnostic deepening is necessary. Finally, further research is needed for evaluating the effectiveness of drugs that can actively induce awakening from anesthesia.
Acknowledgments
The authors thank Dr Alessandra Trocino, Librarian at the Library of NCI G. Pascale Foundation of Naples, Italy, for her excellent bibliographic service and assistance.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rose J, Weiser TG, Hider P, Wilson L, Gruen RL, Bickler SW. Estimated need for surgery worldwide based on prevalence of diseases: a modelling strategy for the WHO Global Health Estimate. Lancet Glob Health. 2015;3:
2. Kelz MB, Sun Y, Chen J, et al. An essential role for orexins in emergence from general anesthesia. Proc Natl Acad Sci U S A. 2008;105(4):1309–1314. doi:10.1073/pnas.0707146105
3. Cascella M, Bimonte S, Muzio MR. Towards a better understanding of anesthesia emergence mechanisms: research and clinical implications. World J Methodol. 2018;8:9–16.
4. Tarnal V, Vlisides PE, Mashour G. The Neurobiology of Anesthetic Emergence. J Neurosurg Anesthesiol. 2016;28:250–255.
5. Cascella M. Emergence from anesthesia: a winding way back. Anaesthesiol Intensive Ther. 2018;50:168–169.
6. Ferreira AL, Correia R, Vide S, et al. Patterns of Hysteresis Between Induction and Emergence of Neuroanesthesia Are Present in Spinal and Intracranial Surgeries. J Neurosurg Anesthesiol. 2020;32:82–89.
7. Chen L, Li S, Zhou Y, et al. Neuronal mechanisms of adenosine A2A receptors in the loss of consciousness induced by propofol general anesthesia with functional magnetic resonance imaging (fMRI) [published online ahead of print, 2020 Aug 12]. J Neurochem. 2020;10.
8. Imas OA, Ropella KM, Ward BD, Wood JD, Hudetz AG. Volatile anesthetics disrupt frontal-posterior recurrent information transfer at gamma frequencies in rat. Neurosci Lett. 2005;387:145–150.
9. Akeju O, Loggia ML, Catana C, et al. Disruption of thalamic functional connectivity is a neural correlate of dexmedetomidine-induced unconsciousness. Elife. 2014;3:e04499. doi:10.7554/eLife.04499
10. Scheib CM. Brainstem Influence on Thalamocortical Oscillations during Anesthesia Emergence. Front Syst Neurosci. 2017;11:66. doi:10.3389/fnsys.2017.00066
11. de Lecea L, Huerta HR. Hypocretin (orexin) regulation of sleep-to-wake transitions. Front Pharmacol. 2014;5:16. doi:10.3389/fphar.2014.00016
12. Yang C, Zhang L, Hao H, Ran M, Li J, Dong H. Serotonergic neurons in the dorsal raphe nucleus mediate the arousal-promoting effect of orexin during isoflurane anesthesia in male rats. Neuropeptides. 2019;75:25–33. doi:10.1016/j.npep.2019.03.004
13. Cascella M, Bimonte S, Amruthraj NJ. Awareness during emergence from anesthesia: features and future research directions. World J Clin Cases. 2020;8(2):
14. Moore AD, Anghelescu DL. Erratum to: emergence Delirium in Pediatric Anesthesia. Paediatr Drugs. 2017;19(3):267. doi:10.1007/s40272-017-0227-3
15. Xará D, Silva A, Mendonça J, Abelha F. Inadequate emergence after anesthesia: emergence delirium and hypoactive emergence in the Postanesthesia Care Unit. J Clin Anesth. 2013;25(6):
16. Evered L, Silbert B, Knopman DS, et al. Recommendations for the nomenclature of cognitive change associated with anaesthesia and surgery-2018. Br J Anaesth. 2018;121(5):
17. Pavlin DJ, Rapp SE, Polissar NL, Malmgren JA, Koerschgen M, Keyes H. Factors affecting discharge time in adult outpatients. Anesth Analg. 1998;87:816–826. doi:10.1097/00000539-199810000-00014
18. Zelcer J, Wells DG. Anaesthetic-Related Recovery Room Complications. Anaesth Intens Care. 1987;15(2):168–174. doi:10.1177/0310057X8701500209
19. Maeda S, Tomoyasu Y, Higuchi H, Ishii-Maruhama M, Egusa M, Miyawaki T. Independent predictors of delay in emergence from general anesthesia. Anesthesia Progress. 2015;62(1):
20. Wiinholdt D, Eriksen SAN, Harms LB, et al. Inadequate emergence after non-cardiac surgery—A prospective observational study in 1000 patients. Acta Anaesthesiol Scand. 2019;63(9):1137–1142. doi:10.1111/aas.13420
21. Tzabazis A, Miller C, Dobrow MF, Zheng K, Brock-Utne JG. Delayed emergence after anesthesia. J Clin Anesth. 2015;27(4):353–360. doi:10.1016/j.jclinane.2015.03.023
22. Kissin I, Vinik HR, Castillo R, Bradley EL. Alfentanil potentiates midazolam-induced unconsciousness in subanalgesic doses. Anesth Analg. 1990;71(1):
23. Elyassi AR, Long RP, Bejnarowicz RP, Schoneboom BA. Possible gabapentin and ketamine interaction causing prolonged central nervous system depression during post-operative recovery following cervical laminoplasty: a case report. J Med Case Rep. 2011;5(1):167. doi:10.1186/1752-1947-5-167
24. Liu M, Hu X, Liu J. The effect of hypothermia on isoflurane MAC in children. Anesthesiology. 2001;94(3):
25. Bowie MW, Slattum PW. Pharmacodynamics in older adults: A review. Am J Geriatr Pharmacother. 2007;5(3):263–303. doi:10.1016/j.amjopharm.2007.10.001
26. Fredman B, Lahav M, Zohar E, Golod M, Paruta I, Jedeikin R. The effect of midazolam premedication on mental and psychomotor recovery in geriatric patients undergoing brief surgical procedures. Anesth Analg. 1999;89(5):1161–1166. doi:10.1213/00000539-199911000-00014
27. Buchanan FF, Myles PS, Leslie K, Forbes A, Cicuttini F. Gender and recovery after general anesthesia combined with neuromuscular blocking drugs. Anesth Analg. 2006;102(1):291–297. doi:10.1213/01.ANE.0000181321.55422.C6
28. Campesi I, Fois M, Franconi F. Sex and gender aspects in anesthetics and pain medication. Handb Exp Pharmacol. 2012;214:
29. Yonekura H, Murayama N, Yamazaki H, Sobue KA. Case of Delayed Emergence After Propofol Anesthesia: genetic Analysis. AA Case Rep. 2016;7(11):
30. Kansaku F, Kumai T, Sasaki K, et al. Individual differences in pharmacokinetics and pharmacodynamics of anesthetic agent propofol with regard to CYP2B6 and UGT1A9 genotype and patient age. Drug Metab Pharmacokinet. 2011;26:532–537.
31. Aranake A, Mashour GA, Avidan MS. Minimum alveolar concentration: ongoing relevance and clinical utility. Anaesthesia. 2013;68:512–522. doi:10.1111/anae.12168
32. Ebert TJ, Robinson BJ, Uhrich TD, Mackenthun A, Pichotta PJ. Recovery from sevoflurane anesthesia: a comparison to isoflurane and propofol anesthesia [published correction appears in Anesthesiology 1999 Feb;90(2):644]. Anesthesiology. 1998;89(6):1524–1531. doi:10.1097/00000542-199812000-00032
33. Lin TC, Lu CC, Hsu CH, Wu GJ, Lee MS, Ho ST. Duration effect of desflurane anesthesia and its awakening time and arterial concentration in gynecologic patients. Clinics. 2013;68(10):
34. Thomsen JL, Nielsen CV, Eskildsen KZ, Demant MN, Gätke MR. Awareness during emergence from anesthesia: significance neuromuscular monitoring in patients with butyrylcholinesterase deficiency. Br J Anaesth. 2015;115:Si78–Si88.
35. Swift A, Wilson M. Reversal of the effects of clonidine using naloxone. Anaesth Rep. 2019;7:4–6.
36. Ohtani N, Kida K, Shoji K, Yasui Y, Masaki E. Recovery profiles from dexmedetomidine as a general anesthetic adjuvant in patients undergoing lower abdominal surgery. Anesth Analg. 2008;107:1871–1874. doi:10.1213/ane.0b013e3181887fcc
37. Makarem J, Larijani AH, Eslami B, Jafarzadeh A, Karvandian K, Mireskandari SM. Risk factors of inadequate emergence following general anesthesia with an emphasis on patients with substance dependence history. Korean J Anesthesiol. 2020;73:302–310. doi:10.4097/kja.19214
38. Kalra S, Bajwa SJ, Baruah M, Sehgal V. Hypoglycaemia in anesthesiology practice: diagnostic, preventive, and management strategies. Saudi J Anaesth. 2013;7(4):
39. Khare A, Meena S, Sethi P, Bafna U, Gill N. Delayed Recovery From General Anaesthesia due to Severe Hypoglycemia in a Non-diabetic Adult. J Coll Physicians Surg Pak. 2015;25(8):627.
40. Leung AA, McAlister FA, Rogers SO, Pazo V, Wright A, Bates DW. Preoperative Hyponatremia and Perioperative Complications. Arch Intern Med. 2012;172(19):1474–1481. doi:10.1001/archinternmed.2012.3992
41. Cohen S, Hunter CW, Yanni B, Striker P, Hijazi RH. Central anticholinergic syndrome strikes again. J Clin Anesth. 2006;18:399–400.
42. Frost EA. Differential diagnosis of delayed awakening from general anesthesia: a review. Middle East J Anaesthesiol. 2014;22(6):537–548.
43. Han D, Connelly NR, Weintraub A, Kanev P, Solis E. Conversion locked-in syndrome after implantation of a spinal cord stimulator. Anesth Analg. 2007;104:163–165. doi:10.1213/01.ane.0000250363.40356.b8
44. Numan T, Slooter AJC, van der Kooi AW, et al. Functional connectivity and network analysis during hypoactive delirium and recovery from anesthesia. Clin Neurophysiol. 2017;128:914–924. doi:10.1016/j.clinph.2017.02.022
45. Chemali JJ, Van Dort CJ, Brown EN, Solt K. Active emergence from propofol general anesthesia is induced by methylphenidate. Anesthesiology. 2012;116(5):
46. Carr ZJ, Vells B, Wood BR, et al. A double blind randomized placebo controlled pilot study of single-dose preoperative modafinil for functional recovery after general anesthesia in patients with obstructive sleep apnea. Medicine. 2018;97(39):e12585. doi:10.1097/MD.0000000000012585
47. Wang Q, Fong R, Mason P, Fox AP, Xie Z. Caffeine accelerates recovery from general anesthesia. J Neurophysiol. 2014;111:1331–1340. doi:10.1152/jn.00792.2013
48. Fong R, Wang L, Zacny JP, et al. Caffeine Accelerates Emergence from Isoflurane Anesthesia in Humans: A Randomized, Double-blind, Crossover Study. Anesthesiology. 2018;129:912–920. doi:10.1097/ALN.0000000000002367
49. Kenny JD, Taylor NE, Brown EN, Solt K. Dextroamphetamine (but Not Atomoxetine) Induces Reanimation from General Anesthesia: implications for the Roles of Dopamine and Norepinephrine in Active Emergence. PLoS One. 2015;10(7):e0131914.
50. Alkire MT, McReynolds JR, Hahn EL, et al. Thalamic microinjection of nicotine reverses sevoflurane-induced loss of righting reflex in the rat. Anesthesiology. 2007;107:264–272.
51. Kenny JD, Chemali JJ, Cotten JF, et al. Physostigmine and Methylphenidate Induce Distinct Arousal States During Isoflurane General Anesthesia in Rats. Anesth Analg. 2016;123:1210–1219. doi:10.1213/ANE.0000000000001234
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.