")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Defective DNA repair mechanisms in prostate cancer: impact of olaparib
Authors De Felice F , Tombolini V , Marampon F, Musella A, Marchetti C
Received 6 December 2016
Accepted for publication 13 January 2017
Published 1 March 2017 Volume 2017:11 Pages 547—552
DOI https://doi.org/10.2147/DDDT.S110264
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Francesca De Felice,1 Vincenzo Tombolini,1 Francesco Marampon,2 Angela Musella,3 Claudia Marchetti3
1Department of Radiotherapy, Policlinico Umberto I, “Sapienza” University of Rome, Rome, 2Department of Biotechnological and Applied Clinical Sciences, Laboratory of Radiobiology, University of L’Aquila, L’Aquila, 3Department of Gynecological and Obstetrical Sciences and Urological Sciences, “Sapienza” University of Rome, Rome, Italy
Abstract: The field of prostate oncology has continued to change dramatically. It has truly become a field that is intensely linked to molecular genetic alterations, especially DNA-repair defects. Germline breast cancer 1 gene (BRCA1) and breast cancer 2 gene (BRCA2) mutations are implicated in the highest risk of prostate cancer (PC) predisposition and aggressiveness. Poly adenosine diphosphate ribose polymerase (PARP) proteins play a key role in DNA repair mechanisms and represent a valid target for new therapies. Olaparib is an oral PARP inhibitor that blocks DNA repair pathway and coupled with BRCA mutated-disease results in tumor cell death. In phase II clinical trials, including patients with advanced castration-resistant PC, olaparib seems to be efficacious and well tolerated. Waiting for randomized phase III trials, olaparib should be considered as a promising treatment option for PC.
Keywords: prostate cancer, metastatic disease, castration resistant, BRCA, DNA-repair, PARP, olaparib
Introduction
Prostate cancer (PC) is a frequently diagnosed cancer worldwide, with 14% of total new cancer cases and 6% of the total cancer deaths in males.1 PC is characterized by a wide spectrum of clinical behavior that spans from indolence to a very aggressive and lethal disease.2 Durable control of advanced PC represents a public health need, and an in-depth analysis of the genomic landscape seems to be paramount. In fact, PC is also one of the most heritable human malignancies, suggesting that approximately 50% of the inter-individual variation in PC risk is due to genetic factors. Family history of PC is presently viewed as a strong risk factor for the disease development.3 It has been observed that PC shares genetic variants with other types of familial malignancies, especially with breast and ovarian cancers (OCs).4 Genome-wide association studies have identified more than 70 common variants explaining about 30% of the excess familial PC risk.5 Deleterious mutations in DNA repair pathways, including breast-cancer susceptibility gene 1 (BRCA1) and breast-cancer susceptibility gene 2 (BRCA2) mutation carries, predispose to a more aggressive clinical course disease and worst cancer-specific survival.6,7 For instance, patients with germline mutations in BRCA2 have a 8.6-fold increased risk of developing PC by age 65, with an absolute risk of 15%.8 However a “BRCAness” phenotype should be delineate, including a large spectrum of somatic mutation in genes involved in DNA repair processes, to research a treatment tailored to “BRCAness” status.
In fact, men with metastatic PC and DNA-repair gene mutations have been reported to exhibit sustained responses to inhibitors of poly adenosine diphosphate ribose polymerase (PARP) and platinum-based chemotherapy.9 Normal cells are less subjected to DNA damage than tumor cells and, therefore, less influenced by inhibitors of DNA repair mechanisms. The nuclear PARP enzymes are physiologically involved in multiple aspects of DNA repair and transcription regulation. In BRCA1/2-deficient cells, and thus in cells with defective DNA repair mechanisms, PARP inhibition translates into selective cell death. Consistent with this assumption, the PARP inhibitor olaparib (Lynparza™; AstraZeneca) has been tested in several solid tumor types occurring in patients with germline mutation in BRCA1 and/or BRCA2, and it has recently been approved for treating OCs with BRCA1/2 mutations.10 Based on the efficacy demonstrated in platinum chemotherapy-sensitive OC patients, PARP inhibitor therapy is being rapidly added to PC clinical trial practice, but how to best incorporate it with existing therapies remains an urgent need. This review provides highlights in pharmacology profile, clinical efficacy, tolerability and role in therapy of olaparib, to potentially offer a more tailored treatment approach in PC patients in the near future.
DNA repair mechanisms
DNA damage can be acquired in cells over time through exposure to exogenous chemicals and physical agents or endogenous reactive metabolites including reactive oxygen and nitrogen species. Efficient and correct repair of DNA damage is critical for cellular survival. DNA repair can be grouped into single-strand breaks (SSBs) and double-strand breaks (DSBs). SSBs include base-excision repair, nucleotide excision repair, and mismatch excision repair, whereas DSBs comprise non-homologous end-joining (NHEJ) and homologous recombination (HR) mechanisms.
Single-strand breaks
Most frequently, DNA damage is reduced to SSBs. The repair of base damage is initiated by the glycosylate, a DNA repair enzyme, which recognizes and excises damage bases. The subsequent apyrimidinic site is recognized by an endonuclease enzyme that nicks the DNA adjacent to the lesion. Then an exonuclease removes the abasic site and the gap is patched by DNA polymerase, using the opposite DNA strand as a template.
The nucleotide excision repair is mediated by structure-specific endonucleases, which identify more generalized DNA structural distortion. These repair proteins incise the DNA strand on both sides of the complex lesion, activating the repair process, which is therefore similar to that of base excision.
The specificity of mismatch excision repair is primarily for base–base mismatches and insertion/deletion loops caused by DNA polymerase during DNA replication and recombination. It consists of mismatch recognition and assembly of the repair complex, degradation of the error-containing strand, and repair synthesis.11,12
Double-strand breaks
NHEJ represents the dominant repair pathway. The DNA-dependent protein kinase binds the ends of double-stranded DNA and then recruits the catalytic subunit of DNA-dependent protein kinase to the ends. Thus NHEJ simply restores DNA integrity by joining the two broken ends together.13,14 On the other hand, HR is a more accurate repair process, error-free, and most active in the late S/G2 phases of the cell cycle.15 It involves the use of the intact sister chromatid and a vast number of proteins, including RAD51, BRCA1, BRCA2, and ATM. Repair is initiated by resection of a DSB. Strand invasion is followed by DNA synthesis and the second DSB end is, therefore, captured to resolve in subsequent DNA synthesis and ligation.16
Defective processes linked to PC
Synthetic lethality
The concept of synthetic lethality refers to the combination of two entities to form something new. It occurs when the simultaneous inactivation of two genes results in cellular death.17 In recent years, synthetic lethality has attracted researchers’ attention because it may explain the sensitivity of tumor cells to certain drugs that act in specific DNA repair processes. The large potential of synthetic lethality-based cancer therapy is mainly represented by the combination of germline BRCA1/2 mutations and chemical inhibition of the DNA repair enzyme PARP.
BRCA1 and BRCA2 are tumor suppressor genes that mainly contribute to repair DNA damage. They map on two different chromosomes, 17q21 and 13q12.3, respectively.18 BRCA1 and BRCA2 form complexes that activate the repair of DSBs in response to DNA damage. Secondary to DSBs, BRCA1 is phosphorylated by ataxia-telangiectasia mutated (ATM) kinase and, in cooperation with BRCA2 and RAD51 protein, activates DNA repair through HR.
By contrast, PARP enzyme detects SSBs and recruits different pathways that use the complementary undamaged strand to repair damages and correct mutations. When PARP enzymes are pharmacologically inhibited, the relatively innocuous SSBs cannot be repaired and become lethal when occurring in a cell in which HR pathway of DNA repair is lacking (synthetic lethality). Therefore, germline mutations of BRCA genes or somatic mutations in genes involved in HR, such as ATM and RAD51, can determine loss of DNA repair ability, resulting ultimately in cancer onset.
Focus on PC
It is important to recognize that not only BRCA1 and BRCA2 but also other genes encoding proteins that are involved in DNA repair are essential in generating BRCAness phenotype in PC. Expanding the focus to other DNA repair/recombination genes, both ATM and RAD51 mutations can confer sensitivity to PARP inhibitors. The Stand Up To Cancer–Prostate Cancer Foundation (SU2C-PCF) castration-resistant PC project identified aberrations of DNA-repair genes, including BRCA1, BRCA2, and ATM, at substantially high frequencies (19.3%).19 Mateo et al9 data showed tumor aberrations in DNA-repair genes, mainly BRCA2, ATM, and BRCA1, in 33% of cases. Pritchard et al2 recruited 692 men with documented metastatic PC. DNA-repair gene mutations were identified in 11.8% cases. Of the 20 genes analyzed, pathogenic germline mutations were found in 16 genes, including BRCA2 (5.3%), ATM (1.6%), BRCA1 (0.9%), and RAD51 (0.4%).
Profile of olaparib
Olaparib is a potent PARP inhibitor that induces synthetic lethality in BRCA1/2-deficient tumor cells. In December 2014, the European Medicines Agency approved olaparib to be used in the maintenance therapy of patients affected by BRCA1/2-mutated OC responding to platinum-based chemotherapy; furthermore, in the US, olaparib also has received accelerated approval by the US Food and Drug Administration (US FDA) as a monotherapeutic agent for patients with deleterious or suspected deleterious germline BRCA-mutated advanced OC and who have been treated with three or more prior lines of chemotherapy.10 As observed in OC, PARP inhibitors appears to induce clinical responses in patients with metastatic PC expressing HR DNA-repair defects. In fact, olaparib has been designed to target DNA repair pathway. If PARP enzyme is inhibited, SSB cannot be repaired efficiently and causes cumulative DNA damage. The PARP inhibition, in addition to the deficiency in the HR pathway that occurs in BRCA-mutated PC cells, translates into tumor cells death.
Mechanism of action
Physiologically, PARP is a nuclear enzyme complex discovered in the early 1970s. The PARP family comprises a group of 17 proteins that function in several cellular mechanisms, including transcription, DNA replication, and DNA repair. Of the PARP family, PARP1 and PARP2 are the best-characterized subtypes and their function is paramount to maintain the genomic integrity in the base-excision repair pathway.20 When damage is limited to one of the DNA strands, PARP enzymes are involved to coordinate DNA repair through base excision. It is a zinc-finger DNA-binding protein that detects specifically DNA SSBs. At the site of breakage, PARP catalyzes the transfer of the ADP-ribose moiety from NAD+ substrate to several protein acceptors involved in DNA metabolism and activates the base-excision machinery to repair the SSB.21 Inhibition of PARP enzymes would determine a collapse in the base-excision repair pathway, resulting in the accumulation of DNA damage. In normal cells, PARP inhibition would be of no significant importance because of efficient DSBs repair mechanisms. However, in cells with deficient HR, such as BRCA-mutated PC cells, PARP inhibition would be lethal for tumor cells.
As a consequence, PARP inhibitors would kill tumor cells selectively, with minimal effects on healthy cells with normal gene function.
Pharmacology
Olaparib is available as capsules for oral administration. Following oral administration, olaparib is rapidly absorbed, with peak plasma concentrations (Cmax) typically achieved 1–3 h thereafter.10 Mean apparent volume of distribution, mean apparent plasma clearance, and mean terminal half-life are 40.3 L, 4.55 L/h, and 6.10 h, respectively. Exposure increased proportionally with dose at doses up to 100 mg twice daily but increased in a less than proportional fashion at higher doses.22 Based on safety assessments, the maximum tolerated dose of olaparib is identified as 400 mg twice daily. When given at a dosage of 400 mg twice a day orally, the estimated Cmax ranged from 1.45 to 11.0 μg/mL, with an area under the plasma concentration-time curve from 0–12 h ranged from 6.56 to 122 μg·h/mL. Olaparib is primarily metabolized by cytochrome P-450 isozyme 3A. Drug-related metabolites are eliminated in the urine (35%–50%) and in the feces (12%–60%).
Clinical efficacy in PC
Currently, olaparib is undergoing trials to be used in the treatment of PC. Results of phase II clinical trials showed that patients with BRCA1/2-mutant PC are likely to potentially respond to PARP inhibitors. Details are presented in Table 1. To date, there are no published and ongoing randomized phase III trials evaluating the use of olaparib in PC.
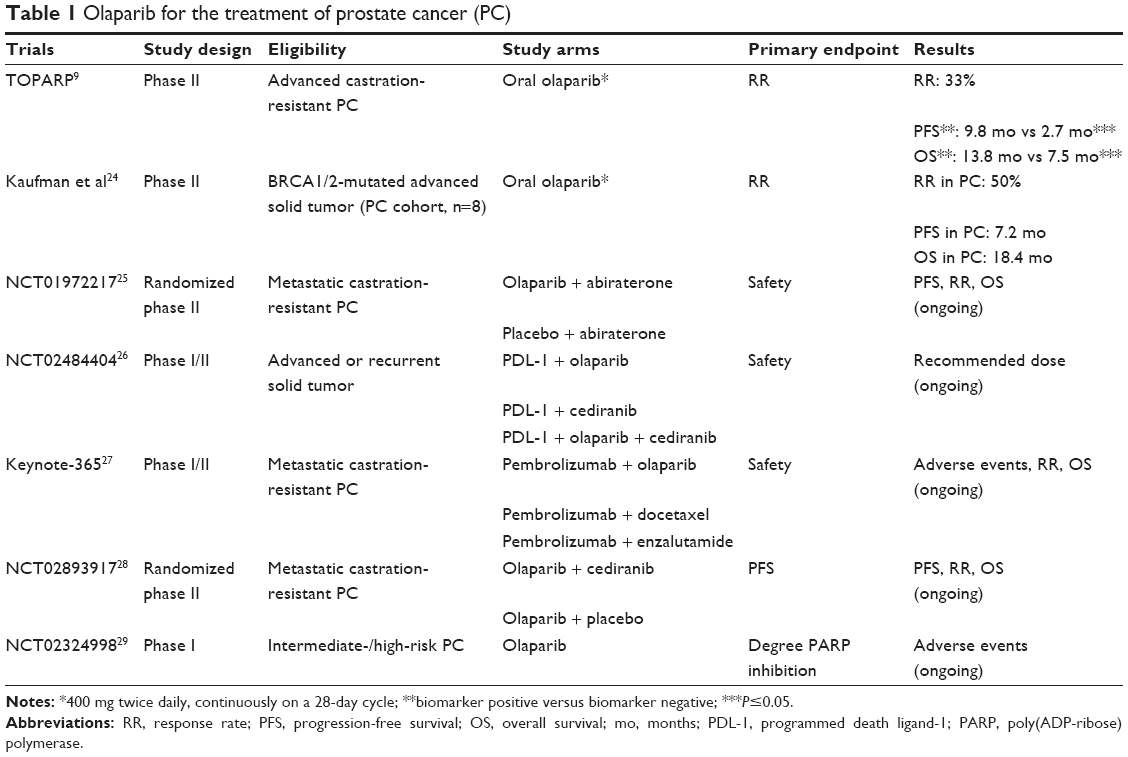 | Table 1 Olaparib for the treatment of prostate cancer (PC) |
The Trial of PARP Inhibition in Prostate Cancer (TOPARP) trial is a two-part adaptive-design phase II trial that aims to evaluate olaparib in patients with advanced castration-resistant PC.23 Overall, 50 patients were enrolled in part A (TOPARP-A) trial.9 The primary outcome was to evaluate the response rate of olaparib tablets at a dose of 400 mg twice a day in this setting of patients. Response was defined as objective response by Response Evaluation Criteria in Solid Tumors (RECIST), prostate-specific antigen (PSA) decline of ≥50%, or a confirmed reduction of circulating tumor cell count from ≥5 cells/7.5 mL of blood at baseline to <5 cells/7.5 mL. Overall, 33%, 22%, and 29% of patients had an objective response, reductions in the PSA level of 50% or more and confirmed reduction in the circulating tumor cell count to <5 cells/7.5 mL, respectively. Patients with aberrations in DNA-repair genes (33%) had a significantly higher response rate (P<0.001) than those with no defects. Of interest, of these patients with DNA repair mutations, 88% showed a response to olaparib, indicating that the specificity for response in this setting of patients was 94%. In addition, olaparib resulted in improved progression-free survival (PFS) (median PFS: 9.8 months versus 2.7 months; P<0.001) and overall survival (OS) (median OS, 13.8 months versus 7.5 months; P=0.05) among patients with DNA-repair defects in their tumor cells. Currently, patients’ recruitment continues for the second part of the trial (TOPARP-B).
A multicenter phase II study was designed to assess the efficacy and safety of olaparib in confirmed genetic BRCA1/2 mutation in different malignant solid tumors.24 In total, 298 patients were enrolled, of whom 8 had metastatic PC. PC patients received oral olaparib 400 mg twice a day. Tumor response rate was assessed according to RECIST and it was 50% (95% confidence interval 15.7–84.3) in those with PC. Median duration of response was 372 days, and median time to onset of response was 54.5 days. PFS and OS were 7.2 months and 18.4 months, respectively.
Ongoing trials
Studies evaluating PARP inhibitors in combination with other PC therapies have been proposed. A randomized phase II trial (NCT01972217) compares the efficacy, safety, and tolerability of olaparib versus placebo when associated with abiraterone treatment in patients with metastatic castration-resistant PC who have received prior docetaxel-based chemotherapy.25
A phase I/II study (NCT02484404) of the anti-programmed death ligand-1 antibody MEDI4736 in combination with olaparib and/or cediranib (vascular endothelial growth factor inhibitor) for different advanced solid tumors, including PC, tests the safety of this drugs combination.26
The Keynote-365 study is a phase Ib/II trial (NCT02861573) that analyzes three different combination therapies in metastatic castration-resistant PC and cohort A will receive olaparib (400 mg twice a day) plus pembrolizumab.27 The purpose of this study is to assess the safety and efficacy of these combination therapies.
A randomized phase II trial (NCT02893917) studies how well olaparib with or without cediranib works in treating patients with metastatic hormone-resistant PC.28 The primary end point is to assess the clinical activity of this drugs’ combination.
Lastly, there is a phase I study (NCT02324998) investigating the feasibility and tolerability of a short course of neoadjuvant treatment with olaparib given prior to radical prostatectomy in patients with early, localized, intermediate, and high-risk PC.29 Primary objective is to determine the pharmacodynamic biomarker effects of olaparib in this setting of patients.
Safety
Tolerability
Globally the tolerability profile of PARP inhibitors is substantially manageable. Hematologic toxicity, including anemia, thrombocytopenia, and neutropenia, and gastrointestinal disorders, primarily nausea and vomiting, are the most commonly reported adverse events. Fatigue is also reported. Rarely, olaparib therapy may increase the risk of developing myelodysplastic syndrome and acute leukemia, but this risk must be further characterized, especially in PC. In fact, there are no cases of myelodysplastic syndrome and acute leukemia reported in PC series. However, at the time of the FDA approval in OC, among 2,618 patients exposed to olaparib, myelodysplastic syndrome and acute leukemia occurred in 0.8% of cases.10
In the TOPARP-A trial, the median duration of olaparib treatment was 12 weeks, and the average delivered-dose intensity was 87%.9 Anemia (20%) and fatigue (12%) were the most frequent grade ≥3 drug-related effects, in keeping with other trials of olaparib. Overall, 26% of cases required a reduction in the dose of olaparib and anemia was the most common indication for dose reduction. Olaparib was permanently discontinued in 6% cases due to adverse events.
Resistance
Not all patients with DNA repair deficiency will respond to olaparib. Several resistance mechanisms that restore defective HR have been proposed, but, at present, which mechanisms contribute to PARP inhibition resistance in PC is still unclear.30 Both ongoing and future trials could detect secondary mutations associated with olaparib resistance.
Patient-focused perspectives
Today, parameters for patient’s quality of life (QoL) should be considered as paramount in the clinical evaluation to significantly improve treatment algorithm.
Olaparib represents an attractive therapy, considering that it has a generally more favorable toxicity profile than cytotoxic chemotherapy. Data regarding the impact of olaparib on QoL are very limited, and further investigations are needed. At this time, QoL data are restricted to secondary endpoints in the phase II trials in OC, in which olaparib versus placebo resulted in equal or better QoL rates.31,32
Survival benefit may be considered as a surrogate of QoL improvement, sparing patients from the symptoms of progressive disease, avoiding additional therapies and their attendant toxicities, and preventing the psychological burden and uncertainty that come with disease progression. However, it should be important to consider QoL assessment in future studies design in PC.
Conclusion
Currently, there is a clinical need and urgency for additional treatments that are well tolerated and that can improve outcomes in PC patients, especially in those with metastatic castrate-resistant disease.
While the use of olaparib is currently restricted to the treatment of OC, ongoing studies will focus on expanding its role in the management of other solid tumors, including PC.
Regarding PC, olaparib warrants further investigation in phase III trials aimed at confirming earlier phase II data. However, if phase III trials fail to show improvement in survival among patients with metastatic castration-resistant PC and DNA-repair defects, definitive conclusions cannot be reached. Based on available data, PARP inhibition seems feasible, but only in patients with appropriate biomarkers status. The preselection of patients based on proper molecular profile should be paramount, as we move toward the new era of targeted therapies. Moreover, the optimal combinations, as well as the sequence of treatments, should have a better understanding. Therefore, it is too early to establish the real impact of olaparib therapy in PC, but if future trials will confirm its efficacy, olaparib might represent an important step toward personalized medicine for PC.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443–453. | ||
Eeles RA. Genetic predisposition to prostate cancer. Prostate Cancer Prostatic Dis. 1999;2(1):9–15. | ||
Leongamornlert D, Saunders E, Dadaev T, et al. Frequent germline deleterious mutations in DNA repair genes in familial prostate cancer cases are associated with advanced disease. Br J Cancer. 2014;110(6):1663–1672. | ||
Castro E, Goh C, Olmos D, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31(14):1748–1757. | ||
Kote-Jarai Z, Leongamornlert D, Saunders E, et al; UKGPCS Collaborators. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br J Cancer. 2011;105(8):1230–1234. | ||
Gallagher DJ, Gaudet MM, Pal P, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res. 2010;16(7):2115–2121. | ||
Dhawan M, Ryan CJ, Ashworth A. DNA repair deficiency is common in advanced prostate cancer: new therapeutic opportunities. Oncologist. 2016;21(8):940–945. | ||
Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. | ||
Musella A, Marchetti C, Gasparri ML, et al. PARP inhibition: a promising therapeutic target in ovarian cancer. Cell Mol Biol (Noisy-le-grand). 2015;61(6):44–61. | ||
Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281(41):30305–30309. | ||
Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98. | ||
Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res. 2011;711(1–2):61–72. | ||
Karlsson KH, Stenerlöw B. Extensive ssDNA end formation at DNA double-strand breaks in non-homologous end-joining deficient cells during the S phase. BMC Mol Biol. 2007;8:97. | ||
Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. 2003;22(37):5792–5812. | ||
Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7(10):739–750. | ||
Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585(1):1–6. | ||
Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95(11):866–871. | ||
Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. | ||
de Murcia JM, Niedergang C, Trucco C, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. 1997;94(14):7303–7307. | ||
Trucco C, Oliver FJ, de Murcia G, Ménissier-de Murcia J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998;26(11):2644–2649. | ||
Marchetti C, Imperiale L, Gasparri ML, et al. Olaparib, PARP1 inhibitor in ovarian cancer. Expert Opin Investig Drugs. 2012;21(10):1575–1584. | ||
Institute of Cancer Research. TOPARP: a Phase II Trial of Olaparib in Patients with Advanced castration Resistant Prostate cancer (TOPARP). Available from: https://clinicaltrials.gov/ct2/show/NCT01682772. NLM identifier: NCT01682772. Accessed January 25, 2017. | ||
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. | ||
AstraZeneca. Ph II study to evaluate olaparib with abiraterone in treating metastatic castration resistant prostate cancer. Available from: https://clinicaltrials.gov/ct2/show/NCT01972217. NLM identifier: NCT01972217. Accessed January 25, 2017. | ||
National Cancer Institute (NCI). Phase I/II study of the anti-programmed death ligand-1 antibody MEDI4736 in combination with olaparib and/or cediranib for advanced solid tumors and advanced or recurrent ovarian, triple negative breast, lung, prostate and colorectal cancers. Available from: https://clinicaltrials.gov/ct2/show/NCT02484404. NLM identifier: NCT02484404. Accessed January 25, 2017. | ||
Merck Sharp & Dohme Corp. Study of pembrolizumab (MK-3475) combination therapies in metastatic castration-resistant prostate cancer (MK-3475-365/KEYNOTE-365). Available from: https://clinicaltrials.gov/ct2/show/NCT02861573. NLM identifier: NCT02861573. Accessed January 25, 2017. | ||
National Cancer Institute (NCI). Olaparib with or without cediranib in treating patients with metastatic hormone-resistant prostate cancer. Available from: https://clinicaltrials.gov/ct2/show/NCT02893917. NLM identifier: NCT02893917. Accessed January 25, 2017. | ||
Cambridge University Hospitals NHS Foundation Trust. Studying the effects of olaparib (± Degarelix) given to men with intermediate/high risk prostate cancer before radical prostatectomy (CaNCaP03). Available from: https://clinicaltrials.gov/ct2/show/NCT02324998. NLM identifier: NCT02324998. Accessed January 25, 2017. | ||
Lavery A, Gilson C, Chowdhury S. PARP inhibitors and stratified treatment of prostate cancer. Expert Rev Anticancer Ther. 2016;16(12):1213–1215. | ||
Ledermann JA, Harter P, Gourley C, et al. Quality of life during olaparib maintenance therapy in platinum-sensitive relapsed serous ovarian cancer. Br J Cancer. 2016;115(11):1313–1320. | ||
Kaye SB, Lubinski J, Matulonis U, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30(4):372–379. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.