")
Back to Journals » International Journal of Nanomedicine » Volume 14
Decoy exosomes as a novel biologic reagent to antagonize inflammation
Authors Duong N , Curley K, Brown A , Campanelli A, Do MA, Levy D, Tantry A , Marriott G, Lu B
Received 4 December 2018
Accepted for publication 9 February 2019
Published 9 May 2019 Volume 2019:14 Pages 3413—3425
DOI https://doi.org/10.2147/IJN.S196975
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Natalie Duong,1 Kevin Curley,1 Annie Brown,1 Alexander Campanelli,1 Mai Anh Do,1 Daniel Levy,1 Adarsh Tantry,1 Gerard Marriott,2 Biao Lu1
1Department of Bioengineering, School of Engineering, Santa Clara University, Santa Clara, CA 95053, USA; 2Department of Bioengineering and Tsinghua-Berkeley Shenzhen Institute, University of California at Berkeley, Berkeley, CA 94720, USA
Background: Exosomes are ubiquitous naturally secreted stable nanovesicles that can be engineered to target and deliver novel therapeutics to treat a host of human diseases.
Methods: We engineered the surfaces of cell-derived nanovesicles to act as decoys in the treatment of inflammation by antagonizing the major proinflammatory cytokine, tumor necrosis factor alpha (TNFα).
Results: Decoy exosomes were generated by displaying the TNFα binding domain of human TNF receptor-1 (hTNFR1) on the outer surface of exosomes using stably transfected HEK293 cells. We developed an efficient method to purify the engineered exosomes from conditioned medium based on sequential centrifugation, ultrafiltration, and precipitation. We characterized decoy exosomes using immune-quantification, nanoparticle tracking analysis, and confocal microscopy to confirm that they retain the correct orientation, size, and shape of naturally produced exosomes. We demonstrated the engineered decoy exosomes specifically antagonize activities of TNFα using an inflammatory reporter cell line.
Conclusions: Decoy exosomes produced in human cells serve as a novel biologic reagent for antagonizing inflammatory signaling mediated by TNFα.
Keywords: Decoy exosomes, Inflammation, CD63, Receptor, TNFα, Drug delivery
Introduction
Inflammation is a hallmark of many debilitating and life-threatening disorders, including inflammatory bowel disease,1 rheumatoid arthritis,2 psoriasis,3 atherosclerosis,4 Alzheimer’s,5,6 sepsis,5 and cancer.7,8 Many of these disorders are chronic and impose a significant burden to family and society due to their high morbidity and mortality, and therapies are costly, both financially and for patient care.9–11 In the United States and Canada alone, it is estimated that more than 1.4 million people suffer from inflammatory bowel disease.12 While many genetic and environmental factors play a prominent role in disease progression,13 the proinflammatory cytokine, TNFα, is recognized as the central player in eliciting inflammatory cascades.12,14,15 Protein therapeutics, such as anti-TNFα antibodies that selectively block the TNFα cascade, have become the mainstay therapy for the management of chronic inflammatory diseases.16–18 Despite the promise of the early studies, concerns about side effects of these protein drugs, including induction of anti-antibodies and immuno-suppression, may limit the applications to disease treatments.19–22 Additionally, the efficacy of antibody-based therapies is limited by their rapid clearance and poor penetration of tissue,23 especially when compared to other biologics, such as exosomes. Thus, there is a pressing demand to introduce improved therapeutic agents to treat inflammatory diseases; to this end, we introduce a new class of decoy exosomes that antagonize the major proinflammatory cytokine, TNFα.
Exosomes are cell-derived nanovesicles that are being engineered by some laboratories to function as disease cell-targeting drug delivery vehicles. Exosomes can be engineered to display specific proteins on their external membrane, for example to target specific receptors on a tumor cell, and to display other functional proteins in their lumen.24–26 Circulating in blood and other body fluids, including synovial joints, engineered exosomes could be used to deliver large amounts of therapeutic reagents to targeted tissues. The intrinsic biocompatibility and high degree of tissue-penetration have led numerous groups to pursue the engineering of exosomes as drug-delivery vehicles to treat diverse human diseases.27 For example, exosomes have been engineered to deliver siRNAs to neurons, microglia, and oligodendrocytes in the brain to affect specific gene knockdowns.28 Exosomes bearing targeting and therapeutic proteins circulate in the body much longer than synthetic vehicles, and are found to be well suited to deliver anti-oncogenic KRAS small RNAs to deep-seated pancreatic cancer cells.29 However, we are unaware of robust and high-fidelity strategies to engineer exosomes with protein therapeutics to treat inflammation. Here we show that engineered exosomes with TNF receptor (TNFR1) can be used to antagonize the proinflammatory cytokine TNFα. We further show the establishment in a proof-of-concept study that demonstrates the effectiveness of exosomes in delivering the “decoy-binding domain” of the TNF-receptor to a cell model of inflammation.
A vital component of this study is the application of an exosome surface display technology developed by our group to load diverse therapeutics and protein reporters to the inside or outside of the exosome membrane surface via genetic fusions.30 Individually, we fuse target proteins to the outer and inner surface of exosomes using the endogenous transmembrane protein CD63, an abundant tetraspanin protein expressed on the surface of exosomes.31,32 In this study, we used tetraspanin CD63 to engineer exosomes bearing a surface-exposed TNFR1. Since these novel exosomes function as agonists in TNF-signaling of inflammation responses in cells, we refer to them as decoy exosomes. Because each decoy exosome displays multiple copies of TNFR1 on its surface, it is proposed to function as a biological sponge that absorbs soluble TNFα in the affected tissue and thereby compromises TNF-mediated inflammation in the exosome-targeted tissue.
The goals of this study are two-fold: 1) to establish human cell lines in permanent culture that are genetically modified with our tetraspanin CD63 surface-display system to produce decoy exosomes bearing genetically transformed TNFα receptors, and 2) to demonstrate these decoy exosomes antagonize TNFα in a cellular model of inflammation. To achieve these goals, we designed and constructed a chimeric gene composed of three geometrically defined protein domains: the transmembrane CD63; the decoy receptor hTNFR1-EC on the outer surface; and the green fluorescent protein (GFP) on the inner surface of the exosomes. GFP functions as a fluorescent reporter of the expression of the chimeric gene and moreover enables imaging of the distribution of decoy exosomes in cell culture or tissue. Using a combination of genetic and cellular methods and analyses, we showed the transfection of HEK293 cells with the engineered constructs produce a fusion protein on the surface of exosomes with correct geometry. Moreover, we used fluorescence microscopy to show HEK293 cells produce large numbers of decoy exosomes that are released to the conditioned medium. Finally, we developed a protocol to purify decoy exosomes from conditioned medium in high-yield and showed that decoy exosomes exhibit a specific anti-TNFα activity on an inflammatory cell reporter system established by our group.33
Because of their intrinsically high tissue-penetrating ability, we hypothesize decoy exosomes are more effective in delivering therapeutic (anti-inflammatory) cargo to target cells compared to the injection of protein-based therapeutics. Additionally, our work provides a proof-of-concept to develop agonist-binding engineered exosomes to treat other diseases, including cancer, cardiovascular, and neuronal disorders.
Material and methods
Materials
We purchased human recombinant TNFα from R&D Systems (Minneapolis, MN) and puromycin hydrochloride from ThermoFisher Scientific (Waltham, MA). We purchased the luciferase assay substrate and cell lysis reagents from Promega (Madison, WI). We purchased the organelle staining reagent LysoTracker Red DND-99, fusion genes for tracking cellular organelles of early (Rab5a-RFP) and late (Rab7a-RFP) endosomes from ThermoFisher Scientific. Finally, we purchased the exosome marker (XPACK-RFP) from System Biosciences (SBI, Palo Alto, CA).
Design and construction of expression vectors
We constructed the fusion protein of CD63-GFP as reported.30 We inserted either TNFR1-EC or RFP at the 2nd extracellular loop of CD63 using a fusion technology.30 We confirmed the fidelity of all constructs by double-stranded DNA sequencing. Details of the coding sequences of the decoy chimeric protein appear in the Additional file 1 (Supplementary sequences). We have previously reported on the RFP-control chimaera.30
Cell culture
We purchased the human embryonic kidney cells (HEK293) from Alstem (Richmond, California). Cells were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM) with 2mM glutamine (Gibco; MA, USA) supplemented with 10% fetal bovine serum (GE Healthcare Life Sciences; Issaquah, WA) and 100 U/mL penicillin/streptomycin (Gibco; MA, USA) at 37°C in 5% CO2.
Transfection, drug selection, and establishment of stable cell lines
Stable cell lines expressing various CD63 fusion proteins were established as previously reported.30 Briefly, we transfected HEK293 with plasmid DNA (1~2.5 µg/well) using either Lipofectamine (ThermoFisher Scientific, Waltham, MA) or FuGENE6 transfection reagent (Promega, Madison, WI) for 24~48 hrs. Next, we transferred the transfected HEK293 cells to a drug selection medium containing 5 µg/mL puromycin. The resultant cell lines stably expressed the fusion gene and remained GFP/RFP-positive for at least 50 days (equivalent to 25 passages). We maintained these stably transfected cells in medium with puromycin although before an experiment, we bathed cells in a puromycin-free medium for at least 2 passages. Stable reporter cell lines that detect TNFα signaling were also similarly established as previously reported.33
Fluorescence-activated cell sorting (FACS)
Cultured HEK293 cells stably expressing the CD63-RFP-GFP or CD63-TNFR1-GFP fusion proteins were analyzed using an Accuri C6 Plus Flow Cytometer (BD Biosciences, San Jose, CA), as reported.34 In brief, cells grown at 80~90% confluency on 3 cm dishes were collected and sorted using both FL1 and FL3A channels to quantify the levels of GFP and RFP intensity – we recorded over 10,000 events for each sample.
Exosome preparation
We prepared exosomes from the conditioned medium as previously reported.34 Briefly, first we transferred the stably transfected cells to serum-free UltraCulture medium (Lonza, Allgendale, NJ), and then we collected the conditioned medium, centrifuged to remove cell debris, and ultra-filtered to remove large extracellular vesicles (>200 nm) using a 0.22 µm filter. We mixed the filtered medium containing mainly exosomes with exosome-precipitation solution ExoQuick-TC (SBI, Palo Alto, CA). We determined the protein content of the resuspended pellet using a NanoDrop Lite (ThermoFisher Scientific, Waltham, MA). We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV190008) with an EV-Track score of 38%.35
Nanoparticle analysis and characterization
Exosomes isolated from engineered cells were subjected to nanoparticle tracking analysis (NTA) using a NanoSight LM10 instrument (Malvern Instruments Ltd, Malven, UK) with a 405 nm and 60 mV laser source. Typically, we used 1 ml of a diluted exosome preparation for laser light scattering study usually making 3 recordings of 60 seconds per sample. We processed the data using the NTA software to determine the size distribution of the exosomes.
Dot-blot immuno-analysis
We used a dot-blot immuno-analysis to identify target proteins using premade dot-blots from SBI (Palo Alto, CA). Each blot contained 12 pre-printed spots and featured 8 antibodies for exosomal markers including CD63, CD81, ALIX, FLOT1, ICAM1, EpCam, ANXAS, and TSG101 as well as cytosolic GM130 to identify any cellular contamination. We used ~300 μg of exosome proteins in each assay according to the immuno-binding and detection protocol in the user manual.
Fluorescence and confocal microscopy
Images of live cells or fluorescently labeled exosomes were recorded using an Olympus fluorescence microscope (Waltham, MA) or Leica TCS SP8 confocal microscope (Buffalo Grove, IL). To show the intracellular localization of fusion proteins, we recorded both fluorescent and phase contrast images from the same field. Any image modifications including brightness and contrast were applied to the entire image using Image J software.
Luciferase assay
Reporter luciferase assays were conducted using a luciferase assay kit according to the manufacturer’s instructions (Promega, Madison, WI). First, we lysed the cells and then recorded the luciferase activity with a microplate reader (BMG LABTECH, Cary, NC) as previously reported.36
Cell models for testing the antagonist activity of the decoy exosomes
We developed two cell models to assay for the antagonist activity of the decoy exosomes: 1) a cell co-culture model to assess the effect of newly released exosomes and 2) an exosome treatment model to assess the effect of purified exosomes. In the cell co-culture experiments, we combined 2.5×103 of inflammation reporter cells with 7.5×103 of either HEK293 cells producing the decoy exosomes (treatment group), RFP-exosome-control (mock control), or the parental HEK293 cells (no treatment control) in a 96-well plate. After 24 hrs of co-culture, we seeded cells in a serum-free Ultra-Culture either in the presence or absence of 1 ng/mL TNFα. After another 24 hr-incubation period, we first lysed the cells and then conducted a luciferase assay on the lysate. For the decoy exosome experiments, we seeded 1×104 of inflammation reporter cells in a 96-well plate overnight. Cells were then switched to serum-free Ultra-Culture in groups with either decoy exosomes, control exosomes, or no exosomes in either the presence or absence of 1 ng/mL TNFα. Cells without TNFα treatment were also included in the study to account for background signals from the reporter cells. After 24 hrs of treatment, we lysed the reporter cells and conducted the luciferase assays on the lysates as previously reported.33
Data collection and statistical analysis
We used the Student’s paired, two-tailed t-test to determine statistical significance of our studies, with P-values <0.05 being considered significant. We expressed the values for all measurements as mean ± standard error.
Results and discussion
Design and construction of a fusion gene encoding the decoy receptor on the outer surface of exosomes
Unlike other nanoscale carriers such as liposomes, biopolymers, and gold nanoparticles,37,38 we produced exosomes within living human cells that harbor a repertoire of endogenous membrane proteins on their surface, including CD63, a hallmark biomarker of exosomes.39,40 We elected to engineer CD63 as a molecular scaffold for our exosome surface display technology – we have previously shown this approach allows one to project specific target-recognizing peptides on the outer and inner surface of the exosome membrane.30 We introduced the extracellular TNFα binding domain of human TNFα receptor 1 (hTNFR1-EC) at the 2nd loop of CD63 in order to project the TNFα receptor on the external face of the exosome. The choice of insertion site of TNFR1 in the 2nd loop but not the 1st loop is due to two major considerations. First, the chosen site is between Alanine 133 and Serine 134, which is both on the outer surface of exosomes and near the most exposed region of CD63.30 Second, the 1st loop is relatively small and tends to be covered by the larger 2nd loop, hence TNFR1 inserted at this loop may become sterically inaccessible to its ligand.39,40 To track and image the distributions of fusion proteins in cells and exosomes, we fused GFP to the C-terminus of CD63, which is located on the inner surface of exosomes (Figure 1A, upper panel). We also constructed an RFP fusion protein (replacing hTNFR1-EC) to produce the control group of fluorescently visible but inactive exosomes (Figure 1A, lower panel). To facilitate the establishment of stable cell lines that secrete these engineered exosomes, we cloned the coding sequence of each fusion into a vector that contained two expression cassettes: the CMV-driven fusion gene and the EF1α-driven puromycin resistance gene for drug selection (Figure 1A). According to this design, the engineered exosomes would display either the hTNFR1-ED (decoy exosome) or RFP (control exosome) at the 2nd loop on the outer surface along with GFP on the inner surface of modified exosomes (Figure 1B). Thus, decoy cells would produce decoy exosomes with hTNFR1-ED and GFP, while control cells would produce control exosomes with no receptor but both RFP and GFP, whereas parental cells would neither express GFP nor RFP.
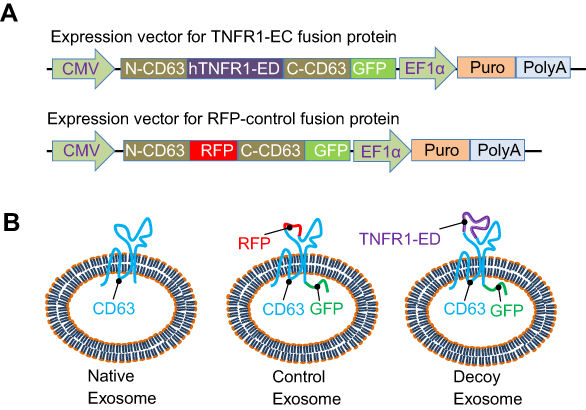 | Figure 1 Surface display of extracellular domain (ED) on the outer surface of decoy exosome. (A) Schematic illustration of vectors expressing a decoy chimera containing the extracellular domain of human tumor necrosis factor receptor 1 (hTNFR1-ED, top) or a control RFP (bottom). (B) The strategy of presenting the decoy receptor on the outer surface of exosomes using a CD63 scaffold. CD63 (blue) anchors the membrane in an M-shaped topology with its two termini located within the lumen (native exosome, Left). The control exosomes have a C-terminal GFP with or without an RFP in the second loop of CD63 (RFP-control exosome, middle). The hTNFR1-ED (purple) is fused in the second loop on the outer surface of the exosome, while a reporter GFP (green) is attached to its C-terminus inside its lumen (decoy exosome, right). Abbreviations: CMV, cytomegalovirus promoter; N-CD63, N-terminus of CD63; hTNFR1-EC, the extracellular domain of the human TNFR1; C-CD63, C-terminus of CD63; EF-1α, elongation factor-1 promoter; Puro, puromycin resistance gene; PolyA, polyadenylation signaling sequences. |
Establishment and characterization of stable cell lines that produce control or decoy exosomes
To permanently express the decoy proteins in human cells, we first established stable cell lines by transfecting HEK293 cells with our expression vector DNA followed by puromycin treatment for more than 40 days (Figure 2A). These stable cells consistently produce the decoy or control proteins, which may be integrated into pre-secreted exosomes in transfected cells.
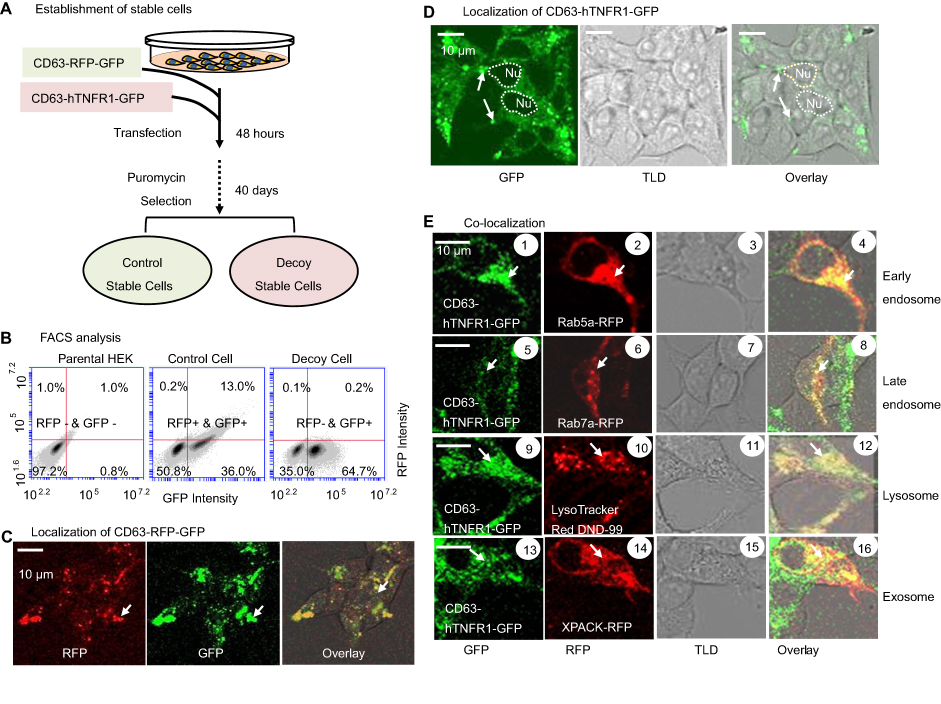 | Figure 2 Establishment and characterization of stable source cell lines. (A) Scheme and timeline for the establishment of stable cell lines for production of either the CD63-RFP-GFP control exosomes or CD63-hTNFR1-GFP decoy exosomes. (B) FACS analysis of parental HEK293, CD63-RFP-GFP control, and CD63-hTNFR1-GFP stable cells. (C, D) Confocal images of the stable cell lines established by genetically transforming the parental HEK293 cells. Both GFP and RFP fusion proteins showed punctuated morphology in the cytoplasm for control CD63-RFP-GFP, while only GFP fusion proteins were expressed in CD63-hTNFR1-GFP cells. (E) Subcellular localization of the fusion proteins. Green fusion proteins were co-localized with red organelle markers for the early (1~4) or late endosomes (5~8), lysosomes (9~12), or exosomes (13~16) in the decoy exosome producing cells. Arrows indicate the organelle structures of endosome/lysosome/MVB. Scale bar: 10 µm. Abbreviation: TLD, transmitted light detector for light imaging. |
Next, we characterized the expression level of each gene fusion in living HEK293 cells using FACS analysis. We analyzed the expression of CD63-RFP-GFP (control cells) or CD63-TNFR1-GFP (decoy cells) fusion proteins in stably transfected cultured cells using a BD Accuri C6 Plus Flow Cytometer. We also analyzed the control group, ie, the parental HEK cell in the same way. As expected, we recorded an increase in the intensity of GFP fluorescence in the decoy cells (Figure 2B, right), as compared to the parental control HEK cells (non-GFP, non-RFP) (Figure 2B, left). We also recorded significant increases in the intensities of RFP and GFP in mock engineered cells compared to the parental control HEK cells (Figure 2B, middle). These FACS results showed we successfully generated stable cells expressing defined fusion genes. In the decoy group, ~64.7% of cells showed a dramatic increase in the level of GFP expression. In the RFP/GFP mock control group, ~49% of control cells expressed GFP and RFP. In contrast, almost all parental HEK293 cells were GFP- and RFP-negative. However, it is noteworthy that the distribution of fluorescence intensity was broad in the stably transfected cells (decoy and control), a common finding in studies that use the transfection protocol described above.
To establish whether the expressed fusion proteins were positioned on the exosomal membrane by CD63, we recorded fluorescent images of GFP and RFP in transfected cells. Confocal images revealed the distributions of both RFP (red) and GFP (green) in the cytosol of control cells identified as nodules (Figure 2C), and furthermore, we found significant co-localization of the emission signals, as evidenced by the strong yellow (merged) color in the overlay image (Figure 2C). In contrast, we found GFP positive nodules alone in the cytosol of decoy cells (Figure 2D), consistent with the finding of the FACS analysis detailed above (Figure 2B).
Exosomes are first generated within the endocytic compartments and then released to the extracellular environment. We conducted a series of co-localization studies to confirm our premise whether the punctuated nodule structures identified in fluorescence images are endocytic compartments. First, we transfected stable decoy cells with a validated endosomal marker Rab5a-RFP, and then we recorded their intracellular localization expression using confocal microscopy followed by colocalization image analysis. As expected, we found the decoy cells expressed CD63-hTNFR1-GFP (Figure 2E, 1, 5) and the endosomal marker Rab5a-RFP (Figure 2E, 2, 6) after 2 days of the transfection. The signals of the two fluorescent proteins exhibited a remarkable degree of overlap in the early endosomes (Figure 2E, 4), and in late endosomes (Figure 2E, 8). Second, we transfected decoy cells with a validated lysosomal marker (LysoTracker Red DND-99) and observed an extensive overlap of green (CD63-hTNFR1-GFP) and red (LysoTracker Red) fluorescent signals – this overlap is apparent in the merged (yellow) images (Figure 2E, 1, 2). These confocal images confirm the co-localization of CD63-hTNFR1-GFP in lysosomal compartments (Figure 2E, 1, 2), a finding that strongly indicates the expressed decoy receptors localize exclusively to exosomes. Finally, we transfected the stable decoy cells with a known exosome marker, XPACK. Similarly, we observed an extensive signal overlap between our fusion proteins (CD63-hTNFR1-GFP, green) and the exosome marker (XPACK-red) (Figure 2E, 1, 6), supporting the conclusion that our CD63-based fusion proteins target and subsequently anchored to the exosome.
Together, the results show stable decoy and control cell lines can produce either decoy receptors or control proteins. The findings of analysis of confocal fluorescence microscope images provide further evidence that CD63-based therapeutic proteins are incorporated into exosomes via the endogenous pathway that involves early and late endosomes as well as lysosomes.
Production and characterization of decoy exosomes
Next, we investigated whether decoy exosomes are released by cells into the culture medium. First, we isolated decoy exosomes from the conditioned medium of stable decoy cells through a combination of differential centrifugation, ultrafiltration, and precipitation, as reported previously (see exosome preparation flowchart, Figure 3A).34 We employed nanoparticle tracking analysis (NTA) (Figure 3B, left panel) to record typical Brownian movement and size distributions of the purified exosomes. In summary, the mean size of 73–75 nm is consistent with that measured for natural exosomes (Figure 3B, right panel).31 To further confirm that our purified nanoparticles were exosomes, we performed an immune dot-blot analysis using a panel of 9 antibodies. The blot revealed positive stains for all 8 exosome markers (CD63, CD81, ALIX, FLOT1, ICAM1, EpCam, ANXAS, and TSG101), whereas the control cytosolic marker (GM130) was negative (Figure 3C). To qualitatively demonstrate that released decoy exosomes are loaded with CD63-TNFR1-GFP, we took confocal images from decoy and control exosome samples. The images showed strong GFP and/or RFP, indicating that the fusion proteins were targeted and anchored on the exosome (Figure 3D). Together, these data demonstrate our stable decoy cells express, integrate and may target decoy receptors into exosomes. Importantly, the cells are found to release decoy exosomes into the surrounding culture medium. We further showed that these cells produce a sufficient number of exosomes to allow for characterizations.
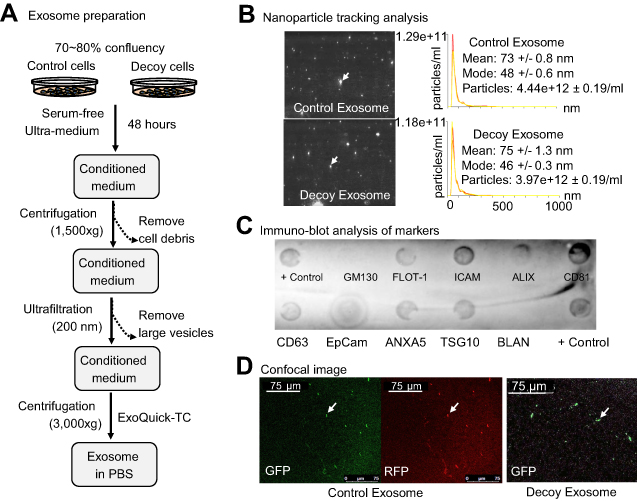 | Figure 3 Exosome preparation and characterization. (A) A centrifuge-ultrafiltration-precipitation protocol used for exosome preparation. (B) Nanoparticle tracking analysis reveals exosomes in solution (left panel) along with particle size and distribution (right panel). (C) A dot-blot immuno-analysis was carried out using premade dot-blots to detect either exosomal makers, including CD63, CD81, ALIX, FLOT1, ICAM1, EpCam, ANXA5, and TSG10, or cytosolic protein GM130 for evaluating cellular contamination if nay. (D) Confocal images of isolated control and decoy exosomes. |
Antagonistic activities of decoy exosomes
In an earlier study, we established a cell-based reporter system to precisely monitor inflammatory signaling mediated by TNFα (Additional File 2, Figure S1A)33 – the assay can detect as little as 1 ng/ml of TNFα. Consistent with our previous observations,33we now show TNFα generates a robust yet unsaturated inflammatory response in our reporter cells (Additional File 2, Figure S1B). In this study, we quantified antagonistic activities of our decoy exosomes using 1 ng/mL of TNFα – this dosage is known to induce inflammatory responses both in vitro and in vivo.41,42 Our reporter cell models are similar to in vitro inflammatory assays, which have been used to study the acute response elicited by various inflammatory cytokines including TNFα as reported.43
First, we tested the antagonistic activity of decoy exosomes using a co-culture experiment (Figure 4A). As demonstrated by the experiments detailed above, decoy cells express the hTNFR1-ED fusion protein in secreted exosomes. After mixing various source cells (parental, decoy, or mock control) with our established TNFα reporter cells for 24 hrs, we treated each co-cultured preparation with either TNFα (1 ng/mL) or vehicle control for an additional 24 hrs. Under this condition, each co-culture showed a significant increase in luciferase activity after TNFα treatment (Figure 4B, Column 1, 2 3 vs Column 4). Among them, the co-culture containing parental HEK293 cells showed a robust response to TNFα with a drastic increase in luciferase activity (~20-fold over vehicle control, Figure 4B, Column 1 vs Column 4). As expected, the luciferase activity decreased significantly in the co-culture containing decoy cells (~41% compared to parental control, P=0.036; Figure 4B, Column 1 vs Column 3), indicating an antagonistic effect of expressed TNFR1-ED towards the native TNFα receptor. In contrast, we only observed a minor decrease in luciferase activity in the co-culture containing mock control cells (~22% compared to parental control, P=0.23; Figure 4B, Column 1 vs Column 2). These data suggest decoy cells (TNFR1-ED) elicit a specific antagonism towards the TNFα-receptors situated on cellular surfaces. Together, our results indicate that forced expression of TNFR1-ED antagonizes TNFα-mediated inflammatory signaling in HEK293 cells.
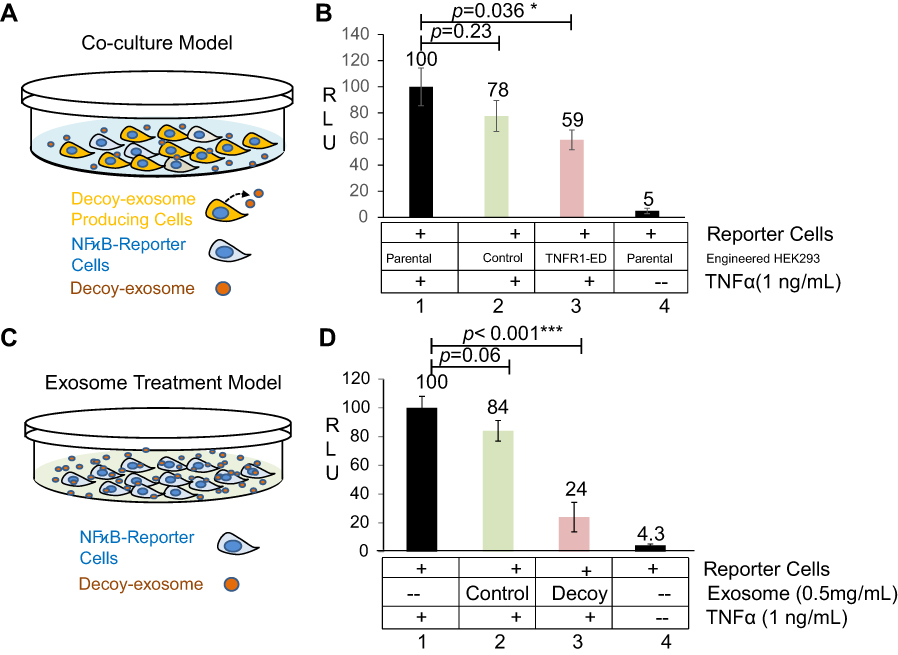 | Figure 4 Efficacy of decoy exosomes on antagonizing TNFα activities in two cell models. (A) Co-culture experimental design for testing newly secreted exosomes and their antagonistic activities against TNFα. (B) Bar graph showing the specific antagonistic activities. Data are presented as mean ± SE, n=9 from 3 independent experiments. (C) Exosome treatment for testing the direct effect of decoy exosomes and their antagonistic activities against TNFα. (D) Bar graph showing the specific antagonistic activities against TNFα. Data are mean ± SE (n=6) from 2 independent experiments with two independent stable lines. The RLU was normalized to the reporter cells, which were the parental cells HEK293 cells without exosomes/engineered HEK293 cells. Column 2: Non-specific effect. Column 4: Background level activities. *Denotes P<0.05, while ***denotes P<0.001, using Student’s t-test.Abbreviation: RLU, relative light units of luciferase activities. |
To further confirm that the observed antagonistic effects are solely due to the decoy exosomes, we treated the inflammation reporter cells directly for 24 hrs with purified exosomes prepared from various source cells (parental, decoy, and mock control), either in the presence or absence of TNFα (1 ng/mL) (Figure 4C). As expected, the reporter cells treated with parental exosomes had a robust inflammatory response to TNFα treatment, showing a marked increase (~23-fold over vehicle control) of luciferase activity (Figure 4D, Column 1 vs 4). In agreement with our co-culture results, the treatment of cells with decoy exosomes resulted in a significant decrease in luciferase activity compared to treatment with parental exosomes (~76%, P<0.001; Figure 4D, Column 1 vs 3). In contrast, treatment of cells with mock control exosomes induced a small and insignificant decrease of luciferase activity (~16%, P=0.06; Figure 4D, Column 1 vs 2). Together, our results collectively demonstrate that decoy exosomes specifically antagonize TNFα, which indicates their potential as novel anti-inflammatory biologic therapeutic agents.
We initially tried two different concentrations of exosomes. At a lower concentration (0.1 mg/ml), the decoy exosomes produced ~24.6% inhibition of TNFα activity (n=6, P=0.55). At a higher concentration (0.5 mg/ml), our decoy exosomes produced ~76% inhibition of TNFα activity (n=6, P<0.05), indicating a high amount of decoy exosomes was required. Because decoy exosomes tended to aggregate if the concentration was beyond 0.5 mg/ml, we did not try a higher concentration to determine the maximum inhibition. It appears that the effective dosage to inhibit >75% of TNFα activity (exosomes, 0.5 mg/ml) is much higher than those of the chimeric bivalent inhibitor (TNFR1-Fc, ~10 ng/ml) or monoclonal antibodies (mAbs, 1~50 µg/ml).44,45 However, one shall be careful not to overly interpret the significance of those in vitro results. Because the higher tissue-penetrating ability can only manifest in animals and patients, the advantage of this exosome-based method over other conventional decoy methods may not be fully appreciated until the decoy exosomes are finally tested in animal models.
The present study is focused on the earlier stage of development, including the development of an engineering strategy and proving in concept of decoy exosomes as a new anti-TNFα reagent. A scale-up production of decoy exosomes from more desirable producing cells may be needed to obtain quality exosomes for animal studies to determine if decoy exosomes can be translated into a new class of clinical biologics for the treatment of rheumatic arthritis, psoriasis, and inflammatory bowel diseases.28,29,46
Conclusion
We developed a molecular engineering approach to generate decoy exosomes that bind specifically to the inflammatory cytokine, TNFα. Our results show that these decoy exosomes antagonize TNFα-elicited signaling in cellular models of inflammation. In the future, our approach can be further exploited to display multiple receptors that are activated by other inflammatory cytokines, including interleukin-1, interleukin-6, and interleukin-12/23 – this feature, we argue, would elicit an even higher and more robust anti-inflammatory response.22,47,48We also note that exosomes may exhibit a greater penetration of inflamed tissue compared to antibodies. Importantly, we anticipate extending the principle of decoy exosomes to different classes of membrane receptors to treat other diseases. For instance, decoy exosomes engineered to compete with the vascular endothelial growth factor (VEGF) could be used as a cancer therapy or for retinal diseases.49,50In summary, our study demonstrates a new avenue of therapeutics using decoy exosomes as a biological sponge to absorb detrimental factors in blood or tissues – decoy exosomes represent a novel class of biologics to treat human diseases, including inflammation, cancer, and cardiovascular disorders.
Data availability
All data generated or analyzed in this study are included in this published article and its Supplementary information files.
Acknowledgment
We thank Dr. Yan Jiang for critically reading and editing the manuscript. This work was supported by internal funds from the School of Engineering, Santa Clara University. GM acknowledges support from the Tsinghua-Berkeley Shenzhen Institute. The funding institute plays no role in the design of the study and collection, analysis, and interpretation of data.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12(Suppl 1):S3–S9.
2. Cross M, Smith E, Hoy D, et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73:1316–1322. doi:10.1136/annrheumdis-2013-204627
3. Schon MP. Advances in psoriasis treatment. Lancet. 2005;366:1333–1335. doi:10.1016/S0140-6736(05)67542-3
4. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–126. doi:10.1056/NEJM199901143400207
5. Rhee C, Gohil S, Klompas M. Regulatory mandates for sepsis care – reasons for caution. N Engl J Med. 2014;370:1673–1676. doi:10.1056/NEJMp1400276
6. Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord. 2000;14(Suppl 1):S47–S53.
7. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi:10.1038/nature07205
8. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008;267:204–215. doi:10.1016/j.canlet.2008.03.028
9. Hay JW, Hay AR. Inflammatory bowel disease: costs-of-illness. J Clin Gastroenterol. 1992;14:309–317.
10. Wimo, A., Guerchet, M, Ali, GC, et al. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimers Dement. 2016;13:1–7. doi:10.1016/j.jalz.2016.07.150
11. Cooper NJ. Economic burden of rheumatoid arthritis: a systematic review. Rheumatology (Oxford). 2000;39:28–33.
12. Nielsen OH, Ainsworth MA. Tumor necrosis factor inhibitors for inflammatory bowel disease. N Engl J Med. 2013;369:754–762. doi:10.1056/NEJMct1209614
13. Straub RH, Schradin C. Chronic inflammatory systemic diseases: an evolutionary trade-off between acutely beneficial but chronically harmful programs. Evol Med Public Health. 2016;2016:37–51. doi:10.1093/emph/eow001
14. Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi:10.1038/nri910
15. Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244–247.
16. Coskun M, Nielsen OH. Tumor necrosis factor inhibitors for inflammatory bowel disease. N Engl J Med. 2013;369:2561–2562. doi:10.1056/NEJMc1312800
17. Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014–2022. doi:10.1056/NEJMoa030409
18. Barton JL. Patient preferences and satisfaction in the treatment of rheumatoid arthritis with biologic therapy. Patient Prefer Adherence. 2009;3:335–344.
19. Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med. 2003;348:601–608. doi:10.1056/NEJMoa020888
20. Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–2806. doi:10.1002/art.22025
21. de Vries MK, Wolbink GJ, Stapel SO, et al. Decreased clinical response to infliximab in ankylosing spondylitis is correlated with anti-infliximab formation. Ann Rheum Dis. 2007;66:1252–1254. doi:10.1136/ard.2007.072397
22. De Benedetti, F., Brunner, HI, Ruperto, N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–2395. doi:10.1056/NEJMoa1112802
23. Kooloos WM, de Jong DJ, Huizinga TW, Guchelaar HJ. Potential role of pharmacogenetics in anti-TNF treatment of rheumatoid arthritis and Crohn’s disease. Drug Discov Today. 2007;12:125–131. doi:10.1016/j.drudis.2006.11.013
24. Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014;29:116–125. doi:10.1016/j.ceb.2014.05.004
25. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. doi:10.1146/annurev-cellbio-101512-122326
26. Mittelbrunn M, Sanchez-Madrid F. Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol. 2012;13:328–335. doi:10.1038/nrm3335
27. Yim, N., Ryu, SW, Choi, K, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein–protein interaction module. Nat Commun. 2016;7:12277. doi:10.1038/ncomms12277
28. Alvarez-Erviti, L., Seow, Y, Yin, H, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. doi:10.1038/nbt.1807
29. Kamerkar, S., LeBleu, VS, Sugimoto, H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi:10.1038/nature22341
30. Stickney Z, Losacco J, McDevitt S, Zhang Z, Lu B. Development of exosome surface display technology in living human cells. Biochem Biophys Res Commun. 2016;472:53–59. doi:10.1016/j.bbrc.2016.02.058
31. Marcus ME, Leonard JN. FedExosomes: engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals (Basel). 2013;6:659–680. doi:10.3390/ph6050659
32. Kowal, J., Arras, G, Colombo, M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113:E968–E977. doi:10.1073/pnas.1521230113
33. Afshari A, Uhde-Stone C, Lu B. Live visualization and quantification of pathway signaling with dual fluorescent and bioluminescent reporters. Biochem Biophys Res Commun. 2014;448:281–286. doi:10.1016/j.bbrc.2014.04.108
34. Meyer, C., Losacco, J, Stickney, Z, et al. Pseudotyping exosomes for enhanced protein delivery in mammalian cells. Int J Nanomedicine. 2017;12:3153–3170. doi:10.2147/IJN.S133430
35. Van Deun, J., Mestdagh, P, Agostinis, P, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14:228–232. doi:10.1038/nmeth.4185
36. Afshari A, Uhde-Stone C, Lu B. A cooled CCD camera-based protocol provides an effective solution for in vitro monitoring of luciferase. Biochem Biophys Res Commun. 2015;458:543–548. doi:10.1016/j.bbrc.2015.01.150
37. Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75:1–18. doi:10.1016/j.colsurfb.2009.09.001
38. Sahay, G., Querbes, W, Alabi, C, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31:653–658. doi:10.1038/nbt.2614
39. Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J. 1997;11:428–442.
40. Pols MS, Klumperman J. Trafficking and function of the tetraspanin CD63. Exp Cell Res. 2009;315:1584–1592. doi:10.1016/j.yexcr.2008.09.020
41. Lu B, Moser A, Shigenaga JK, Grunfeld C, Feingold KR. The acute phase response stimulates the expression of angiopoietin like protein 4. Biochem Biophys Res Commun. 2010;391:1737–1741. doi:10.1016/j.bbrc.2009.12.145
42. Lu B, Lu Y, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. LPS and proinflammatory cytokines decrease lipin-1 in mouse adipose tissue and 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 2008;295:E1502–E1509. doi:10.1152/ajpendo.90323.2008
43. Wang Y, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. Downregulation of liver X receptor-alpha in mouse kidney and HK-2 proximal tubular cells by LPS and cytokines. J Lipid Res. 2005;46:2377–2387. doi:10.1194/jlr.M500134-JLR200
44. Peppel K, Crawford D, Beutler B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J Exp Med. 1991;174:1483–1489.
45. Engelmann H, Aderka D, Rubinstein M, Rotman D, Wallach D. A tumor necrosis factor-binding protein purified to homogeneity from human urine protects cells from tumor necrosis factor toxicity. J Biol Chem. 1989;264:11974–11980.
46. Moreland LW, Heck LW
47. Griffiths, CE, Strober, BE, van de Kerkhof, P, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. 2010;362:118–128. doi:10.1056/NEJMoa0810652
48. Tvete IF, Natvig B, Gåsemyr J, et al. Comparing effects of biologic agents in treating patients with rheumatoid arthritis: a multiple treatment comparison regression analysis. PLoS One. 2015;10:e0137258. doi:10.1371/journal.pone.0137258
49. Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333:328–335. doi:10.1016/j.bbrc.2005.05.132
50. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi:10.1038/nrd1381
Supplementary materials
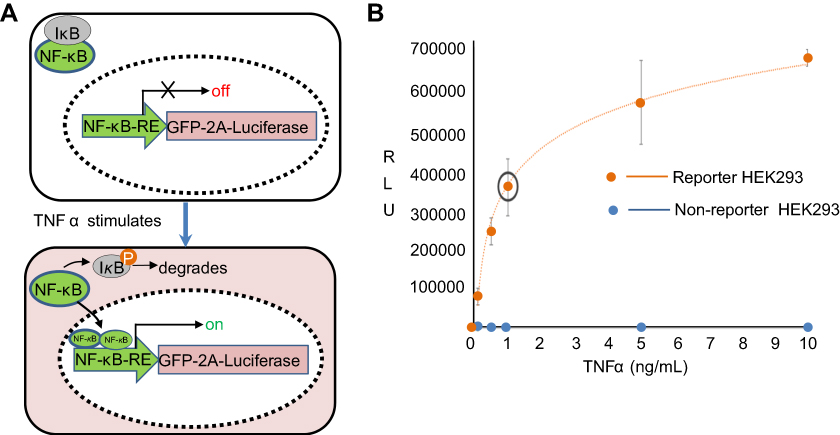 | Figure S1 Reporter cell line and its dose-response to TNFα. (A) Molecular mechanism of the reporter line. A genetically encoded reporter circuit (NF-κB-RE-driven GFP and fire luciferase gene with a self-splicing 2A peptide) was incorporated into the genome of HEK293 cells. In the absence of TNFα, the transcription factor NF-κB remains in cytosol and associates with its inhibitor protein IκB. Therefore, the reporter genes has little expression due to lack of NF-κB binding to its response element (RE). In the presence of TNFα, TNFα binding to its receptor results in the phosphorylation of IκB, which leads to its degradation and a release of NF-κB. The freed NF-κB then enters into nucleus and binds the RE in the promoter of reporter, resulting in an increase of luciferase activities. (B) A dose-response of TNF α. HEK293 cells were stimulated with incremental amount of TNFα for 24 hours. Reporter cells were then collected for the luciferase activities. A dose-dependent response to TNFα was evident (red line) as compared the non-reporter parental HEK293 (blue dots). |
Supplementary sequences
- N-CD63-hTNFR1-EC-C-CD63-GFP coding sequences
- N-CD63-hTNFR1-EC-C-CD63-GFP chimeric protein sequences
1. N-CD63-hTNFR1-EC-C-CD63-GFP coding sequences
ATGGCGGTGGAAGGAGGAATGAAATGTGTGAAGTTCTTGCTCTACGTCCTCCTGCTGGCCTTTTGCGCCTGTGCAGTGGGACTGATTGCCGTGGGTGTCGGGGCACAGCTTGTCCTGAGTCAGACCATAATCCAGGGGGCTACCCCTGGCTCTCTGTTGCCAGTGGTCATCATCGCAGTGGGTGTCTTCCTCTTCCTGGTGGCTTTTGTGGGCTGCTGCGGGGCCTGCAAGGAGAACTATTGTCTTATGATCACGTTTGCCATCTTTCTGTCTCTTATCATGTTGGTGGAGGTGGCCGCAGCCATTGCTGGCTATGTGTTTAGAGATAAGGTGATGTCAGAGTTTAATAACAACTTCCGGCAGCAGATGGAGAATTACCCGAAAAACAACCACACTGCTTTCGAATCTGGCATGGGCCTCTCCACCGTGCCTGACCTGCTGCTGCCACTGGTGCTCCTGGAGCTGTTGGTGGGAATATACCCCTCAGGGGTTATTGGACTGGTCCCTCACCTAGGGGACAGGGAGAAGAGAGATAGTGTGTGTCCCCAAGGAAAATATATCCACCCTCAAAATAATTCGATTTGCTGTACCAAGTGCCACAAAGGAACCTACTTGTACAATGACTGTCCAGGCCCGGGGCAGGATACGGACTGCAGGGAGTGTGAGAGCGGCTCCTTCACCGCTTCAGAAAACCACCTCAGACACTGCCTCAGCTGCTCCAAATGCCGAAAGGAAATGGGTCAGGTGGAGATCTCTTCTTGCACAGTGGACCGGGACACCGTGTGTGGCTGCAGGAAGAACCAGTACCGGCATTATTGGAGTGAAAACCTTTTCCAGTGCTTCAATTGCAGCCTCTGCCTCAATGGGACCGTGCACCTCTCCTGCCAGGAGAAACAGAACACCGTGTGCACCTGCCATGCAGGTTTCTTTCTAAGAGAAAACGAGTGTGTCTCCTGTAGTAACTGTAAGAAAAGCCTGGAGTGCACGAAGTTGTGCCTACCCCAGATTGAGAATGTTAAGGGCACTGAGGACTCAGGCACCACAGGGCTCGATTTAAATTCGATCCTGGACAGGATGCAGGCAGATTTTAAGTGCTGTGGGGCTGCTAACTACACAGATTGGGAGAAAATCCCTTCCATGTCGAAGAACCGAGTCCCCGACTCCTGCTGCATTAATGTTACTGTGGGCTGTGGGATTAATTTCAACGAGAAGGCGATCCATAAGGAGGGCTGTGTGGAGAAGATTGGGGGCTGGCTGAGGAAAAATGTGCTGGTGGTAGCTGCAGCAGCCCTTGGAATTGCTTTTGTCGAGGTTTTGGGAATTGTCTTTGCCTGCTGCCTCGTGAAGAGTATCAGAAGTGGCTACGAGGTGATGatggagagcgacgagagcggcctgcccgccatggagatcgagtgccgcatcaccggcaccctgaacggcgtggagttcgagctggtgggcggcggagagggcacccccaagcagggccgcatgaccaacaagatgaagagcaccaaaggcgccctgaccttcagcccctacctgctgagccacgtgatgggctacggcttctaccacttcggcacctaccccagcggctacgagaaccccttcctgcacgccatcaacaacggcggctacaccaacacccgcatcgagaagtacgaggacggcggcgtgctgcacgtgagcttcagctaccgctacgaggccggccgcgtgatcggcgacttcaaggtggtgggcaccggcttccccgaggacagcgtgatcttcaccgacaagatcatccgcagcaacgccaccgtggagcacctgcaccccatgggcgataacgtgctggtgggcagcttcgcccgcaccttcagcctgcgcgacggcggctactacagcttcgtggtggacagccacatgcacttcaagagcgccatccaccccagcatcctgcagaacgggggccccatgttcgccttccgccgcgtggaggagctgcacagcaacaccgagctgggcatcgtggagtaccagcacgccttcaagacccccatcgccttcgccagatcccgcgctcagtcgtccaattctgccgtggacggcaccgccggacccggctccaccggatctcgcCATCATCATCATCATCATTAA
Abbreviations: N-CD63, N-terminus of CD63 coding sequences; C-CD63, C-terminus of CD63 coding sequences; hTNFR1-EC, human TNFα receptor 1 extracellular domain coding sequences; GFP, green fluorescent protein coding sequences
2. N-CD63-hTNFR1-ED-C-CD63-GFP chimeric protein sequences
MAVEGGMKCVKFLLYVLLLAFCACAVGLIAVGVGAQLVLSQTIIQGATPGSLLPVVIIAVGVFLFLVAFVGCCGACKENYCLMITFAIFLSLIMLVEVAAAIAGYVFRDKVMSEFNNNFRQQMENYPKNNHTAFESGMGLSTVPDLLLPLVLLELLVGIYPSGVIGLVPHLGDREKRDSVCPQGKYIHPQNNSICCTKCHKGTYLYNDCPGPGQDTDCRECESGSFTASENHLRHCLSCSKCRKEMGQVEISSCTVDRDTVCGCRKNQYRHYWSENLFQCFNCSLCLNGTVHLSCQEKQNTVCTCHAGFFLRENECVSCSNCKKSLECTKLCLPQIENVKGTEDSGTTGLDLNSILDRMQADFKCCGAANYTDWEKIPSMSKNRVPDSCCINVTVGCGINFNEKAIHKEGCVEKIGGWLRKNVLVVAAAALGIAFVEVLGIVFACCLVKSIRSGYEVMMESDESGLPAMEIECRITGTLNGVEFELVGGGEGTPKQGRMTNKMKSTKGALTFSPYLLSHVMGYGFYHFGTYPSGYENPFLHAINNGGYTNTRIEKYEDGGVLHVSFSYRYEAGRVIGDFKVVGTGFPEDSVIFTDKIIRSNATVEHLHPMGDNVLVGSFARTFSLRDGGYYSFVVDSHMHFKSAIHPSILQNGGPMFAFRRVEELHSNTELGIVEYQHAFKTPIAFARSRAQSSNSAVDGTAGPGSTGSRHHHHHH
Abbreviations: N-CD63, N-terminus of CD63; C-CD63, C-terminus of CD63; hTNFR1-EC, human TNFα receptor 1-extracellular domain; GFP, green fluorescent protein.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.