")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Decreased Risk of Ischemic Heart Disease in Individuals with Severe Alpha 1-Antitrypsin Deficiency (PiZZ) in Comparison with the General Population
Authors Tanash H, Ekström M , Basil N, Rönmark E, Lindberg A, Piitulainen E
Received 27 January 2020
Accepted for publication 20 April 2020
Published 4 June 2020 Volume 2020:15 Pages 1245—1252
DOI https://doi.org/10.2147/COPD.S247377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Hanan Tanash,1 Magnus Ekström,2 Nawfal Basil,1 Eva Rönmark,3 Anne Lindberg,4 Eeva Piitulainen1
1Departments of Respiratory Medicine, Skåne University Hospital, Lund University, Lund, Sweden; 2Faculty of Medicine, Department of Clinical Sciences Lund, Respiratory Medicine and Allergology, Lund University, Lund, Sweden; 3Department of Public Health and Clinical Medicine, The OLIN Unit, Division of Occupational and Environmental Medicine, Umeå University, Umeå, Sweden; 4Department of Public Health and Clinical Medicine, Division of Medicine, Umeå University, Umeå, Sweden
Correspondence: Hanan Tanash
Department of Respiratory Medicine, Malmö S-205 02, Sweden
Tel +46 40 33 10 00
Fax +46 40 33 62 25
Email [email protected]
Background: Severe alpha-1-antitrypsin deficiency (AATD) is an established risk factor for chronic obstructive pulmonary disease (COPD) and liver disease, but the effect on the incidence of ischemic heart disease (IHD) is not well known. The aim was to evaluate the risk of incident IHD in patients with severe AATD compared with a random sample of the general population, with known smoking habits.
Methods: AAT-deficient individuals, phenotype PiZZ (n=1545), were included in the Swedish National AATD Register. Controls (n=5883) were selected from population-based cohorts in Northern Sweden. Data on IHD and comorbidities were obtained by nationwide cross-linkage with the Swedish National Patient Register. Risk factors for incident IHD were analyzed using Cox regression, adjusted for age, gender, smoking status and the presence of COPD, hypertension, hyperlipidemia and diabetes.
Results: At inclusion, 46% of the PiZZ individuals and 53% of the controls were never-smokers. During follow-up (median 16 years; range 0.2– 23), 8% (n=123) of PiZZ individuals and 12% (n=690) of controls developed IHD. The controls had a significantly higher risk for incident IHD than the PiZZ individuals, with adjusted hazard ratio (HR) of 1.8 (95% CI 1.4– 2.3). The risk was higher for controls in both ever-smokers (HR 2.1; 95% CI 1.5– 2.9) and never-smokers (HR 1.5; 95% CI 1.1– 2.2).
Conclusion: PiZZ individuals have a lower risk of developing incident ischemic heart disease than the control subjects with known smoking habits, who had been randomly selected from population-based cohorts.
Keywords: alpha-1-antitrypsin deficiency, chronic obstructive pulmonary disease, ischemic heart disease, smoking
Introduction
Alpha-1-antitrypsin (AAT) is a glycoprotein and acute-phase protein that is a member of the serine proteinase inhibitor family. Inhibiting the action of proteolytic enzymes (especially neutrophil elastase) is the primary role of AAT in all tissues, including lung parenchyma and the arterial wall.1 This function impedes excessive degradation of elastin and collagen fibers in extracellular matrix and inflammatory cascades that result in inflammation-associated tissue remodeling.2,3 Severe alpha-1-antitrypsin deficiency (AATD), phenotype PiZZ, is an autosomal, co-dominant hereditary condition and a strong risk factor for developing chronic obstructive pulmonary disease (COPD), more specifically panacinar emphysema.1 AATD leads to polymerization of the AAT molecule in the liver cells, leading to its accumulation within the hepatocytes, which also leads to an increased risk of liver disease.4,5 AATD has also been implicated in the pathogenesis of several non-respiratory diseases such as systemic vasculitis, necrotizing panniculitis, and various inflammatory and neoplastic conditions.6,7
The effect of severe AATD on the risk of developing cardiovascular diseases is unclear. From the broad biological activity of AAT, its genetic deficit could be an independent determinant of cardiovascular effects. Previous reports on such an association have been conflicting.8–13 The potential causal relation between AATD and risk of ischemic heart disease (IHD) is important because the prevalence of AATD varies within the European population from 1 in 1500 to 1 in 3500, and because IHD is the leading cause of morbidity and mortality worldwide, with an increased risk in patients with COPD.14–16
The aim of this study was to determine whether individuals with severe AATD included in the Swedish national AATD register, are at increased risk of developing ischemic heart disease when compared with controls from the general population with known smoking habits.
Methods
This was a longitudinal, national, register-based study comparing individuals with severe AATD with randomly selected, population-based controls.
The Study Population and Data Collection
AATD Individuals
The AAT-deficient individuals were identified using the Swedish National AATD Register.17 Inclusion criteria to the register are the age of 18 years a minimum, severe AATD (phenotype PiZZ) and written, informed consent. More than 95% of the AATD patients identified in Sweden agree to participate in the register. The reasons for the analysis of plasma proteins leading to the diagnosis of AAT deficiency are the following: any respiratory disease and/or symptoms, liver disease, the results of family or population screening and other diseases. The subgroup “other diseases” includes renal diseases, joint symptoms, repeated infections other than respiratory tract infection, high sedimentation rate, or other signs and symptoms for which plasma protein analysis has been performed as part of the clinical investigation.
After inclusion in the AATD register, clinical examination, blood samples, including liver function tests and lung function tests, are performed at the patient’s home clinic/hospital, and the results are reported by the attending physician to register by means of a questionnaire every 2 years.
Population-Based Controls
The controls were selected from three population-based cohorts within the epidemiological research program, the Obstructive Lung disease in Northern Sweden (OLIN) studies, as previously described in detail.18 The control cohorts consisted of randomly selected adults from the population register for the northern part of Sweden, invited to participate in postal questionnaire surveys in 1992 (n=4851, aged 20–69 years), in 1996 (n=7420, 20–70 years) and in 2006 (n=6165, 20–69 years).19,20 The response rates were 85%, 85% and 77%, respectively. The questionnaire included questions on smoking habits, respiratory diseases and symptoms. Smoking habits were divided into never-smoking, ex-smoking (quitted smoking at least 12 months previously) and current smoking. Ever-smokers were defined as ex-smokers or current smokers.
PiZZ subjects have been included in the AATD register continuously since 1991, and the OLIN study included three cohorts at different time points. Thus, we randomly selected 2000 subjects from each of the OLIN cohort; a total of 6000 controls. One control was excluded due to an incorrect Swedish personal identification number. Vital status was obtained from the Swedish National Register of Causes of Death.
Data on diagnoses of ischemic heart disease and other conditions were obtained from the Swedish National Patient Register (SNPR). The SNPR covers more than 99% of all hospitalizations since 1987 and about 80% of all hospital-based outpatient care since 2001 nationwide, including at outpatient clinics for cardiology.21
Diagnoses were coded according to the 9th (before 1996) and 10th WHO International Classification of Disease (ICD) system. ICD codes were categorized as (ICD-9; ICD-10): ischemic heart disease (IHD) (410–414; I20-I25) including angina pectoris (413; I20), acute myocardial infarction (410; I21), subsequent myocardial infarction (411; I22), certain current complications following acute myocardial infarction (412; I23), other acute ischemic heart diseases (411; I24) and chronic ischemic heart disease (414; I25); hyperlipidemia (272.4; E78.5); hypertension (401–405; I109, I12-I15); diabetes mellitus (250; E10-E15). Diagnosis of COPD was also based on ICD codes (490–496; J449).
Ethical Considerations
The AAT register and the OLIN studies were approved by the Regional Ethical Review Board, Lund, Sweden, the Ethical Review Board at Umeå University and by the Swedish Data Inspection Board. All participants provided informed, written consent. This study was approved by the Regional Ethical Review Board, Lund, Sweden (Dnr 2014/427).
Statistical Analysis
The primary study endpoint was a new onset of IHD (incident IHD). Patients who had IHD or had undergone lung transplantation before inclusion in the study were excluded, to yield the population at risk (PAR). Baseline data were tabulated using frequencies and percentages of categorical variables and means with standard deviation (SD) for normally distributed, continuous variables. Comparisons of continuous variables with normal distribution were analyzed using ANOVA.
All participants were followed from the date of inclusion in the AAT register or the OLIN studies (baseline) to the date of the first onset of IHD during follow-up, the date of lung transplantation, the date of death, or study end (January 1, 2015).
Cox regression was performed to evaluate risk factors of developing incident IHD as events and were expressed as hazard ratios (HR) with 95% confidence intervals (CI).
The model was adjusted for risk factors at inclusion: age, gender, smoking status and the presence of COPD, hypertension, hyperlipidemia and diabetes.
Adjusted Cox analyses were also performed separately in ever-smokers and non-smokers, as well as for patients with or without COPD. Statistical significance was defined as a two-sided p < 0.05. Statistical analyses were performed with the Statistical Package for the Social Sciences (SPSS), version 24.0 (IMB Corp, Armonk, NY, USA).
Results
By January 2015, a total of 1595 AAT-deficient individuals had been included in the AATD register. We excluded 10 individuals with a previous lung transplant and 40 individuals with a history of IHD, leaving a population at risk (PAR) in total of 1545 PiZZ individuals. Of the initial 5999 controls, 116 were excluded due to IHD at baseline, leaving a PAR of 5883 controls. The clinical characteristics of the study population at inclusion are shown in Table 1. The controls were significantly younger than the AAT-individuals. The median follow-up time was 16 (range 0.2–23) years.
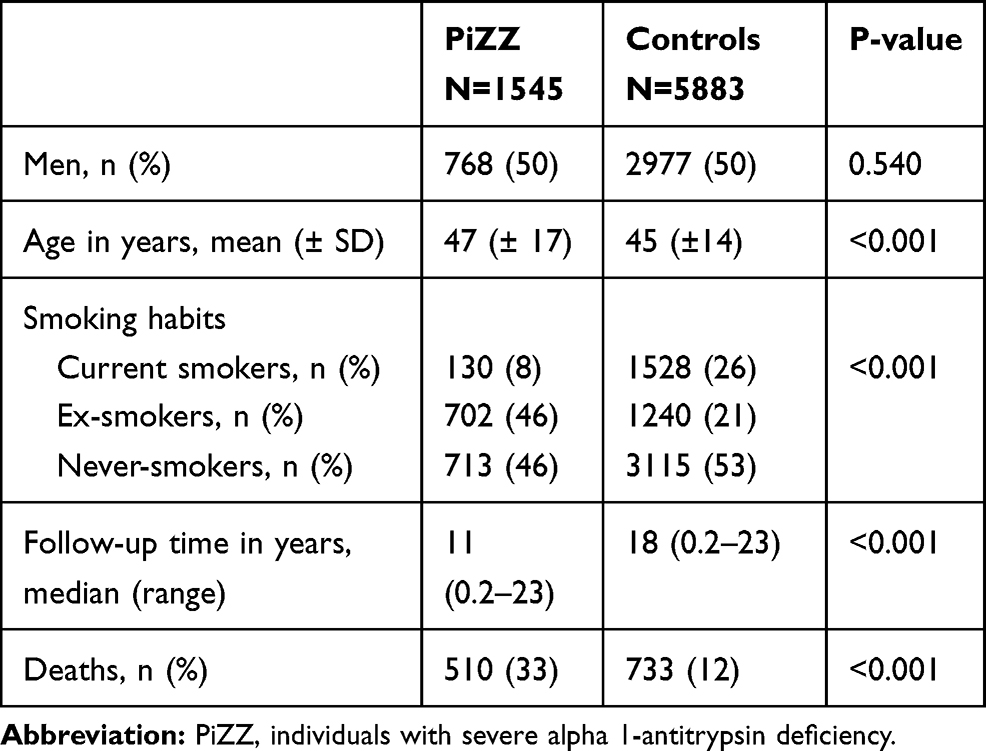 |
Table 1 Clinical Characteristics of the Population at Risk for Individuals with Severe Alpha-1-Antitrypsin Deficiency and for Population-Based Controls at Inclusion |
At inclusion, 46% of the PiZZ individuals and 4% of the controls had a diagnosis of COPD. There was no significant difference in the prevalence of hypertension, diabetes mellitus or hyperlipidemia between the PiZZ individuals and the controls. The difference was also insignificant in the smoking subgroups (Table 2).
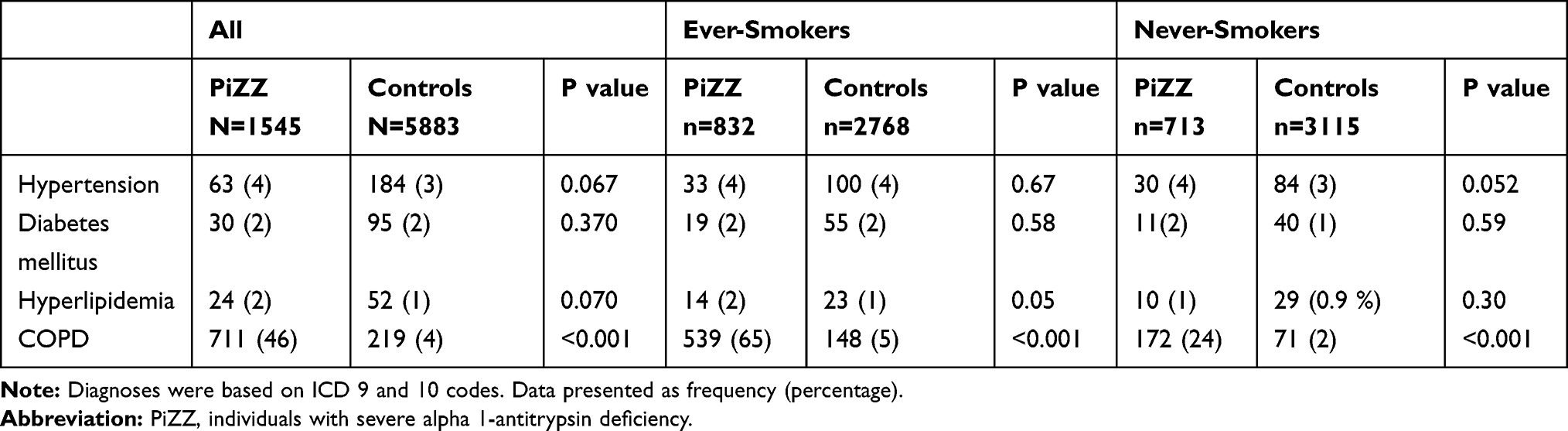 |
Table 2 Prevalence of Comorbidities at Inclusion for the PiZZ Individuals and the Controls; in the Total Population and Stratified by Smoking Habits |
Incidence of IHD
In the AATD group, 123 (8%) subjects developed incident IHD during the follow-up, yielding an incidence rate of 6.9 (95% CI 5.7–8.28) per 1000 person-years. Among the controls, 690 (12%) developed IHD during the follow-up, corresponding to an incidence rate of 8.0 (95% CI 7.4–8.6) per 1000 person-years. The cumulative incidence of IHD in the individuals with severe AATD and the controls is shown in Figure 1.
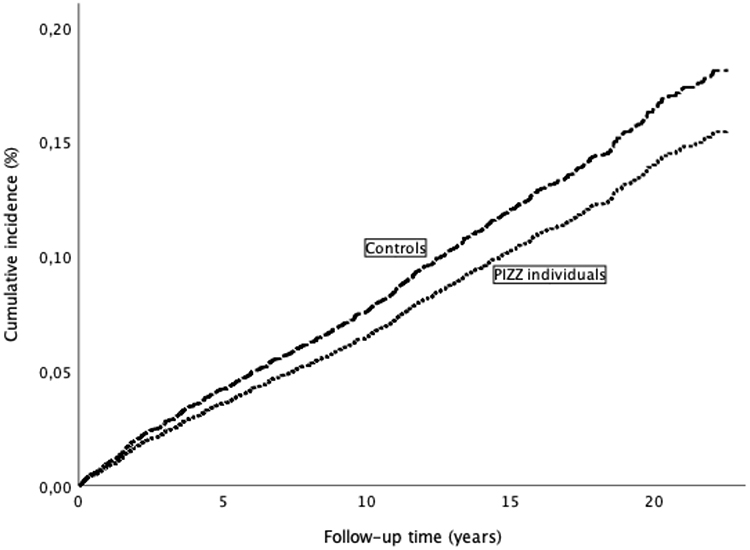 |
Figure 1 Cumulative incidence of ischemic heart disease in individuals with severe alpha-1-antitrypsin deficiency (PiZZ) and controls from the general population. |
The incidence of IHD was higher in the ever-smoking controls compared with the ever-smoking PiZZ individuals (15% vs 9%; p <0.001), while the difference between the never-smoking controls and never-smoking PiZZ individuals was not significant (9% vs 6%; p =0.108).
In multivariate analysis, AATD status, gender, age at inclusion, smoking habits and the presence of COPD, hypertension, diabetes and hyperlipidemia were all independent risk factors for incident IHD (Table 3). The controls had a significantly higher risk of developing IHD than AAT-deficient individuals, adjusted hazard ratio (HR) of 1.8 (95% CI 1.4–2.3).
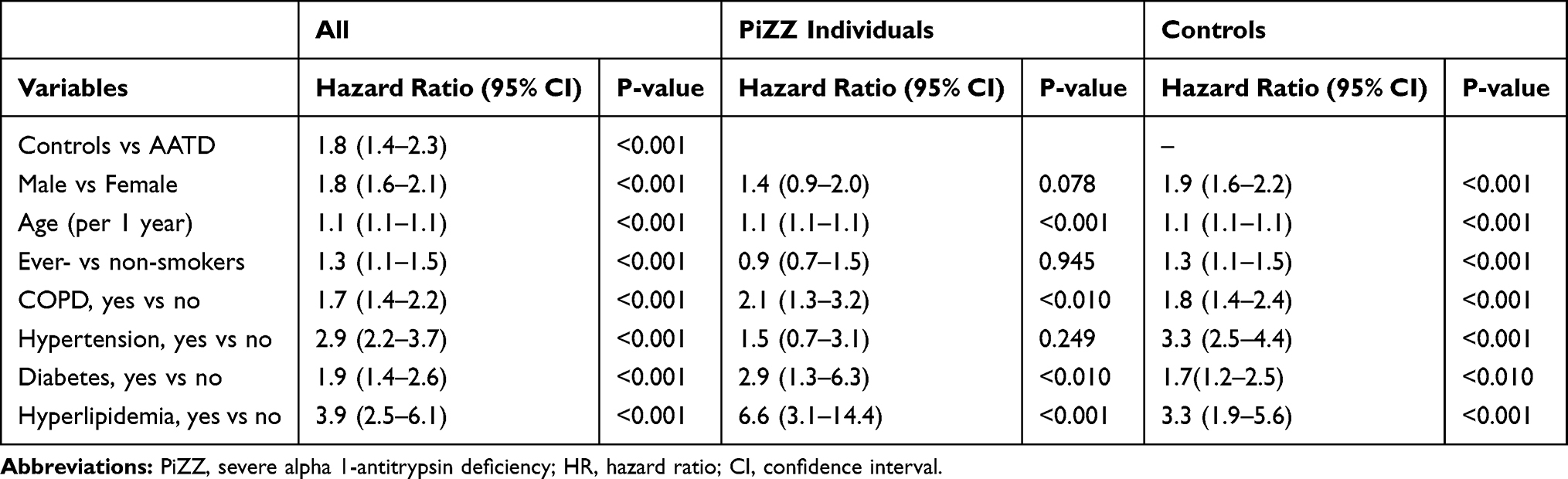 |
Table 3 Multivariate Analysis of Risk Factors for Incident Ischemic Heart Disease in the Total Study Population and in Stratified Analyses in AAT-Deficient Individuals and Controls from the General Population |
When the PiZZ individuals and the controls were analyzed separately, male sex, ever-smoking and hypertension were not associated with the risk of developing IHD in PiZZ individuals (Table 3).
When an adjusted Cox analysis was performed separately for smoking subgroups, both ever- and never-smoking AAT-deficient individuals had a significantly lower risk of developing IHD than the controls (Table 4).
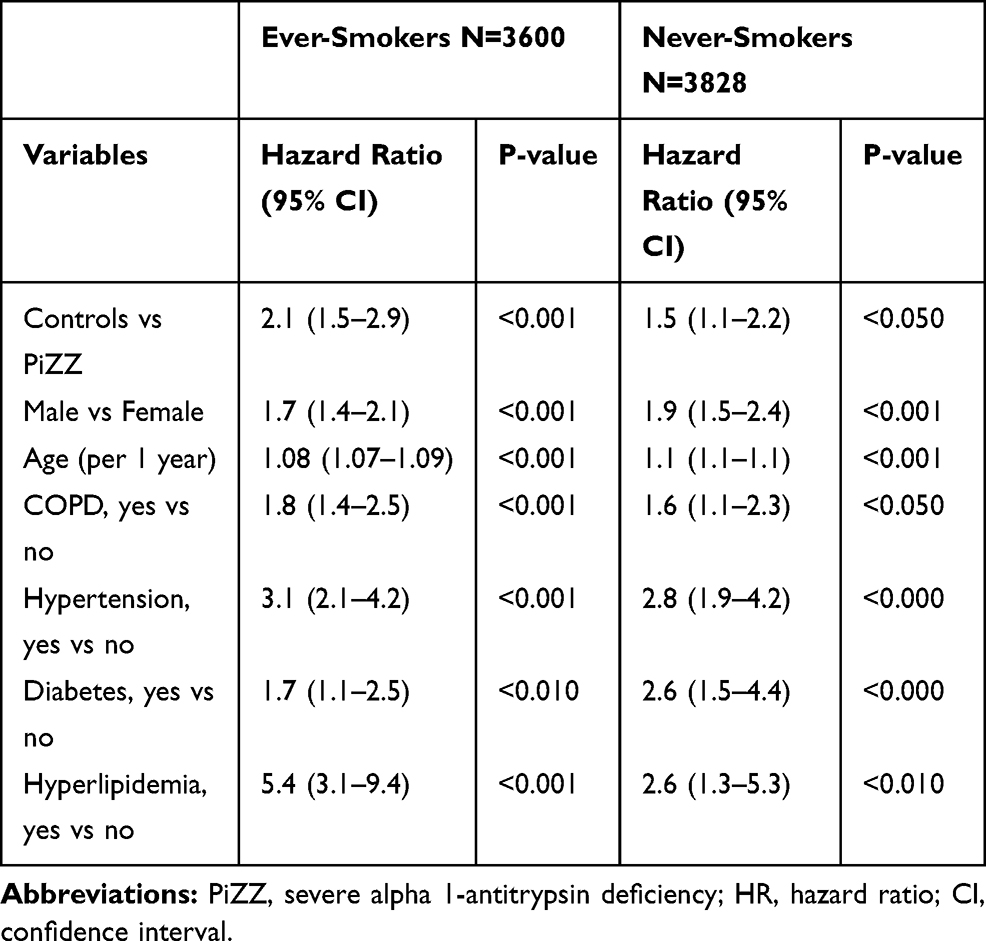 |
Table 4 Multivariate Analysis of Risk Factors for Incident Ischemic Heart Disease (HR; 95% CI) in the Study Population as a Whole, Stratified by Smoking Habits |
As COPD is common among PiZZ individuals and might influence the risk of developing IHD, the adjusted association was analyzed separately in patients with COPD and those without COPD. The PiZZ individuals with COPD had a higher risk of developing IHD compared with the PiZZ individuals without COPD, with an unadjusted (crude) risk ratio (HR) of 10.0 (95% CI 5.2–19.2). The risk was still significantly higher after adjustment for potential confounders (gender, age at baseline, smoking habits and the presence of diabetes, hypertension and hyperlipidemia) with HR of 3.56 (95% CI 1.84–6.91). For controls with COPD, the unadjusted HR was 3.30 (95% CI 2.71–4.03) and the adjusted was RR 1.61 (95% 1.32–1.97).
Discussion
This is the largest longitudinal study of individuals with severe AATD (PiZZ) to date and shows that PiZZ individuals have a lower risk of developing ischemic heart disease than the general population, independent of age, gender, smoking habits and comorbidities such as hypertension, diabetes mellitus and hyperlipidemia. The main finding of this study is that PiZZ status is independently associated with future IHD risk.
The results of our study are in accordance with our previous findings of reduced mortality due to IHD in individuals with severe AAT deficiency compared with the Swedish general population,22 in which similar results were found in both smoking and never-smoking PiZZ individuals. In that study, we were not able to compare the risk with that in smokers and never-smokers in the general population, because no data on individual smoking habits in the Swedish general population are available. In the present study, the controls were a random sample of subjects from the Swedish general population with known smoking habits, which allowed comparisons between the smoking subgroups.
One possible explanation for the lower risk of developing IHD in the AAT-deficient individuals than in the controls is that a low AAT-level may rather protect against than promote cardiovascular disease because the prevalence of predisposing factors/conditions for cardiovascular diseases, such as hypertension and hyperlipidemia are the same among AAT-deficient individuals and subjects in the general population. Another interesting hypothesis proposed for the reduced risk of IHD in AAT-deficient individuals is that a low serum concentration of AAT, or the Z mutation of the AAT molecule, may provide biochemical or genetic protection against IHD.23 Dichtl et al have shown that AAT deficiency leads to fewer cleaved fragments of AAT in atherosclerotic plaques and thereby reduces atherosclerotic inflammation, and risk of IHD. Dahl et al have demonstrated that AAT-deficient individuals have lower blood pressure than the Danish general population.8 As hypertension is a major risk factor for both CVD and IHD, a lower level of blood pressure in AAT-deficient individuals than in the general population might reduce the risk of developing CVD and IHD.
Data on 2645 COSYCONET patients (the German COPD and SYstemic consequences-COmorbidities NETwork) have also shown a significantly lower prevalence of cardiovascular comorbidities in AAT-deficient patients with COPD than in non-AATD COPD patients.9 However, this study included only 139 patients with AATD and COPD, and 110 (79%) of them were on AAT augmentation therapy. Furthermore, the information on AATD, medication, and laboratory measurement of the serum AAT concentration was self-reported, while the genotyping data for the SERPINA1 gene were not available for the majority of the patients. In our study, the number of participants was more than tenfold higher, Pi phenotyping was available in all PiZZ subjects, and none of the participants was being treated with augmentation therapy.
In the retrospective, longitudinal, observational study, Greulich analyzed data on 4 million insurants in Germany. ICD-10 codes were used to assess the prevalence of comorbidities in AAT-deficient patients with COPD compared to non-AAT-deficient patients with COPD.24 The author found that ischemic heart disease was less frequent in the AATD group, while hypertension and diabetes were more frequent in AAT-deficient patients. This study included both homozygous and heterozygous AATD.
On the other hand, some studies on AATD and cardiovascular diseases have reported conflicting results.10–13 Vizzardi et al have observed that individuals with AATD, examined by echocardiography, had larger aortic diameters, greater aortic stiffness and less aortic distensibility compared with healthy controls matched for age, gender and body mass index.11 An abnormal stiffness of the abdominal aortic wall due to a reduction of elastic fibers and a significant reduction of AAT in the vascular wall in the case of an acutely dissected ascending aorta in humans has been reported by Schachner et al.12 Duckers et al have reported increased aortic stiffness in a small group of PiZZ individuals with COPD in comparison with age- and gender-matched controls with normal lung function, indicating an increased risk of developing cardiovascular disease.13 No patients with usual COPD (without AATD) were included in the study, and it therefore remains unclear whether the increased aortic stiffness was related to AATD or COPD per se. However, all these studies have included only small, and thus selected, samples of individuals with AATD.
In general, it has been well recognized that patients with COPD have an increased risk of cardiovascular diseases.16,25 This association has been explained by common risk factors, most notably smoking.25,26 We also found that in the PiZZ individuals, COPD was an independent risk factor for developing IHD both in PiZZ subjects and in controls. However, smoking was an independent risk factor for developing IHD only in the controls, not in the PiZZ subjects, which is difficult to explain. It is possible that the smoking burden was lower in the AAT-deficient subjects than in the controls. At inclusion (baseline), the proportion of current smokers was lower among the PiZZ individuals than among the controls. Furthermore, most PiZZ individuals stop smoking immediately after having been diagnosed with severe AAT deficiency. Thus, their total smoking burden might be lower than that of the controls.
A strength of our study is the long follow-up time of up to 23 years, and the opportunity to compare the risk of IHD between PiZZ individuals and a random sample of subjects from the general population with known smoking habits. The PiZZ phenotype was confirmed by Pi phenotyping in all AAT-deficient individuals at a central, certified laboratory. Because the laboratory reports the personal data of all diagnosed PiZZ subjects to the AATD register, more than 95% of all adult PiZZ individuals in Sweden are included in the register. As the prevalence of the PiZZ phenotype is 1/1600 in Sweden, about 25% of all adult PiZZ individuals are included in the register. The Swedish personal identification number offers a unique opportunity to study the risk of disease in individuals with AATD by cross-linkage between data from all Swedish registers with no patient lost to follow-up.
There are also some limitations in our study. First, the controls were not recruited specifically for this study. They were taken from a separate study of a random population sample in the northern part of Sweden, whereas the PiZZ subjects in the AAT deficiency register were recruited nationwide. Secondly, the PiZZ subjects included in the AAT deficiency register do not represent a random sample of PiZZ subjects in Sweden, even if a high proportion of them, 24%, were identified by screening. The third limitation is the fact that the Swedish National Patient Register (SNPR) includes only the diagnoses that are reported for in- and out-patients at hospitals. The diagnoses made at the clinics of general practices are not reported to the SNPR system. Thus, diagnoses of COPD, hypertension, diabetes, hyperlipidemia that do not need investigations by a specialist at a hospital are not included in the analyses. As an example, the diagnosis of COPD might be underestimated in the controls compared with the PiZZ individuals, because the majority of the PiZZ individuals are referred, treated and followed-up by chest physicians or specialists in internal medicine at hospitals. Fourth, we did not have any information on the period of smoking in the control subjects, and we were therefore not able to define the number of pack-years, which is needed to assess the total smoking burden. Probably, the AAT-deficient subjects had a lower smoking burden than the controls measured as the mean number of pack-years, because most of these subjects stopped smoking directly after they had received the diagnosis of AAT deficiency. Fifth, our analyses lack data on medication, which may be useful to increase the capture of risk factors such as hypertension and diabetes in the analysis.
In conclusion, our results indicate that AAT-deficient individuals have a lower risk of developing incident ischemic heart disease than the controls with known smoking habits randomly identified from the general population. The Z mutation of the AAT molecule may provide protection against ischemic heart disease. Further prospective studies on the risk of IHD in AAT-deficient individuals are needed.
Acknowledgments
We would like to acknowledge the help we received from the OLIN staff who collected and reported their findings to us. We would also like to acknowledge the help we received from all the Swedish doctors who reported appropriate data to the Swedish AATD register. The authors also wish to thank Lars Jehpsson for statistical advice.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
HT was supported by unrestricted grants from the Swedish Heart-Lung Foundation, the Skåne University Hospital and the Swedish Society of Medicine.EP was supported by unrestricted grants from the Swedish Heart-Lung Foundation, ME was supported by unrestricted grants from the Swedish Society of Medicine and the Swedish Heart-Lung Foundation.
Disclosure
Dr Anne Lindberg reports personal fees from Boehringer-Ingelheim, personal fees from AstraZeneca, personal fees from Novartis, personal fees from GlaxoSmithKline, outside the submitted work. The authors report no other conflict of interest. The authors alone are responsible for the drafting as well as the content of this study.
References
1. ATS/ERS. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. doi:10.1164/rccm.168.7.818
2. Janciauskiene SM, Bals R, Koczulla R, et al. The discovery of alpha1-antitrypsin and its role in health and disease. Respir Med. 2011;105:1129–1139. doi:10.1016/j.rmed.2011.02.002
3. Fulop T, Khalil A, Larbi A. The role of elastin peptides in modulating the immune response in aging and age-related diseases. Pathol Biol. 2012;60(1):28–33. doi:10.1016/j.patbio.2011.10.006
4. Eriksson S, Carlson S, Velez R. Risk of cirrhosis and primary liver cancer in alpha-l-antitrypsin deficiency. N Engl J Med. 1986;314:736–739. doi:10.1056/NEJM198603203141202
5. Tanash H, Piitulainen E. Liver disease in adults with severe alpha 1-antitrypsin deficiency. J Gastroenterol. 2019;6:541–548. doi:10.1007/s00535-019-01548-y
6. Segelmark M, Elzouki AN, Wieslander J, Eriksson S. The PiZ gene of alpha 1-antitrypsin as a determinant of outcome in PR3-ANCA-positive vasculitis. Kidney Int. 1995;48:844–850. doi:10.1038/ki.1995.360
7. Edmonds BK, Hodge JA, Rietschel RL. Alpha 1-antitrypsin deficiency- associated panniculitis: case report and review of the literature. Pediatr Dermatol. 1991;8:296–299. doi:10.1111/j.1525-1470.1991.tb00937.x
8. Dahl M, Tybjaerg-Hansen A, Sillesen H, Jensen G, Steffensen R, Nordestgaard BG. Blood pressure, risk of ischemic cerebrovascular and ischemic heart disease, and longevity in alpha(1)-antitrypsin deficiency: the Copenhagen City Heart Study. Circulation. 2003;107:747–752. doi:10.1161/01.CIR.0000049642.20484.5D
9. Fähndrich S, Biertz F, Karch A, et al. Cardiovascular risk in patients with alpha-1-antitrypsin deficiency. Respir Res. 2017;18(1):171. doi:10.1186/s12931-017-0655-1
10. Pezzini A, Magoni M, Corda L, et al. Alpha-1-antitrypsin deficiency-associated cervical artery dissection: report of three cases. Eur Neurol. 2002;47(4):201–204. doi:10.1159/000057899
11. Vizzardi E, Corda L, Pezzali N, et al. Elastic properties of the ascending aorta in patients with alpha1-antitrypsin deficiency (Z homozygotes). Heart. 2012;98(18):1354–1358. doi:10.1136/heartjnl-2012-302144
12. Schachner T, Golderer G, Sarg B, et al. The amounts of alpha 1 antitrypsin protein are reduced in the vascular wall of the acutely dissected human ascending aorta. Eur J Cardiothorac Surg. 2010;37(3):684–690. doi:10.1016/j.ejcts.2009.07.025
13. Duckers JM, Shale DJ, Stockley RA, et al. Cardiovascular and musculoskeletal co-morbidities in patients with alpha-1-antitrypsin deficiency. Respir Res. 2010;11:173–179. doi:10.1186/1465-9921-11-173
14. Luisetti M, Seersholm N. Alpha1-antitrypsin deficiency. 1: epidemiology of alpha1-antitrypsin deficiency. Thorax. 2004;59:164–169. doi:10.1136/thorax.2003.006494
15. Nowbar AN, Howard JP, Finegold JA, et al. 2014 global geographic analysis of mortality from ischaemic heart disease by country, age and income: statistics from World Health Organisation and United Nations. Int J Cardiol. 2014;174:293–298. doi:10.1016/j.ijcard.2014.04.096
16. Divo M, Cote C, de Torres JP, et al. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(2):155–161. doi:10.1164/rccm.201201-0034OC
17. Piitulainen E, Tanash HA. The clinical profile of subjects included in the Swedish National Register on individuals with severe alpha 1-antitrypsin deficiency. COPD. 2015;1:36–41. doi:10.3109/15412555.2015.1021909
18. Tanash HA, Ekström M, Rönmark E, Lindberg A, Piitulainen E. Survival in individuals with severe alpha 1-antitrypsin deficiency (PiZZ) in comparison with a general population with known smoking habits. Eur Respir J. 2017;50(3):1700198. doi:10.1183/13993003.00198-2017
19. Rönmark E, Warm K, Bjerg A, Backman H, Hedman L, Lundbäck B. High incidence and persistence of airborne allergen sensitization up to age 19 years. Allergy. 2017;72(5):723–730. doi:10.1111/all.13053
20. Larsson LG, Lindberg A, Franklin KA, Lundbäck B. Symptoms related to obstructive sleep apnoea are common in subjects with asthma, chronic bronchitis and rhinitis in a general population. Respir Med. 2001;95:423–429. doi:10.1053/rmed.2001.1054
21. Ludvigsson JF, Andersson E, Ekbom A, et al. External review and validation of the Swedish national inpatient register. BMC Public Health. 2011;11(1):450. doi:10.1186/1471-2458-11-450
22. Tanash HA, Ekström MP, Wagner P, Piitulainen E. Cause-specific mortality in individuals with severe alpha 1-antitrypsin deficiency (PiZZ) in comparison with the general population in Sweden. Int J Chron Obstruct Pulmon Dis. 2016;11:1663–1669. doi:10.2147/COPD.S109173
23. Dichtl W, Moraga F, Ares MPS, et al. The carboxyl-terminal fragment of 1-antitrypsin is present in atherosclerotic plaques and regulates inflammatory transcription factors in primary human monocytes. Mol Cell Biol Res Commun. 2000;4:50–61. doi:10.1006/mcbr.2000.0256
24. Greulich T, Nell C, Hohmann D, et al. The prevalence of diagnosed α1-antitrypsin deficiency and its comorbidities: results from a large population-based database. Eur Respir J. 2017;49(1):1600154. doi:10.1183/13993003.00154-2016
25. Carter P, Lagan J, Fortune C, et al. Association of cardiovascular disease with respiratory disease. J Am Coll Cardiol. 2019;73(17):2166–2177. doi:10.1016/j.jacc.2018.11.063
26. Jönsson A, Fedorowski A, Engström G, Wollmer P, Hamrefors V. High prevalence of undiagnosed COPD among patients evaluated for suspected myocardial ischaemia. Open Heart. 2018;5(2):e000848. doi:10.1136/openhrt-2018-000848
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.