")
Back to Journals » International Journal of General Medicine » Volume 14
Defective Allele of the Neuronal Nitric Oxide Synthase Gene Increases Insulin Resistance During Acute Phase of Myocardial Infarction
Authors Nóbrega OT , Campos-Staffico AM, Oliveira EK , Munhoz DB, Moura FA , Carvalho LSF , Soares AASM , Gomes CM , Tonet-Furioso AC , Sposito AC
Received 23 May 2021
Accepted for publication 30 June 2021
Published 20 July 2021 Volume 2021:14 Pages 3669—3676
DOI https://doi.org/10.2147/IJGM.S313661
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Otávio T Nóbrega,1 Alessandra M Campos-Staffico,2 Elayne Kelen Oliveira,3 Daniel B Munhoz,3 Filipe A Moura,3 Luis Sérgio F Carvalho,4 Alexandre Anderson SM Soares,1,5 Ciro M Gomes,1,6 Audrey C Tonet-Furioso,7 Andrei C Sposito1,3 On behalf of Brasilia Heart Study and the Brasilia Heart Study Group
1Programa de Pós-Graduação em Ciências Médicas, Faculdade de Medicina, Universidade de Brasília (UnB), Brasília, DF, Brazil; 2Departamento de Farmácia, Faculdade de Ciências da Saúde, Universidade de Brasília (UnB), Brasília, DF, Brazil; 3Departamento de Cardiologia, Faculdade de Ciências Médicas, Universidade Estadual de Campinas (Unicamp), Campinas, SP, Brazil; 4Centro de Inovação, Ensino e Pesquisa, Instituto de Gestão Estratégica em Saúde do Distrito Federal, Brasília, DF, Brazil; 5Serviço de Cardiologia, Instituto Biocárdios, Brasília, DF, Brazil; 6Laboratório de Dermatomicologia, Faculdade de Medicina, Universidade de Brasília (UnB), Brasília, DF, Brazil; 7Programa de Pós-Graduação em Gerontologia, Universidade Católica de Brasília (UCB-DF), Taguatinga, DF, Brazil
Correspondence: Andrei C Sposito
Laboratório de Biologia Vascular e Aterosclerose (AteroLab), Departamento de Cardiologia, Faculdade de Ciências Médicas, Universidade Estadual de Campinas (Unicamp), 126 Rua Tessália Vieira de Camargo, Campinas, 13083-887, SP, Brazil
Tel +55 19 3521 9590
Fax +55 19 3289 4107
Email [email protected]
Background: Glycemic disorders are strong predictors of mortality in ST-elevation myocardial infarction (STEMI) patients, and disruption in nitric oxide (NO) production is associated with insulin-resistant states. We evaluated whether a defective allele of the neuronal nitric oxide synthase (nNOS) gene (NOS1) might influence insulin response and blood-glucose balance during the acute phase of STEMI and if post-infarction total plasma-NO levels and vasodilation scores varied across nNOS genotypes.
Methods: Consecutive patients with STEMI (n=354) underwent clinical evaluations and genotyping for the promoter variation rs41279104. In-hospital clinical and blood evaluations were performed at admission and five days after STEMI, with glycemic, insulinemic, and disposition indices assessed at the same times. Flow-mediated dilation (FMD) was assessed by reactive hyperemia on the 30th day.
Results: Homozygotes for the defective allele (A) showed lower glycemia and insulin sensitivity on day 1 while showing the highest β-cell function and no changes in the circulating NO pool, which is compatible with hyperresponsive β cells counteracting the inherent glucose-resistant state of AA patients. At day 5, glycemic scores had shifted to indicate greater insulin sensitivity among A homozygotes, paralleled by a significant yet poor increase in NO bioavailability compared to that among G carriers. All in all, defective homozygotes showed greater insulin resistance at admission that had reversed by 5 days after STEMI. Even so, A carriers developed lower FMD scores compared to G homozygotes after the acute phase.
Conclusion: A defective nNOS allele (and due decline in NO production) seemed to elicit a hyperinsulinemia response to compensate for an insulin-resistant state during the acute phase of STEMI and to be associated with poor endothelial function after the acute phase.
Keywords: nitric oxide, insulin, blood glucose, vasodilation, polymorphism, myocardial infarction
Introduction
Myocardial infarction (MI) is a stressful condition that predisposes individuals to a significant increase in blood-glucose levels. Hyperglycemia prevails in up to 50% of all patients with ST-elevation MI (STEMI), though only 20%–25% have been diagnosed with diabetes mellitus.1 Mechanistically, insulin resistance causes proinflammatory and procoagulant states that can enhance myocardial injury from excessive coronary constriction, endothelial dysfunction, and platelet aggregation.2 As such, hyperglycemia is an independent predictor of mortality in STEMI,3 with clinical and observational data confirming that normalization of blood glucose is associated with improved outcomes and survival.4
Nitric oxide (NO) is a key molecule during ischemic disorders, due to its beneficial effects in controlling myocardial contractility, limiting cardiac remodeling after MI, and promoting vasodilation.5 However, NO production plays a central role not only in ischemic conditions but also in insulin metabolism. Pharmacological inhibition of murine NO production induces in vivo insulin resistance in skeletal and cardiac muscles and adipose tissue.6 Similarly, disruptions in endothelial and neuronal NO synthase (nNOS) genes (NOS3 and NOS1, respectively) also cause insulin resistance in mice.7,8 In humans, diminished NO production has been associated with insulin-resistant status, such as type 2 diabetes mellitus, obesity, hypertension, and dyslipidemia.9–11
Several single-nucleotide polymorphisms (SNPs) have been explored by observational studies throughout the globe on the subject of insulin resistance,12,13 and based on the contribution of these, systematic reviews and meta-analysis studies can be built. No genome-wide study has so far addressed the genetics of glycemic homeostasis in the immediate post-MI phase. nNOS is a constitutive enzyme mainly expressed in the central nervous system that is implicated in autonomic regulation of heart rhythm, microcirculation, and contractility.14 Given that the nNOS promoter region SNP rs41279104 decreases gene expression by 30% in vitro and 50% in vivo,15,16 we decided to investigate whether this surrogate for NO production might influence glycemic and insulinemic scores during the in-hospital acute phase of STEMI and post-STEMI endothelial function.
Methods
Participants and Study Design
The Brazilian Heart Study (BHS), an observational prospective cohort registered at ClinicalTrials.gov (NCT02062554)17 comprising 354 participants. Briefly, consecutive STEMI patients admitted to the Acute Coronary Care Unit at the Hospital de Base do Distrito Federal (Brazil) who met these inclusion criteria were enrolled: ≤24 hours from the onset of MI symptoms, ST-segment elevation ≥1 mm (frontal plane) or 2 mm (horizontal) in contiguous leads, and myocardial necrosis evidenced by scores ≥99th percentile of the reference limit for creatine kinase (CK)–MB (25 U/L) and troponin I (0.04 ng/mL), followed by a decline in both. The main exclusion criteria were any cognitive impairment that precluded responses to medical queries and/or the occurrence of neoplastic disease, chronic renal failure, severe liver disease, or heart failure (based on available clinical, laboratory, and/or image evaluations) concomitant with the STEMI episode.
A complete medical evaluation was performed upon admission (D1), with patients followed in hospital until the fifth day (D5) after MI. Attending physicians decided on each medical treatment, including the choice of reperfusion therapy, without influence from the investigation team. Functional endothelial vascular reactivity was assessed 30 days after STEMI.
Clinical Evaluation
In the first 24 hours after the onset of symptoms, a standardized interview was performed to assess medical history, all drugs currently used, and lifestyle factors. To assist these assessments, blood samples were taken. Hypertension was defined either by use of antihypertensive drugs prior to hospital admission or by repeatedly elevated blood pressure >140/90 mmHg during the hospital stay. Diabetes was defined by use of hypoglycemic drugs and/or insulin, fasting blood glucose ≥126 mg/dL, or glycated hemoglobin (HbA1c) ≥6.5%. Current smoking was defined as one or more cigarettes a day for >1 year before the coronary event. Former smoking was defined as smoking cessation for at least 6 months prior to hospitalization. Sedentary lifestyle was defined as not practicing physical activities >30 minutes over at least 4 days a week. Drugs with vasodilation effects were defined by in-hospital use of calcium-channel blockers, angiotensin-receptor blockers, and/or angiotensin converting–enzyme inhibitors. Nitrates were not included by ruling out from analyses participants exposed to these drugs during hospitalization. Medical and biochemical assessments were repeated as clinically needed during the hospital stay, with glycemic scores being necessarily reassessed at D5.
Biochemical Analyses
Blood samples were drawn at admission and a second round of clinical biochemistry assays done on D5 after a 12-hour overnight fast. Samples in EDTA were centrifuged at 4,500 rpm for 15 minutes at 5°C, and plasma was used in an automatic chemical analyzer to yield assessments in duplicate of glucose, HbA1c, insulin, C-peptide, triglycerides, total cholesterol, high density–lipoprotein cholesterol, creatinine, ultrasensitive C, troponin, and CK-MB with reagents from Roche Diagnostics (Mannheim, Germany), Bio-Rad Laboratories (Hercules, USA), or Dade Behring (Marburg, Germany). Low density–lipoprotein cholesterol was calculated by the Friedewald formula. Estimated glomerular filtration rate (mL/min/1.73 m2) was calculated by the Chronic Kidney Disease–Epidemiology Collaboration equation: 141 × min (SCr × 0.0113/k, 1)α × max (SCr × 0.0113/k, 1)−1.209 × 0.993Age × 1.018 (if female) × 1.159 (if black), where SCr is serum creatinine, k is 0.7 for females and 0.9 for males, and α is −0.329 for females and −0.411 for males.
Glucose Homeostasis-Model Assessment
The Homeostasis Model Assessment (HOMA) Calculator version 2.2.2, was used to assess insulin sensitivity (HOMA2S) based on plasma insulin and β-cell function (HOMA2B) and plasma C-peptide18 in all enrolled participants. The disposition index (DI) was calculated as (HOMA2B × HOMA2S)/100.
DNA Extraction and Analysis
Genomic DNA was obtained from peripheral blood leukocytes. The rs41279104 NOS1 SNP was genotyped using a PCR–restriction fragment-length polymorphism method originally published elsewhere.19 Briefly, GenBank was searched for the NOS1 gene (NG_011991.2) and a pair of primers was designed to amplify a 150 bp fragment with the SNP of interest (−84 G/A, forward 5ʹ-CTGACTGCCCTTGTCTCTCC-3ʹ and reverse 5ʹ-GCGACTGGGGTTTAATTGAC-3ʹ). Each reaction (25 µL) was composed of 100 ng DNA, 25 mM Tris-HCl pH 8.3, 75 mM KCl, 1.5 mM MgCl2, 0.2 µM of each primer, 0.2 µM of each dNTP, 0.25 mg/mL ovalbumin and 0.5 U Taq polymerase (Phoneutria, Minas Gerais, Brazil). Amplification had an initial denaturation at 94°C for 2 minutes, followed by 36 cycles of denaturation at 94°C for 40 seconds, annealing at 63°C for 45 seconds, and extension at 72°C for 50 seconds, followed by a final extension at 72°C for 5 minutes. Enzymatic digestion was carried out at 37°C overnight in 15 µL using 0.1 U Fnu4HI (New England BioLabs, USA) to yield two fragments (92 and 58 bp) visible under electrophoresis in 2.2% agarose gel.
Measurement of Total NO/Nitrite/Nitrate
For quantification of total serum levels of NO, the Griess diazotization reaction based on conversion of nitrate to nitrite (NO2−) by nitrate reductase was employed, performed following the manufacturer’s instructions (R&D Systems), with readings done by colorimetric detection in a Biotek ELX 800 (DeMorellis) device, set at a 540 nm wavelength.
Brachial Artery Reactivity
Endothelial-dependent vasodilation was assessed by ischemia-induced reactive hyperemia on the 30th day post-STEMI after 12-hour overnight fasting and 24 hours after withdrawal of vasoactive drugs.20 Briefly, after 10 minutes of rest, longitudinal images of the brachial artery were taken by an experienced physician blinded to the patient’s history using ultrasound with a high-resolution linear transducer (IE33, 3–9 MHz transducer, Philips Medical Systems) synchronized with electrocardiogram monitoring and participants in a supine position. A blood-pressure cuff placed on the forearm was inflated to 50 mmHg above the systolic pressure for 5 minutes and then deflated. The flow-mediated dilation (FMD) was scanned for 2 minutes and the change in diameter calculated in comparison to the resting state.
Statistical Analyses
Assumptions for parametric models (linearity, normality of distribution, and equal variance) were checked using histograms, normal-probability plots, and residual scatterplots by means of the Kolmogorov–Smirnov test. Normally distributed and skewed baseline data were compared using ANOVA or the Kruskal–Wallis test, respectively, with Tukey’s post hoc test if necessary. ANCOVA was used to adjust the association of the SNP rs41279104 with glycemic and insulinemic scores, as well as with NO levels and brachial artery reactivity for confounding factors, such as age, sex, diabetes mellitus, peak CK-MB, and in-hospital use of drugs as appropriate to each case. Changes between D1 and D5 were additionally adjusted for baseline levels to attenuate the effect of regression toward the mean. Categorical data were tested using the #x1D712;2 test. Two-sided p<0.05 was considered statistically significant, with analyses performed using SPSS 25.0.
Results
There were no significant differences in baseline traits, such as age, sex, anthropometry, and clinical history, or in terms of in-hospital laboratory profile and treatment practices between genotypes (Table 1). More importantly, cases previously diagnosed with diabetes mellitus, arterial hypertension, and other metabolic disorders were equally distributed across genotypic groups.
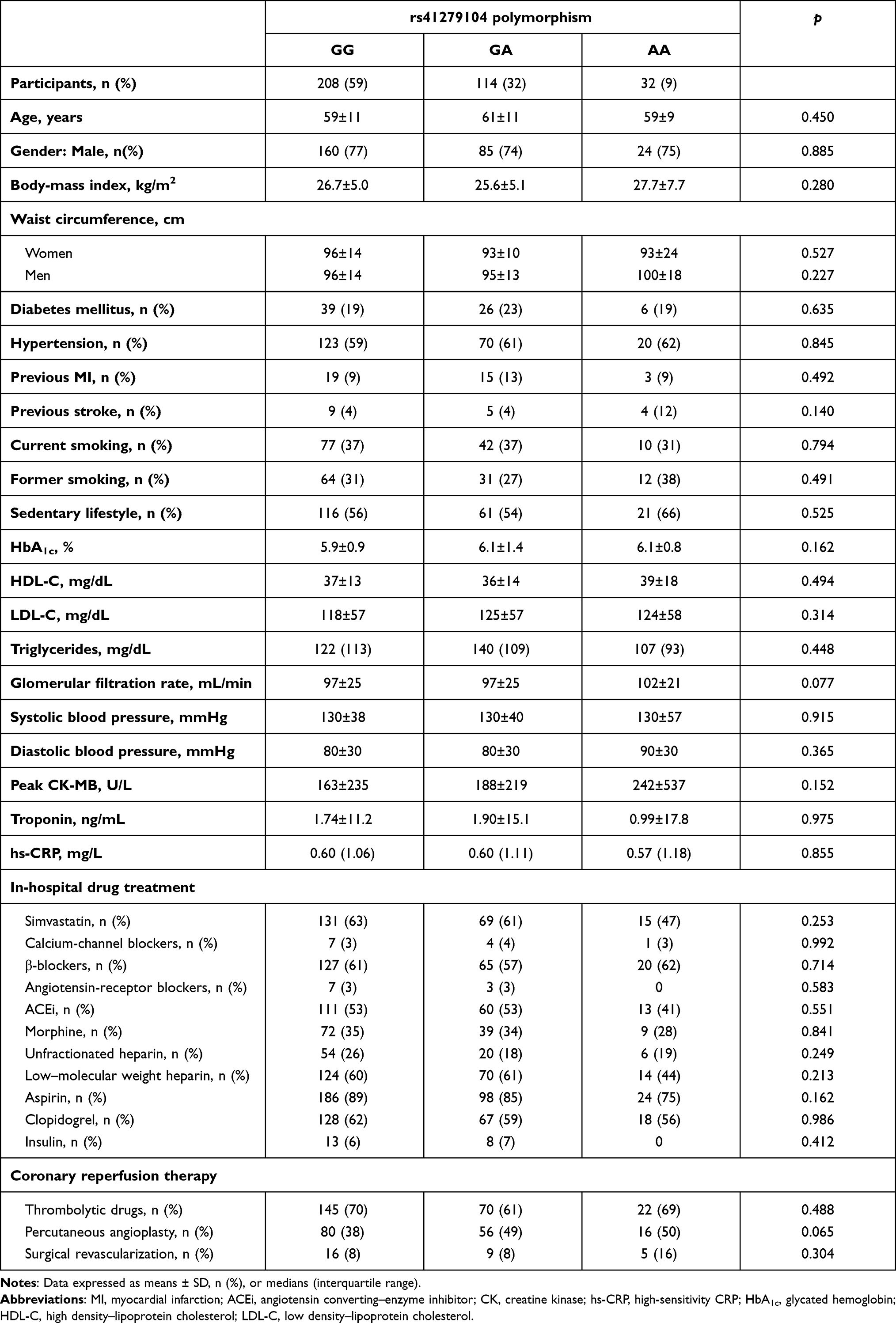 |
Table 1 Baseline data |
Glucose Metabolism and Insulin Response
Homozygotes for the defective allele (A) showed lower glycemia and insulin sensitivity at D1 compared to carriers of the G allele (Table 2). Accordingly, the pancreatic function (HOMA2B) of defective homozygotes — the blood glucose of which was the lowest throughout all the acute phase of STEMI — showed the highest β-cell function on D1, followed by a significant drop within 5 days, in contrast to the positive change observed among G carriers in the period (ΔD5–D1). This is compatible with hyperresponsive β cells counteracting the reduced peripheral insulin sensitivity of AA patients at STEMI onset. Still among A homozygotes, this scenario radically shifts at D5 when HOMA2S and HOMA2B assume scores diametrically opposed to those at D1. Also, G carriers had undergone lower drops in blood glucose by the end of the acute phase of MI (ΔD5–D1) than AA patients.
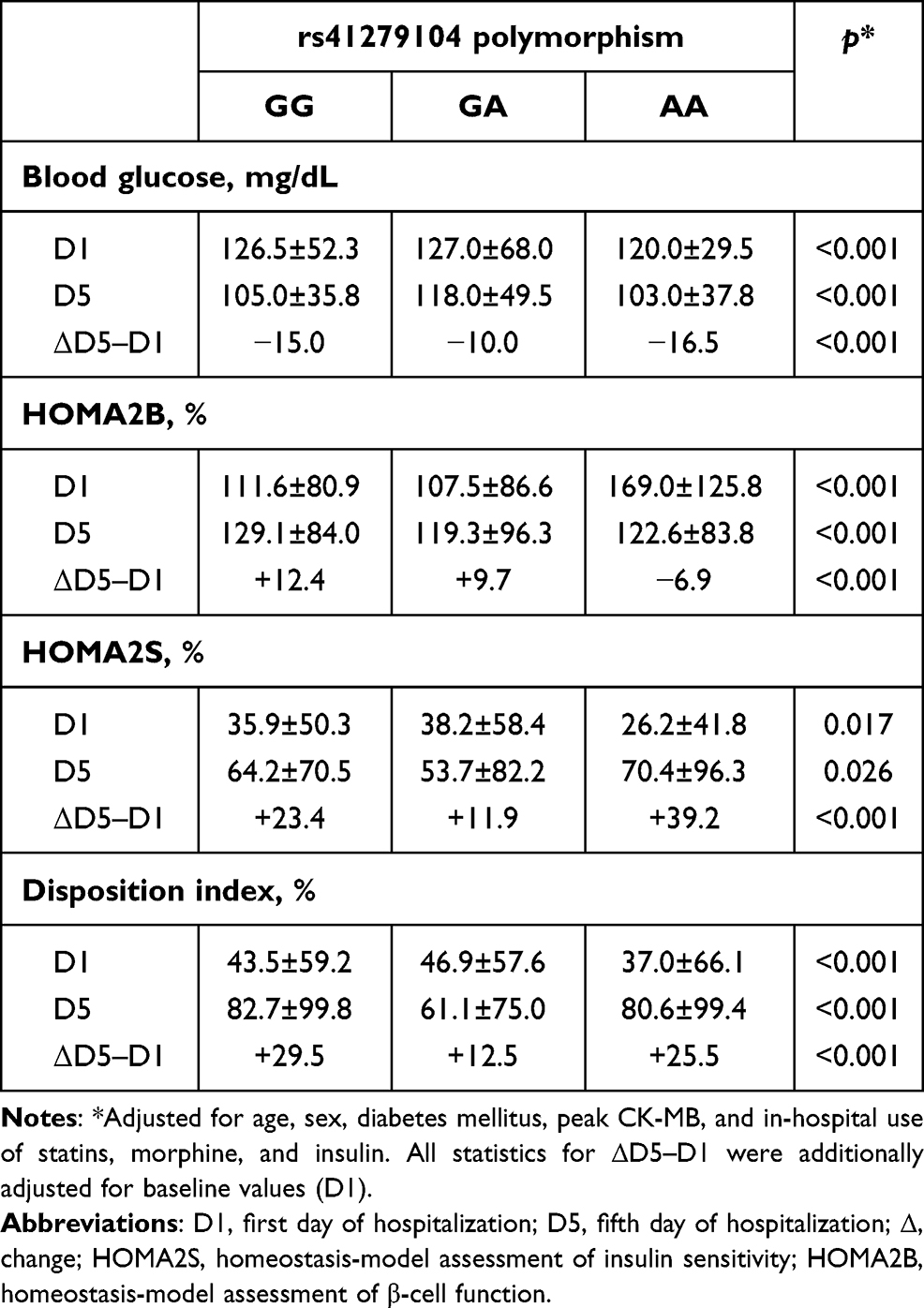 |
Table 2 Comparative analysis of glycemic characteristics across nNOS genotypes |
Insulin secretion and insulin sensitivity are connected via a negative-feedback loop, where pancreatic β cells compensate for changes in whole-body sensitivity by an inversely proportional change in secretion (y = constant/x).21 The DI — the product of HOMA2B and HOMA2S — tends to remain roughly constant in healthy conditions, and deviation is observed when β-cell function is lost and/or insulin-mediated mechanisms to overcome insulin resistance fail, as was the case among A homozygotes at D1 (influenced by the very low scores of HOMA2S), followed by a recovery of the index by D5. Conversely, G carriers had higher index scores at admission, resulting in smaller changes in DI in the period (ΔD5–D1).
NO/Nitrite/Nitrate Levels and Flow-Mediated Dilation
No significant differences were observed in NO levels at STEMI onset (D1). However, defective homozygotes showed the lowest NO levels compared to GG and GA patients at D5 (Tukey post hoc p=0.038 and 0.042, respectively) and the smallest increase in NO levels among all genotypic groups (Table 3). Also, FMD values obtained 30 days after STEMI showed that G homozygotes reached higher dilation scores compared to heterozygotes and AA homozygotes (Tukey post hoc p=0.030 and 0.005, respectively).
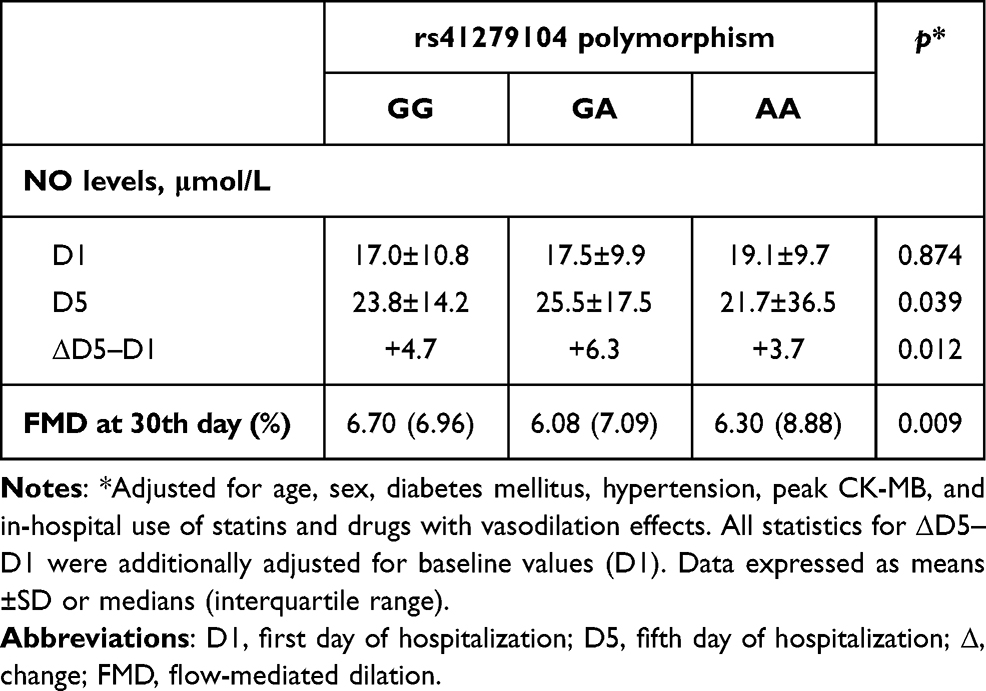 |
Table 3 Comparative analysis of plasma NO levels and flow-mediated vasodilation across nNOS genotypes |
Discussion
Insulin resistance is undeniably related to NO production, an assumption corroborated by epidemiological studies that have associated type 2 diabetes mellitus, obesity, hypertension, and dyslipidemia with reduced NO production.9–11 Although this association is supported by a large body of evidence, the role of neuronal NOS in insulin resistance remains elusive. The present study explored this matter by evaluating a defective isoform of the nNOS gene — the expression of which in vivo is reduced by up to 50% — in actual clinical settings of a major, life-threatening vascular event. During this research, we found that both availability of and sensitivity to insulin varied differently across genotypes and post-MI moments.
Insulin is a well-known vasodilator, and its vasodilatory effect is mediated by NO generation.22 Therefore, the peak production of insulin at D1 observed herein appears to be a compensatory mechanism for the resistant baseline state among the defective homozygotes. It is possible that the unnoticed changes in NO levels at D1 could have occurred from the minor role of the nNOS isoform (compared to eNOS) to account for the systemic pool,23 and the fact that insulin requires a relatively long time (4 hours) to activate NOS so as to contribute to systemic availability.24 Therefore, this phenomenon of relatively high glucose uptake among defective nNOS producers in the immediate hours after STEMI probably relied on an NO-independent mechanism based on hyperinsulinemia. A burst in insulin secretion may compensate for naturally reduced nNOS expression and activity in the paraventricular nucleus, where low NO production upregulates the sympathetic tone via catecholamine release and α-adrenergic activation,25,26 usually leading to an insulin-resistant state27 that tends to be aggravated in a context of myocardial injury.
Interestingly, the pattern of high glucose uptake observed among A homozygotes at D1 was sustained in the course of the acute post-MI phase, mostly attributable to gains in peripheral insulin sensitivity, with a possible contribution by an increase in total NO bioavailability, but not solely, since the NO reservoir was not enhanced to the extent reached among G carriers. As a matter of fact, glycemic scores at D5 and time-course changes (ΔD5–D1) among defective patients were likely to have derived from a scenario of endothelial dysfunction objectively attested by NO shortage, with eNOS — the major isoform — unable to compensate for the endogenously defective nNOS. All in all, this scenario of whole-body endocrine adaptation in glycemic balance is consistent with an impaired NO-generation system among defective (A) homozygotes.
In sum, the interplay between glucose balance and alleles of nNOS during the acute phase of STEMI revealed that AA individuals exhibited endocrine compensation for the inherent inability to produce adequate amounts of NO compared to G carriers. After all, defective homozygotes showed more severe insulin resistance on admission than other genotypes on the DI, which appeared to be compensated within 5 days and possibly due to the hyperinsulinemia response at STEMI onset. In the long run, defective homozygotes were able to recover from endothelial dysfunction, since their flow-mediated dilation was equal to that of heterozygotes (p=0.190) and mildly lower in clinical terms than G homozygotes (p=0.005). Since FMD scores are strong predictors of secondary MI events28 and depend on NO bioavailability,29 our results call for research on long-term post-STEMI outcomes among carriers of defective nNOS, as well as watchful surveillance of the clinical evolution of patients that respond via hyperinsulinemia.
Important limitations need to be acknowledged when interpreting our findings. Firstly, the study exclusively enrolled STEMI patients with a great extent of myocardial injury, which prevents extrapolations to other acute coronary syndromes. Secondly, the observational design precludes any causality inference. Thirdly, the sample size was limited: STEMI is an uncommon, lethal coronary disease, and the possibility of selection bias cannot be ruled out. These limitations may have favored over- or underestimation of the magnitude of the associations found and may also have influenced the lack of statistical significance in some of the analyses. However, the uniqueness of the study population and its consistency with biological processes in the acute phase of MI make it relevant and plausible. Moreover, our data did not seem biased by an unleveled distribution of diabetic cases or by consumption of pharmaceutical products across genotypic groups.
Conclusion
The assumed decline in NO production mediated by the defective nNOS polymorphic gene is associated with an insulin-resistant state during the acute phase of STEMI (which tends to be compensated by a hyperinsulinemia response) and with poor endothelial function (expressed by low FMD scores) after the acute phase of STEMI.
Study Design
The STROBE statement, accessible via the EQUATOR network, was used as a guideline for reporting this observational study and improving the consistency and transparency of the data presented and interpreted herein.
Data Sharing Statement
Deidentified participants data can be made available at any time upon reasonable request sent to the corresponding author: [email protected].
Ethics Approval
The study was carried out in accordance with the Declaration of Helsinki and was approved by the Ethics Committee in Research of the State Department of Health of the Brazilian Federal District (official letters 082/2006 and 354/2011).
Consent to Participate
All participants were enrolled after informed consent had been signed by the patient or next of kin.
Consent for Publication
All authors reviewed the final version of the manuscript, and concur with its submission in the present format.
Acknowledgments
The authors are indebted to the core staff of the Brasilia Heart Study for providing the environment for the analyses herein: Osório LR Almeida, José Carlos Quinaglia e Silva, Riobaldo MR Cintra, Ana Laura R Araújo, Simone N Santos, Valéria N Figueiredo, Joalbo M Andrade, Otávio R Coelho, and Wilson Nadruz Jr.
Funding
This research was supported with grants 471016/2011-0 (CNPq) and 193.000.032-2012 (FAPDF), with a stipend to AC Tonet-Furioso (CAPES, finance code 001) and fellowships for productivity in research to AC Sposito and OT Nóbrega from CNPq.
Disclosure
The authors declare they have no conflicts of interest for this work.
References
1. Wahab NN, Cowden EA, Pearce NJ, et al. Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era? J Am Coll Cardiol. 2002;40(10):1748–1754. doi:10.1016/S0735-1097(02)02483-X
2. Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113(15):1888–1904. doi:10.1161/CIRCULATIONAHA.105.563213
3. Zeller M, Steg PG, Ravisy J, et al. Prevalence and impact of metabolic syndrome on hospital outcomes in acute myocardial infarction. Arch Intern Med. 2005;165(10):1192–1198. doi:10.1001/archinte.165.10.1192
4. Kosiborod M, Inzucchi SE, Krumholz HM, et al. Glucose normalization and outcomes in patients with acute myocardial infarction. Arch Intern Med. 2009;169(5):438–446. doi:10.1001/archinternmed.2008.593
5. Rastaldo R, Pagliaro P, Cappello S, et al. Nitric oxide and cardiac function. Life Sci. 2007;81(10):779–793. doi:10.1016/j.lfs.2007.07.019
6. Roy D, Perreault M, Marette A. Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. Am J Physiol. 1998;274(4):E692–699. doi:10.1152/ajpendo.1998.274.4.E692
7. Duplain H, Burcelin R, Sartori C, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104(3):342–345. doi:10.1161/01.CIR.104.3.342
8. Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes. 2000;49(5):684–687. doi:10.2337/diabetes.49.5.684
9. Gilligan DM, Guetta V, Panza JA, Garcia CE, Quyyumi AA, Cannon RO
10. Zeiher AM, Drexler H, Wollschlager H, Just H. Modulation of coronary vasomotor tone in humans. Progressive endothelial dysfunction with different early stages of coronary atherosclerosis. Circulation. 1991;83(2):391–401. doi:10.1161/01.CIR.83.2.391
11. Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97(11):2601–2610. doi:10.1172/JCI118709
12. Ferreira AP, Ferreira CB, Souza VC, et al. Risk of glycemic disorder in elderly women adjusted by anthropometric parameters and cytokine genotypes. Rev Assoc Med Bras. 2011;57(5):565–569. doi:10.1590/S0104-42302011000500016
13. Alharbi KK, Abudawood M, Khan IA. Amino-acid amendment of arginine-325-tryptophan in rs13266634 genetic polymorphism studies of the SLC30A8 gene with type 2 diabetes-mellitus patients featuring a positive family history in the Saudi population. J King Saud Univ Sci. 2021;33(1):101258. doi:10.1016/j.jksus.2020.101258
14. Zhang YH, Jin CZ, Jang JH, Wang Y. Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology. J Physiol. 2014;592(15):3189–3200. doi:10.1113/jphysiol.2013.270306
15. Saur D, Vanderwinden JM, Seidler B, Schmid RM, De Laet MH, Allescher HD. Single-nucleotide promoter polymorphism alters transcription of neuronal nitric oxide synthase exon 1c in infantile hypertrophic pyloric stenosis. Proc Natl Acad Sci U S A. 2004;101(6):1662–1667. doi:10.1073/pnas.0305473101
16. Cui H, Nishiguchi N, Yanagi M, et al. A putative cis-acting polymorphism in the NOS1 gene is associated with schizophrenia and NOS1 immunoreactivity in the postmortem brain. Schizophr Res. 2010;121(1–3):172–178. doi:10.1016/j.schres.2010.05.003
17. Machado-Silva W, Alfinito-Kreis R, Carvalho LS, et al. Endothelial nitric oxide synthase genotypes modulate peripheral vasodilatory properties after myocardial infarction. Gene. 2015;568(2):165–169. doi:10.1016/j.gene.2015.05.042
18. Caumo A, Perseghin G, Brunani A, Luzi L. New insights on the simultaneous assessment of insulin sensitivity and beta-cell function with the HOMA2 method. Diabetes Care. 2006;29(12):2733–2734. doi:10.2337/dc06-0070
19. Reif A, Herterich S, Strobel A, et al. A neuronal nitric oxide synthase (NOS-I) haplotype associated with schizophrenia modifies prefrontal cortex function. Mol Psychiatry. 2006;11(3):286–300. doi:10.1038/sj.mp.4001779
20. Corretti MC, Anderson TJ, Benjamin EJ, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39(2):257–265. doi:10.1016/S0735-1097(01)01746-6
21. Kahn SE, Prigeon RL, McCulloch DK, et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes. 1993;42(11):1663–1672. doi:10.2337/diab.42.11.1663
22. Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94(3):1172–1179. doi:10.1172/JCI117433
23. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):
24. Kashyap SR, Roman LJ, Lamont J, et al. Insulin resistance is associated with impaired nitric oxide synthase activity in skeletal muscle of type 2 diabetic subjects. J Clin Endocrinol Metab. 2005;90(2):1100–1105. doi:10.1210/jc.2004-0745
25. Turini P, Thalmann S, Jayet PY, et al. Insulin resistance in mice lacking neuronal nitric oxide synthase is related to an alpha-adrenergic mechanism. Swiss Med Wkly. 2007;137(49–50):700–704.
26. Lu QB, Feng XM, Tong N, et al. Neuronal and endothelial nitric oxide synthases in the paraventricular nucleus modulate sympathetic overdrive in insulin-resistant rats. PLoS One. 2015;10(10):e0140762. doi:10.1371/journal.pone.0140762
27. Esler M, Rumantir M, Wiesner G, Kaye D, Hastings J, Lambert G. Sympathetic nervous system and insulin resistance: from obesity to diabetes. Am J Hypertens. 2001;14(11 Pt 2):304S–309S. doi:10.1016/S0895-7061(01)02236-1
28. Guazzi M, Reina G, Gripari P, Tumminello G, Vicenzi M, Arena R. Prognostic value of flow-mediated dilatation following myocardial infarction. Int J Cardiol. 2009;132(1):45–50. doi:10.1016/j.ijcard.2007.10.036
29. Kannenkeril D, Bosch A, Kolwelter J, et al. Dependency of flow-mediated vasodilatation from basal nitric oxide activity. Clin Physiol Funct Imaging. 2021;41(4):310–316. doi:10.1111/cpf.12696
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.