")
Back to Journals » Journal of Blood Medicine » Volume 10
Dehydrated hereditary stomatocytosis: clinical perspectives
Authors Frederiksen H
Received 30 March 2019
Accepted for publication 14 June 2019
Published 4 July 2019 Volume 2019:10 Pages 183—191
DOI https://doi.org/10.2147/JBM.S179764
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Henrik Frederiksen
Department of Haematology, Odense University Hospital, Odense, Denmark
Abstract: Dehydrated hereditary stomatocytosis (DHSt) is a nonimmune congenital hemolytic disorder characterized by red blood cell (RBC) dehydration and lysis. It has been a recognized diagnostic entity for almost 50 years, and autosomal dominant inheritance has long been suspected, but it was not until 2011 that the first genetic alterations were identified. The current study reviews 73 articles published during 1971–2019 and focuses on clinical perspectives of the disease. All but one of the published clinical data in DHSt were either single case reports or case series. From these, it can be seen that patients with DHSt often have fully or partially compensated hemolysis with few symptoms. Despite this, iron overload is an almost universal finding even in patients without or with only sporadic blood transfusions, and this may lead to organ dysfunction. Other severe complications, such as thrombosis and perinatal fluid effusions unrelated to fetal hemoglobin concentration, may also occur. No specific treatment for symptomatic hemolysis exists, and splenectomy should be avoided as it seems to aggravate the risk of thrombosis. Recently, treatment with senicapoc has shown activity against RBC dehydration in vitro; however, it is not known if this translates into relevant clinical effects. In conclusion, despite recent advances in the understanding of pathophysiology in DHSt, options for clinical management have not improved. Entering data into international registries has the potential to fill gaps in knowledge and eventually care of these rare patients.
Keywords: xerocytosis, hemolysis, iron overload, perinatal, thrombosis, review
Introduction
Congenital red blood cell (RBC) disorders are the most prevalent hereditary diseases worldwide, where chronic hemolysis is a leading or contributing cause of symptoms and complications. Disorders of the RBC membrane encompass a major subgroup of congenital RBC disorders where chronic hemolysis that may lead to anemia can have profound consequences for patients. However, even when chronic hemolysis is partially or fully compensated by increased erythropoiesis in the bone marrow, the accelerated RBC destruction and the underlying disorder may have important health consequences. Dehydrated hereditary stomatocytosis (DHSt), also designated hereditary xerocytosis, is a congenital hemolytic disorder with a distinct, albeit variable, phenotype. The disorder was first described in 1971,1 and by transmission pattern in affected families, an autosomal dominant inheritance was suspected.2,3 In 2012 and 2013, three groups almost simultaneously mapped the genetic alterations to the PIEZO1 gene4.–6 PIEZO1 mutations are found on chromosome 16 in the majority of patients, mainly located in the highly conserved COOH end of the gene.3–6 Although patients rarely suffer from severe anemia or dependency of regular transfusions,3,7 recognition of the DHSt diagnosis is crucial for acknowledgment of immediate and future complications, for advisable and contraindicated management, and for planning of adequate follow-up, starting already in utero. This literary review focuses on clinical aspects of the DHSt diagnosis to facilitate the correct diagnosis and for contemporary clinical management of patients.
Materials and methods
The relevant literature for this literary review was identified via the Pubmed database using the search term Dehydrated Stomatocytosis OR Xerocytosis. The literature search was conducted 21 September, 2018 and resulted in 94 hits – all of which were retrieved and reviewed. Reference lists from the included studies were also reviewed and resulted in the inclusion of three additional studies. The articles were published during 1971–2019, since one article was indexed before final publication. During the review process, a study including 126 patients was published and therefore also included.8 Only eight included articles were published before 2000, and the vast majority was published after 2010.The review focuses on clinical aspects of DHSt, and therefore, studies including detailed pathophysiology and genetic findings are described in general terms, aiming at improving awareness of the clinical correlates and their importance. Mainly studies including original data were included in the review. The results were abstracted from the retrieved publications and divided into sections of DHSt frequency, pathophysiology in DHSt, diagnosing DHSt, as well as general symptoms and findings. Separate sections include the results of studies describing complications related to iron overload, splenectomy, perinatal issues, management of DHSt, and finally a section on potential benefits from carrying a PIEZO1 mutation. Some patients are reported in multiple publications.5,6,8,9 Although the author intended not to describe results from the same patients more than once in the same section, this cannot be completely avoided.
Results
For the sections on DHSt, 73 articles were found to be relevant, and additional eight articles were included to discuss the findings.10–15 All studies that included descriptions of patients’ clinical findings or course were based on single cases or case series, the largest study included 126 patients from 64 families.8 One study included findings from an entire large kindred with many affected family members.3
Frequency of DHSt
Based on the sporadic clinical diagnoses, DHSt is thought to be rare, although the population prevalence is unknown. Prevalence estimates of 1:50,000 have been suggested.10 In a recent study within a biochemical database of blood sample results from 2014 to 2016 among 48,000,000 North American patients, a possible DHSt diagnosis was suggested in patients who were found to have compensated hemolysis with elevated ferritin and middle cell hemoglobin concentration (MCHC).16 Based on various combinations of biochemical results, the authors suggest that the 3-year period prevalence of DHSt was 1:8,000 among US adults, emphasizing that a large fraction of patients probably remains undiagnosed.16 Also, a recent study reports on high allele frequency of a specific PIEZO1 mutation in persons of African descent.17
Pathophysiology of hemolysis in DHSt
The life span of erythrocytes depends on their intrinsic hydration, volume, and elasticity, particularly when passing through the splenic sinusoids and capillaries. All these properties are ensured by an intact and well-functioning membrane. However, maintenance of RBC hydration depends also on the content of intracellular proteins and salts where cations play a major role.18–20 Several physiological pathways determine the homeostasis of salts and water in RBCs. As in other cells, the RBC intracellular concentrations of sodium are low, of potassium are high, and vice versa in plasma. These gradients are maintained through active ATP-driven transport exchanging intracellular sodium for extracellular potassium and by diffusion through ion channels. Since water molecules follow the ion movements, the cation balance across the RBC membrane plays a major role in cell hydration. Therefore, when normal cation flux mechanisms are dysregulated, RBC hydration is also affected. Passing through the vasculature, RBCs are exposed to considerable mechanical forces that influence their normal physiology and hydration. In DHSt, a dysfunctional membrane protein eventually leads to a potassium leak out of the RBC that exceeds the inward flux of sodium and the accompanying net loss of water results in RBC dehydration, shrinkage, fragility, and hemolysis. In most patients, the molecular basis for the RBC dehydration is a result of a mutation in the PIEZO1 gene encoding the large transmembrane PIEZO1 cation channel.4,21 A large number of different PIEZO1 mutations have been described in DHSt.4–7,21–26 However, a similar phenotype has also been observed with mutations in the KCNN4 gene affecting the KCa3.1 protein, also known as the Gardos channel.27–30 The PIEZO1 is a nonselective mechanosensitive channel that allows permeation of cations such as sodium, potassium, magnesium, and calcium. The channel is activated by pressure or stretch of the cell membrane that RBCs are repeatedly exposed to, eg, when passing through capillaries and splenic sinusoids. The PIEZO1 mutations that have been found in patients with DHSt result in a gain of function that leads to delayed inactivation of the ion channel.31–33 During the extended opening of PIEZO1, an increased Ca2+ RBC influx again activates the opening of Gardos channel, which promotes K+ efflux, leading to the net loss of total RBC cations and dehydration.23,34,35 Patients with clinical DHSt, but without PIEZO1 gene alterations, may harbor mutations in the KCNN4 gene, which encodes the Ca2+-dependent potassium selective Gardos channel.27–30 The Gardos channel is, under normal conditions, inactive until stimulated with, eg, Ca2+ where activation leads to opening and selective K+ efflux. It is mainly the c.1055G > A KCNN4 missense mutations that have been found in DHSt patients, although other mutations have been described.27–30 Similar to PIEZO1 alterations in DHS, the Gardos channel mutations also lead to an activated state with augmented potassium loss from RBCs.28 The dehydrated cells are less deformable and therefore more vulnerable to lysis, and consequently, RBC life span is shortened. Co-inheritance of hemoglobin disorder traits, such as hemoglobin C and possibly others, may promote RBC dehydration.36
Diagnosing DHSt
Patients with DHSt typically present with a partially or fully compensated nonimmune hemolysis, and a significant proportion is diagnosed late in life.5,7,8,22,24,37–40 Patients show standard laboratory signs of hemolysis with normal or only slightly reduced hemoglobin concentration, reticulocytes elevated 2–6 times the upper limit of normal, and slightly elevated RBC middle cell volume, lactate dehydrogenase (LDH), bilirubin, as well as reduced haptoglobin.3,5,7,8,22,41,42 Distinct laboratory findings in DHSt are signs of RBC dehydration with elevated MCHC and reduced osmotic fragility.3,4,22,37,42,43 Intracellular RBC cation concentration shows low intracellular potassium and high sodium and can be determined using readily available point-of-care equipment.21,22,24,37,43–45 Despite the name of the diagnosis, only occasional RBCs with the characteristic slit like stoma – stomatocytes – are found in peripheral blood smear (Figure 1), where target cells also can be observed.46,47 The number of stomatocytes in the peripheral blood smear may in fact be comparable between patients and unaffected family members,3 and the blood smear may even be relatively normal particularly with KCNN4 mutations.8,24,37 Bone marrow histology is signified by a marked erythroid hyperplasia, which may have dysplastic features and mislead to a diagnosis of dyserythropoietic anemia or even myelodysplastic syndrome.26,29,44,48,49 The result of osmotic gradient ektacytometry is generally with characteristic left-shifted curves,5,41,42,45,50 although they may be normal in patients with KCNN4 mutations.27,28,50 Genetic analysis usually reveals a mutation in the PIEZO1 or KCNN4 genes.3–8,25,29,38 As in other diagnoses with hemolysis, the glycosylated hemoglobin percentage is decreased in DHSt, which can be misinterpreted as a sign of hypoglycemia.51
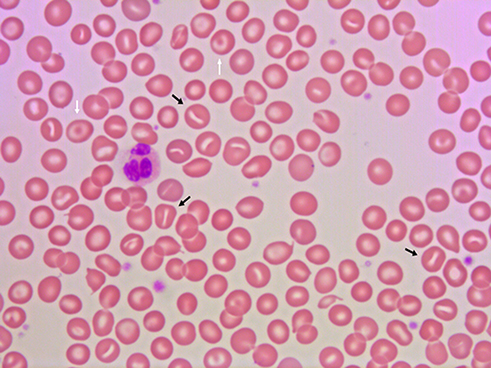 |
Figure 1 Peripheral blood smear from a patient with dehydrated stomatocytosis. The smear reveals stomatocytes (black arrows) and target cells (white arrows). |
General symptoms of hemolysis and clinical findings
Although the first suspicion of DHSt is usually raised in patients with a direct antiglobulin test negative hemolysis, patients often have no or only mild signs and symptoms of a classical hemolytic disorder.24,52 Because of the vague or unspecific symptoms, patients with DHSt may be diagnosed late in life or following complications. For example, a patient suffered iron overload complications from the age of 32 years and later thoracic paravertebral masses of extramedullary hematopoiesis before being diagnosed with DHSt at the age of 82.39
Only a minority of patients suffer from symptomatic anemia, and few require regular blood transfusions, whereas transfusion following a hemolytic crisis is seen in 15–20%.3,7,8,21,37 Intermittent jaundice is frequent, and similarly cholelithiasis, which may also be asymptomatic.3,8,22,24,45 Splenomegaly is frequently observed using imaging, but an enlarged spleen can occasionally also be palpable.7,8,22
Elevated potassium values may be found after blood sampling. This reflects that some patients with DHSt have a temperature-dependent potassium loss from RBCs. Therefore, cooling in blood tubes may lead to in vitro only elevated potassium values, however, with completely normal values in patients – so-called pseudohyperkalemia.42,53 It is important to recognize this phenomenon so that unnecessary and potentially harmful interventions such as changing dose of angiotensin-converting enzyme inhibitor, glucose-insulin infusion, and other treatments can be avoided.53 Pseudohyperkalemia may also delay DHSt diagnosis if indices of hemolysis are not investigated.42
A phenotypic discrepancy between patients with PIEZO1 compared to KCNN4 mutations has been suggested with a lesser degree of RBC dehydration in the latter group.7,8,28,50 This does seem to translate into some clinical differences between these two groups were hemoglobin levels and risks of thrombosis and fetal edema seems to be highest in patients with PIEZO1 mutations.7,8 Different PIEZO1 mutations do not consistently convey phenotypic clinical differences,7 and even among DHSt-affected members of the same family, phenotypic differences can be observed.3 The clinical heterogeneity among patients with the same PIEZO1 mutation is emphasized in a large Canadian kindred with 29 family members affected by DHSt.3 In this family, 46% were or had been anemic, 17% had received blood transfusions, 45% were or had been jaundiced, and 41% had symptomatic gall stones.3 However, some genotype–phenotype correlation in DHSt cannot be excluded. Recently, a de novo PIEZO1 variant occurring in a patient also affected by another PIEZO1 mutation was described.38 Compared to other family members with DHSt but without the additional genetic variant, this patient suffered augmented potassium and water loss from RBCs, more severe anemia, and transfusion dependency, suggesting that additional genetic alterations in PIEZO1-mutated patients may increase severity.38
Iron overload
Only a minority of DHS patients require regular transfusions and despite this hyperferritinemia, high transferrin saturation or clinical iron overload is very frequent in DHSt.3,7,21,22,29,39,44,54–57 In the previously mentioned kindred with 29 patients, only 5 (17%) had ever received a blood transfusion and yet all had elevated ferritin levels, in 7 (24%) patients even exceeding 900 µg/L.3 Complications to iron overload in DHSt have been described. A 19-year-old woman, who had been sporadically transfused since childhood, developed heart failure, and a biopsy verified liver fibrosis several years prior to the DHSt diagnosis.54 A MRI T2* scan revealed iron overload in heart, liver, and also pancreas, after which she started iron chelating treatment, normalizing iron deposits and organ function54 Clinical cardiac iron overload was also suspected in a 25-year-old DHSt patient who had been irregularly transfused. She suddenly died, and autopsy revealed elevated myocardial iron.21 Liver or pancreas involvement with diabetes has also been found in patients aged 22–55 years – in most patients, several years prior to the DHSt diagnosis.9,39,57 Iron chelation therapy has been applied in some patients;21,39,54 however, due to the well-compensated hemolysis, also therapeutic phlebotomies have been found to be feasible in others.9,22,56,57
The mechanism behind iron overload in DHSt is unknown. In hemolytic anemias characterized by an insufficient erythropoiesis such as thalassemias, the hormone, erythroferrone, suppresses the release of hepcidine, which eventually leads to elevated iron absorption and iron overload.12 Even though erythropoiesis is not insufficient, but rather stimulated in DHSt, hepcidin levels have also been found to be low in DHSt, which may, in part, explain the almost universally observed iron overload.22
Thrombosis after splenectomy
In patients with DHSt, many episodes of immediate or late thrombotic complications have been described following splenectomy.7,9,27–29,58–60 However, there is a lack of studies comparing thrombotic risk in splenectomized DHSt patients with DHSt patients who have their spleen in situ, and in some patients, a splenectomy is described without further detail of potential complications.61,62 Almost all thromboses have been venous thromboembolic events, such as lower extremity deep venous thrombosis (DVT), pulmonary embolism (PE), and portal venous thrombosis occurring up to 30 years after surgery.7,55 However, also superficial venous thrombi, peripheral arterial thrombosis, ischemic stroke, and intracardic mural thrombi have been found.9,59 Some of the thrombotic events have been reported to be fatal.9 Both venous thrombosis provoked by immobilization, pregnancy, or contraceptive pills and unprovoked episodes have been described, and some patients have developed thrombi despite ongoing anticoagulant treatment.6,9 In one study, all PIEZO1-mutated DHSt patients who were splenectomized suffered a severe venous thrombotic complication, and furthermore, splenectomy did not improve their anemia.7 In another study including 11 patients from seven families, two were splenectomized of which one developed a provoked DVT and a PE.6 Approximately, ten DHSt patients who have later been found to harbor a KCNN4 mutation have been reported to have been splenectomized during the course of their disease, none of whom suffered thrombotic complications.8,27–29 One of these KCNN4-mutated patients experienced improved hemoglobin levels and became free of blood transfusions following surgery,27 whereas another patient had no clinical benefit from splenectomy.29 Several patients have been described to develop chronic pulmonary hypertension and right-sided heart failure following PE, and one patient was even heart and lung transplanted due to this complication.9,59 Due to the lack of improvement in the degree of anemia for most patients and the putative severe risk of thrombotic complications, splenectomy should be avoided in the treatment of hemolytic anemia in DHSt regardless of the underlying mutation. This also applies if a DHSt patient is considered for splenectomy for any other indication.59
Obstetric and perinatal issues of DHSt
Perinatal edema and hydrops fetalis have been reported in several case studies of patients with DHSt.6,34,45,52,60,62–75 Perinatal edema may even be the first sign of DHSt.8 The degree of edema is unrelated to the fetal hemoglobin concentration and is also unresponsive to in utero blood transfusion through cordocentesis.60,62,66,73,75 Rarely, hydrops fetalis in DHSt may be fatal.35 All patients with perinatal edema seem to harbor a PIEZO1 mutation.8
In some pregnancies, the fetal edema has been seen with concurrent polyhydramnios.60,63,64 However, the amount of amniotic fluid may also be normal despite severe fetal edema.68,74 In most patients, fluid effusions present as fetal ascites with marked abdominal distension, but also pericardial effusion, pleural effusions, or generalized hydrops fetalis can be seen.60,62–64,66,68–71,73–75 In order to prevent lung hypoplasia due to elevated diaphragm, delivery complications, and sequelae to abdominal distension, therapeutic fetal ascites paracentesis has been undertaken repeatedly in many of the pregnancies and without described complications.60,63,64,66,68,69,71,75 In some cases, the volume of ascites fluid that was removed by each paracentesis reached 800–2,300 mL.66,68,69 Recently, a case study described the complete resolution of ascites and pleural effusion in a fetus with DHSt after application of intrauterine peritoneo-amniotic and thoracic-amniotic shunts followed by drainage of 1,000 mL amniotic fluid.71 In almost all patients, perinatal fluid effusion resolves spontaneously and completely after birth. However, patients may suffer from respiratory distress immediately after birth, and the abdomen can have a “prune-like” appearance due to poorly developed abdominal musculature after distension.60,62,69,74
It seems like a paradox that mutations that result in dehydration of RBCs cause fluid effusions and that this is seen only perinatally. However, PIEZO1 expression is increased in lymphatic vessels in the peritoneum in fetal patients with mutations compared to healthy individuals, and this expression is absent in lymphatic vessel endothelium in adult DHSt patients.6 Of note, “loss-of-function” PIEZO1 mutations have been identified in patients with congenital lymphatic dysplasia, leading to primary lymphoedema, and RBCs from these patients also show some phenotypic similarities with DHSt erythrocytes.67,72
Management of patients with DHSt
Contemporary management of DHSt first of all requires making the correct diagnosis for the patient with nonimmune hemolysis. For this, elevated MCHC, and specific RBC K+ and Na+ deviations combined with standard laboratory signs of hemolysis are straightforward clues to ask for conclusive testing with osmotic gradient ektacytometry and/or mutational screening. There is no specific treatment for hemolysis, and if patients have symptomatic anemia, transfusion therapy is the only relevant option.
Splenectomy is contraindicated regardless of the clinical setting, and even in a critical situation like splenic rupture, other attempts for hemostasis should be pursued, if at all possible. Recognition of iron overload during follow-up in regularly transfused, in sporadically transfused, and in patients who have never received blood transfusion is crucial in order to avoid organ failure through chelation therapy or therapeutic phlebotomies. It is not possible to make recommendations of ferritin thresholds for starting iron removal based on existing DHSt publications. In hereditary hemochromatosis (HH) where iron overload may have a similar pathophysiology, the risk of organ damage with ferritin levels below 1,000 µg/L has in some population-based studies been found to be extremely low.76 However, recent guidelines of HH suggest to start phlebotomy already at ferritin levels above 300 µg/L in men and 200 µg/L in women.77 A pragmatic approach in DHSt could be to monitor patients suspected to have clinical relevant iron overload with T2* MRI and to use phlebotomy under close hemoglobin monitoring as the primary therapy in well-compensated patients.
In pregnancy with a fetus at risk for DHSt, regular ultrasound is warranted. This is to discover and monitor prenatal ascites or hydrops fetalis or other signs of fluid effusion so that paracentesis can be instituted if needed. Children with a parent with DHSt should be screened for signs of hemolysis during childhood.
In sickle cell disease (SCD), RBCs are also liable to cation leaks and dehydration through activation of the Gardos channel, and this may eventually lead to hemoglobin S polymerization and painful crisis.78 Senicapoc is a selective Gardos channel blocker and has been investigated as an intervention to reduce painful crisis in a SCD phase III trial.11 Although the trial was terminated early since interim analyses revealed that the primary end point – a reduction in the frequency of painful crisis – was unlikely to be met, the intervention group had statistically significant improvements in markers of hemolysis.11 Sickle cell patients treated with senicapoc had a mean increase in hemoglobin concentration of 5.9 g/L, a mean reduction in LDH of 58.7 U/L, and the treatment was well tolerated.11 In vitro RBCs from DHSt patients with either KCNN4 or PIEZO1 mutation types have been found to become less cation leaky and dehydrated after treatment with senicapoc.35,79,80 However, it is currently unknown whether this translates into a clinically relevant effect in patients with DHSt with symptomatic hemolysis and anemia.
Potential benefits in patients with PIEZO1 mutations
It is well established that the carrier state of other RBC disorders, such as SCD, conveys protection against malaria.15 In vitro studies of RBC hydration also suggest that dehydrated cells are less susceptible to invasion by Plasmodium falciparum and that this applies both to RBCs from healthy donors and to patients with DHSt.81 In order to study this in vivo, a mouse model for human DHSt has recently been developed.17 The human gain-of-function mutation - R to H at position 2,456 in PIEZO1 has been knocked in mice (PIEZO1GOF) constructing a mouse DHSt-like phenotype with mild anemia, hemolysis, reticulocytosis, increased mean corpuscular hemoglobin, reduced osmotic fragility, and splenomegaly. Further, electron microscopy of mouse RBCs showed stomatocytes. Susceptibility to Plasmodium infection was tested using this model. After injection of P. berghie infected erythrocytes PIEZO1GOF, mice lived significantly longer, had lower percentage parasitemia, and were less affected by cerebral malaria than PIEZO1 wild-type (PIEZO1WT) mice.17 In functional studies, the RBC dehydration seemed to be responsible for the improved outcomes in PIEZO1GOF knockin mice.17 Interestingly, a three-nucleotide deletion E756del in PIEZO1 has subsequently been identified with an allele frequency of 9–23% in persons of African ancestry compared to <1% in persons of non-African descent.17 Electron microscopy of RBCs from persons with this deletion reveals frequent echinocytes and stomatocytes, and their RBCs were generally found to be dehydrated and with decreased osmotic fragility. When subjected to P. falciparum in vitro, the RBCs with this novelly identified PIEZO1 deletion also showed lower percentage parasitic invasion. Altogether, these results suggest that PIEZOGOF mutations through the DHSt phenotype convey protection against malaria and complications. Also, populations with endemic malaria develop a positive selection of a hitherto unknown and very common PIEZO1 mutation with a DHSt-like phenotype.17
Discussion
This literature review on clinical perspectives in DHSt emphasizes that almost all current knowledge is based on single case reports or case series. Despite this shortcoming in data, it is beyond discussion that patients often have a relatively mild clinical course with normal or only moderately reduced hemoglobin, rarely transfusion dependency, however with important exceptions. Patients may have symptomatic anemia or suffer potentially severe complications from iron overload, thrombosis following splenectomy, and perinatal fluid effusions. Although perinatal edema seems to be a rare complication even for patients with DHSt, it may also be that PIEZO1 mutations lead to an increased risk of spontaneous abortions emphasizing that the published risk of this complication may be conservative. All complications and late effects are preventable or manageable, and it therefore remains essential to diagnose DHSt in time to avoid adverse effects for patients and their offspring and also to improve quality of life.
DHSt has been a recognized diagnostic entity during the past almost 50 years, but it was not until 2011 that the first genetic alterations were identified.3,4 Since then, advances mainly in laboratory research have improved knowledge on genetics and pathophysiology considerably.5,13,23,31,33 However, clinical management beyond transfusion therapy for the rare DHSt patients with symptomatic anemia remains to be improved. Treatment with senicapoc to prevent RBC dehydration may be feasible and beneficial; however, recruitment for a phase II or phase III trial to investigate this rare subgroup of patients will be challenging, and no such trial is currently registered at the Clinicaltrials.gov website. For the previous advances in finding and understanding the effects of the mutations associated with DHSt, international research collaborations on kindreds from multiple countries were necessary.3,4,6 The best observations on the clinical heterogeneity in DHSt are from the largest of these kindreds.3 In order to improve management for these rare patients, both with and without medical intervention, recruitment into an international registry study such as the Rare Anaemia Disorders European Epidemiological Platform (https://www.eurobloodnet.eu/radeep) is needed. In another rare hemolytic disorder, paroxysmal nocturnal hemoglobinuria (PNH), an international registry, was facilitated through both international collaboration and adequate funding.13 The PNH registry now includes more than 3,500 patients with PNH, allowing for research on clinical presentation, complications, and prognosis on an unprecedented scale.13,14
Conclusion
Currently, the best clinical advice for managing patients with DHSt is to identify them correctly among other patients with nonimmune hemolytic disorders. Failure to do so may put the patients at risk for severe complications and late effects. International collaboration for registries on these rare patients is needed to fill the gaps in knowledge and ultimately to improve care.
Abbreviation list
DHSt, dehydrated stomatocytosis; RBC, red blood cell; DAT, direct antiglobulin test; LDH, lactate dehydrogenase; MCHC, middle cell hemoglobin concentration; DVT, deep venous thrombosis; PE, pulmonary embolism; SCD, sickle cell disease.
Acknowledgments
The author thanks Vickie Svane Kristensen for her help with retrieving references and for proofreading and consultant, PhD Birgitte Preiss for providing photo material for the figure.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Miller DR, Rickles FR, Lichtman MA, La Celle PL, Bates J, Weed RI. A new variant of hereditary hemolytic anemia with stomatocytosis and erythrocyte cation abnormality. Blood. 1971;38(2):184–204.
2. Stewart GW, Ellory JC. A family with mild hereditary xerocytosis showing high membrane cation permeability at low temperatures. Clin Sci (Lond). 1985;69(3):309–319.
3. Houston BL, Zelinski T, Israels SJ, et al. Refinement of the hereditary xerocytosis locus on chromosome 16q in a large Canadian kindred. Blood Cells Mol Dis. 2011;47(4):226–231. doi:10.1016/j.bcmd.2011.08.001
4. Zarychanski R, Schulz VP, Houston BL, et al. Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood. 2012;120(9):1908–1915. doi:10.1182/blood-2012-04-422253
5. Albuisson J, Murthy SE, Bandell M, et al. Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat Commun. 2013;4:1884. doi:10.1038/ncomms2899
6. Andolfo I, Alper SL, De Franceschi L, et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013;121(19):
7. Andolfo I, Russo R, Rosato BE, et al. Genotype-phenotype correlation and risk stratification in a cohort of 123 hereditary stomatocytosis patients. Am J Hematol. 2018;93(12):1509–1517. doi:10.1002/ajh.25276
8. Picard V, Guitton C, Thuret I, et al. Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos-channelopathy: A retrospective series of 126 patients. Haematologica. 2019. doi:10.3324/haematol.2018.205328
9. Stewart GW, Amess JA, Eber SW, et al. Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br J Haematol. 1996;93(2):303–310.
10. Andolfo I, Russo R, Gambale A, Iolascon A. New insights on hereditary erythrocyte membrane defects. Haematologica. 2016;101(11):1284–1294. doi:10.3324/haematol.2016.142463
11. Ataga KI, Reid M, Ballas SK, et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). Br J Haematol. 2011;153(1):92–104. doi:10.1111/j.1365-2141.2010.08520.x
12. Kautz L, Jung G, Du X, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood. 2015;126(17):2031–2037. doi:10.1182/blood-2015-07-658419
13. Schrezenmeier H, Muus P, Socie G, et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica. 2014;99(5):922–929. doi:10.3324/haematol.2013.093161
14. Socie G, Schrezenmeier H, Muus P, et al. Changing prognosis in paroxysmal nocturnal haemoglobinuria disease subcategories: an analysis of the International PNH Registry. Intern Med J. 2016;46(9):1044–1053. doi:10.1111/imj.13160
15. Uyoga S, Macharia AW, Ndila CM, et al. The indirect health effects of malaria estimated from health advantages of the sickle cell trait. Nat Commun. 2019;10(1):856. doi:10.1038/s41467-019-08775-0
16. Kaufman HW, Niles JK, Gallagher DR, et al. Revised prevalence estimate of possible Hereditary Xerocytosis as derived from a large U.S. Laboratory database. Am J Hematol. 2018;93(1):E9–E12. doi:10.1002/ajh.24923
17. Ma S, Cahalan S, LaMonte G, et al. Common PIEZO1 allele in African populations causes RBC dehydration and attenuates plasmodium infection. Cell. 2018;173(2):443–455 e412. doi:10.1016/j.cell.2018.02.047
18. Caulier A, Rapetti-Mauss R, Guizouarn H, Picard V, Garcon L, Badens C. Primary red cell hydration disorders: pathogenesis and diagnosis. Int J Lab Hematol. 2018;40 Suppl 1:68–73. doi:10.1111/ijlh.12820
19. Glogowska E, Gallagher PG. Disorders of erythrocyte volume homeostasis. Int J Lab Hematol. 2015;37 Suppl 1:85–91. doi:10.1111/ijlh.12357
20. Badens C, Guizouarn H. Advances in understanding the pathogenesis of the red cell volume disorders. Br J Haematol. 2016;174(5):674–685. doi:10.1111/bjh.14197
21. Glogowska E, Schneider ER, Maksimova Y, et al. Novel mechanisms of PIEZO1 dysfunction in hereditary xerocytosis. Blood. 2017;130(16):1845–1856. doi:10.1182/blood-2017-05-786004
22. Archer NM, Shmukler BE, Andolfo I, et al. Hereditary xerocytosis revisited. Am J Hematol. 2014;89(12):1142–1146. doi:10.1002/ajh.23799
23. Cinar E, Zhou S, DeCourcey J, Wang Y, Waugh RE, Wan J. Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc Natl Acad Sci U S A. 2015;112(38):11783–11788. doi:10.1073/pnas.1507309112
24. Sandberg MB, Nybo M, Birgens H, Frederiksen H. Hereditary xerocytosis and familial haemolysis due to mutation in the PIEZO1 gene: a simple diagnostic approach. Int J Lab Hematol. 2014;36(4):e62–e65. doi:10.1111/ijlh.12172
25. Shefer Averbuch N, Steinberg-Shemer O, Dgany O, et al. Targeted next generation sequencing for the diagnosis of patients with rare congenital anemias. Eur J Haematol. 2018;101(3):297–304. doi:10.1111/ejh.13097
26. Park J, Jang W, Han E, et al. Hereditary dehydrated stomatocytosis with splicing site mutation of PIEZO1 mimicking myelodysplastic syndrome diagnosed by targeted next-generation sequencing. Pediatr Blood Cancer. 2018;65(7):e27053. doi:10.1002/pbc.v65.7
27. Fermo E, Bogdanova A, Petkova-Kirova P, et al. ‘Gardos Channelopathy’: a variant of hereditary Stomatocytosis with complex molecular regulation. Sci Rep. 2017;7(1):1744. doi:10.1038/s41598-017-01591-w
28. Rapetti-Mauss R, Lacoste C, Picard V, et al. A mutation in the Gardos channel is associated with hereditary xerocytosis. Blood. 2015;126(11):1273–1280. doi:10.1182/blood-2015-04-642496
29. Andolfo I, Russo R, Manna F, et al. Novel Gardos channel mutations linked to dehydrated hereditary stomatocytosis (xerocytosis). Am J Hematol. 2015;90(10):921–926. doi:10.1002/ajh.24117
30. Glogowska E, Lezon-Geyda K, Maksimova Y, Schulz VP, Gallagher PG. Mutations in the Gardos channel (KCNN4) are associated with hereditary xerocytosis. Blood. 2015;126(11):1281–1284. doi:10.1182/blood-2015-07-657957
31. Moroni M, Servin-Vences MR, Fleischer R, Sanchez-Carranza O, Lewin GR. Voltage gating of mechanosensitive PIEZO channels. Nat Commun. 2018;9(1):1096. doi:10.1038/s41467-018-03502-7
32. Bae C, Gottlieb PA, Sachs F. Human PIEZO1: removing inactivation. Biophys J. 2013;105(4):880–886. doi:10.1016/j.bpj.2013.07.019
33. Bae C, Gnanasambandam R, Nicolai C, Sachs F, Gottlieb PA. Xerocytosis is caused by mutations that alter the kinetics of the mechanosensitive channel PIEZO1. Proc Natl Acad Sci U S A. 2013;110(12):E1162–E1168. doi:10.1073/pnas.1219777110
34. Cahalan SM, Lukacs V, Ranade SS, Chien S, Bandell M, Patapoutian A. Piezo1 links mechanical forces to red blood cell volume. Elife. 2015;4:1–12. doi:10.7554/eLife.07370
35. Rapetti-Mauss R, Picard V, Guitton C, et al. Red blood cell Gardos channel (KCNN4): the essential determinant of erythrocyte dehydration in hereditary xerocytosis. Haematologica. 2017;102(10):e415–e418. doi:10.3324/haematol.2017.171389
36. Yang E, Voelkel EB, Lezon-Geyda K, Schulz VP, Gallagher PG. Hemoglobin C trait accentuates erythrocyte dehydration in hereditary xerocytosis. Pediatr Blood Cancer. 2017;64(8). doi:10.1002/pbc.26444
37. Vives Corrons JL, Besson I, Aymerich M, et al. Hereditary xerocytosis: a report of six unrelated Spanish families with leaky red cell syndrome and increased heat stability of the erythrocyte membrane. Br J Haematol. 1995;90(4):817–822.
38. Andolfo I, Manna F, De Rosa G, et al. PIEZO1-R1864H rare variant accounts for a genetic phenotype-modifier role in dehydrated hereditary stomatocytosis. Haematologica. 2018;103(3):e94–e97. doi:10.3324/haematol.2017.180687
39. Imashuku S, Muramatsu H, Sugihara T, et al. PIEZO1 gene mutation in a Japanese family with hereditary high phosphatidylcholine hemolytic anemia and hemochromatosis-induced diabetes mellitus. Int J Hematol. 2016;104(1):125–129. doi:10.1007/s12185-016-1970-x
40. Nolan GR. Hereditary xerocytosis. A case history and review of the literature. Pathology. 1984;16(2):151–154.
41. Llaudet-Planas E, Vives-Corrons JL, Rizzuto V, et al. Osmotic gradient ektacytometry: a valuable screening test for hereditary spherocytosis and other red blood cell membrane disorders. Int J Lab Hematol. 2018;40(1):94–102. doi:10.1111/ijlh.12746
42. Beaurain G, Mathieu F, Grootenboer S, et al. Dehydrated hereditary stomatocytosis mimicking familial hyperkalaemic hypertension: clinical and genetic investigation. Eur J Haematol. 2007;78(3):253–259. doi:10.1111/j.1600-0609.2006.00811.x
43. Shmukler BE, Vandorpe DH, Rivera A, Auerbach M, Brugnara C, Alper SL. Dehydrated stomatocytic anemia due to the heterozygous mutation R2456H in the mechanosensitive cation channel PIEZO1: a case report. Blood Cells Mol Dis. 2014;52(1):53–54. doi:10.1016/j.bcmd.2013.07.015
44. Del Orbe Barreto R, Arrizabalaga B, De la Hoz Rastrollo AB, et al. Hereditary xerocytosis, a misleading anemia. Ann Hematol. 2016;95(9):1545–1546. doi:10.1007/s00277-016-2716-9
45. Risinger M, Glogowska E, Chonat S, et al. Hereditary xerocytosis: diagnostic considerations. Am J Hematol. 2018;93(3):E67–E69. doi:10.1002/ajh.24996
46. Huisjes R, van Solinge WW, Levin MD, van Wijk R, Riedl JA. Digital microscopy as a screening tool for the diagnosis of hereditary hemolytic anemia. Int J Lab Hematol. 2018;40(2):159–168. doi:10.1111/ijlh.12758
47. McGrath KM, Collecutt MF, Gordon A, Sawers RJ, Faragher BS. Dehydrated hereditary stomatocytosis–a report of two families and a review of the literature. Pathology. 1984;16(2):146–150.
48. Paessler M, Hartung H. Dehydrated hereditary stomatocytosis masquerading as MDS. Blood. 2015;125(11):1841. doi:10.1182/blood-2014-07-591040
49. Iolascon A, De Falco L, Borgese F, et al. A novel erythroid anion exchange variant (Gly796Arg) of hereditary stomatocytosis associated with dyserythropoiesis. Haematologica. 2009;94(8):1049–1059. doi:10.3324/haematol.2008.002873
50. Zaninoni A, Fermo E, Vercellati C, et al. Use of laser assisted optical rotational cell analyzer (LoRRca MaxSis) in the diagnosis of RBC membrane disorders, enzyme defects, and congenital dyserythropoietic anemias: a monocentric study on 202 patients. Front Physiol. 2018;9:451. doi:10.3389/fphys.2018.00451
51. Jokinen CH, Swaim WR, Nuttall FQ. A case of hereditary xerocytosis diagnosed as a result of suspected hypoglycemia and observed low glycohemoglobin. J Lab Clin Med. 2004;144(1):27–30. doi:10.1016/j.lab.2004.04.004
52. Velasco-Rodriguez D, Alonso-Dominguez JM, Ruiz E, Acedo N, Gonzalez-Fernandez FA, Villarrubia J. Dehydrated red blood cells in a peripheral blood film: a case of hereditary xerocytosis. Br J Haematol. 2014;166(3):309. doi:10.1111/bjh.12933
53. Gore DM, Layton M, Sinha AK, et al. Four pedigrees of the cation-leaky hereditary stomatocytosis class presenting with pseudohyperkalaemia. Novel profile of temperature dependence of Na+-K+ leak in a xerocytic form. Br J Haematol. 2004;125(4):521–527. doi:10.1111/j.1365-2141.2004.04944.x
54. Assis RA, Kassab C, Seguro FS, et al. Iron overload in a teenager with xerocytosis: the importance of nuclear magnetic resonance imaging. Einstein (Sao Paulo). 2013;11(4):528–532.
55. Fermo E, Vercellati C, Marcello AP, et al. Hereditary xerocytosis due to mutations in PIEZO1 gene associated with heterozygous pyruvate kinase deficiency and beta-thalassemia trait in two unrelated families. Case Rep Hematol. 2017;2017:2769570.
56. Orvain C, Da Costa L, Van Wijk R, et al. Inherited or acquired modifiers of iron status may dramatically affect the phenotype in dehydrated hereditary stomatocytosis. Eur J Haematol. 2018;101(4):566–569. doi:10.1111/ejh.13135
57. Syfuss PY, Ciupea A, Brahimi S, et al. Mild dehydrated hereditary stomatocytosis revealed by marked hepatosiderosis. Clin Lab Haematol. 2006;28(4):270–274. doi:10.1111/j.1365-2257.2006.00774.x
58. Perel Y, Dhermy D, Carrere A, et al. Portal vein thrombosis after splenectomy for hereditary stomatocytosis in childhood. Eur J Pediatr. 1999;158(8):628–630.
59. Jais X, Till SJ, Cynober T, et al. An extreme consequence of splenectomy in dehydrated hereditary stomatocytosis: gradual thrombo-embolic pulmonary hypertension and lung-heart transplantation. Hemoglobin. 2003;27(3):139–147.
60. Grootenboer-Mignot S, Cretien A, Laurendeau I, et al. Sub-lethal hydrops as a manifestation of dehydrated hereditary stomatocytosis in two consecutive pregnancies. Prenat Diagn. 2003;23(5):380–384. doi:10.1002/pd.598
61. Stewart AK, Kedar PS, Shmukler BE, et al. Functional characterization and modified rescue of novel AE1 mutation R730C associated with overhydrated cation leak stomatocytosis. Am J Physiol Cell Physiol. 2011;300(5):C1034–C1046. doi:10.1152/ajpcell.00447.2010
62. Vicente-Gutierrez MP, Castello-Almazan I, Salvia-Roiges MD, et al. Nonimmune hydrops fetalis due to congenital xerocytosis. J Perinatol. 2005;25(1):63–65. doi:10.1038/sj.jp.7211200
63. Ami O, Picone O, Garcon L, et al. First-trimester nuchal abnormalities secondary to dehydrated hereditary stomatocytosis. Prenat Diagn. 2009;29(11):1071–1074. doi:10.1002/pd.2342
64. Basu AP, Carey P, Cynober T, et al. Dehydrated hereditary stomatocytosis with transient perinatal ascites. Arch Dis Child Fetal Neonatal Ed. 2003;88(5):F438–439. doi:10.1136/fn.88.5.f438
65. Beneteau C, Thierry G, Blesson S, et al. Recurrent mutation in the PIEZO1 gene in two families of hereditary xerocytosis with fetal hydrops. Clin Genet. 2014;85(3):293–295. doi:10.1111/cge.12147
66. Entezami M, Becker R, Menssen HD, Marcinkowski M, Versmold HT. Xerocytosis with concomitant intrauterine ascites: first description and therapeutic approach. Blood. 1996;87(12):5392–5393.
67. Fotiou E, Martin-Almedina S, Simpson MA, et al. Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non-immune hydrops fetalis. Nat Commun. 2015;6:8085. doi:10.1038/ncomms9085
68. Grootenboer S, Barro C, Cynober T, et al. Dehydrated hereditary stomatocytosis: a cause of prenatal ascites. Prenat Diagn. 2001;21(13):1114–1118.
69. Halma J, Petrikin J, Daniel JF, Fischer RT. Dehydrated hereditary stomatocytosis presenting as severe perinatal ascites and cholestasis. J Pediatr Gastroenterol Nutr. 2019;68(3):e52–e53. doi:10.1097/MPG.0000000000002077
70. Jenner B, Brockelsby J, Thomas W. Dehydrated hereditary stomatocytosis causing fetal hydrops and perinatal ascites. Br J Haematol. 2018;182(5):620. doi:10.1111/bjh.15462
71. Le Vaillant C, Riteau AS, Eveillard M, Beneteau C. Dehydrated hereditary stomatocytosis: prenatal management of ascites and pleural effusions. Taiwan J Obstet Gynecol. 2018;57(2):323–324. doi:10.1016/j.tjog.2018.02.026
72. Lukacs V, Mathur J, Mao R, et al. Impaired PIEZO1 function in patients with a novel autosomal recessive congenital lymphatic dysplasia. Nat Commun. 2015;6:8329. doi:10.1038/ncomms9329
73. Ogburn PL
74. Poret H, Simon EG, Herve P, Perrotin F. Dehydrated hereditary stomatocytosis and recurrent prenatal ascites. J Obstet Gynaecol. 2013;33(5):527. doi:10.3109/01443615.2013.781142
75. Sanchez M, Palacio M, Borrell A, et al. Prenatal diagnosis and management of fetal xerocytosis associated with ascites. Fetal Diagn Ther. 2005;20(5):402–405. doi:10.1159/000086820
76. Asberg A, Hveem K, Thorstensen K, et al. Screening for hemochromatosis: high prevalence and low morbidity in an unselected population of 65,238 persons. Scand J Gastroenterol. 2001;36(10):1108–1115.
77. Adams P, Altes A, Brissot P, et al. Therapeutic recommendations in HFE hemochromatosis for p.Cys282Tyr (C282Y/C282Y) homozygous genotype. Hepatol Int. 2018;12(2):83–86. doi:10.1007/s12072-018-9855-0
78. Ellory JC, Robinson HC, Browning JA, Stewart GW, Gehl KA, Gibson JS. Abnormal permeability pathways in human red blood cells. Blood Cells Mol Dis. 2007;39(1):1–6. doi:10.1016/j.bcmd.2007.02.011
79. Rivera A, Vandorpe DH, Shmukler BE, et al. Erythrocytes from hereditary xerocytosis patients heterozygous for KCNN4 V282M exhibit increased spontaneous Gardos channel-like activity inhibited by senicapoc. Am J Hematol. 2017;92(6):E108–E110. doi:10.1002/ajh.24716
80. Rapetti-Mauss R, Soriani O, Vinti H, Badens C, Guizouarn H. Senicapoc: a potent candidate for the treatment of a subset of hereditary xerocytosis caused by mutations in the Gardos channel. Haematologica. 2016;101(11):e431–e435. doi:10.3324/haematol.2016.149104
81. Tiffert T, Lew VL, Ginsburg H, Krugliak M, Croisille L, Mohandas N. The hydration state of human red blood cells and their susceptibility to invasion by Plasmodium falciparum. Blood. 2005;105(12):4853–4860. doi:10.1182/blood-2004-12-4948
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.