")
Back to Journals » International Journal of Nanomedicine » Volume 16
Damaging Tumor Vessels with an Ultrasound-Triggered NO Release Nanosystem to Enhance Drug Accumulation and T Cells Infiltration
Authors Xu Y, Liu J, Liu Z, Chen G, Li X, Ren H
Received 3 December 2020
Accepted for publication 16 March 2021
Published 1 April 2021 Volume 2021:16 Pages 2597—2613
DOI https://doi.org/10.2147/IJN.S295445
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Yan Xu, Jiwei Liu, Zhangya Liu, Guoguang Chen, Xueming Li, Hao Ren
School of Pharmaceutical Science, Nanjing Tech University, Nanjing, Jiangsu, People’s Republic of China
Correspondence: Xueming Li; Hao Ren
Nanjing Tech University, School of Pharmaceutical Science and Pharmaceutical Engineering, No. 30 Puzhu South Road, Nanjing, 211816, China
Email [email protected]; [email protected]
Introduction: Limited by tumor vascular barriers, restricted intratumoural T cell infiltration and nanoparticles accumulation remain major bottlenecks for anticancer therapy. Platelets are now known to maintain tumor vascular integrity. Therefore, inhibition of tumor-associated platelets may be an effective method to increase T cell infiltration and drug accumulation at tumor sites. Herein, we designed an ultrasound-responsive nitric oxide (NO) release nanosystem, SNO-HSA-PTX, which can release NO in response to ultrasound (US) irradiation, thereby inhibiting platelet function and opening the tumor vascular barrier, promoting drug accumulation and T cell infiltration.
Methods: We evaluated the ability of SNO-HSA-PTX to release NO in response to US irradiation. We also tested the effect of SNO-HSA-PTX on platelet function. Plenty of studies including cytotoxicity, pharmacokinetics study, biodistribution, blood perfusion, T cell infiltration, in vivo antitumor efficacy and safety assessment were conducted to investigate the antitumor effect of SNO-HSA-PTX.
Results: SNO-HSA-PTX with US irradiation inhibited tumor-associated platelets activation and induced openings in the tumor vascular barriers, which promoted the accumulation of SNO-HSA-PTX nanoparticles to the tumor sites. Meanwhile, the damaged vascular barriers allowed oxygen-carrying hemoglobin to infiltrate tumor regions, alleviating hypoxia of the tumor microenvironment. In addition, the intratumoral T cell infiltration was augmented, together with chemotherapy and NO therapy, which greatly inhibited tumor growth.
Discussion: Our research designed a simple strategy to open the vascular barrier by inhibiting the tumor-associated platelets, which provide new ideas for anti-tumor treatment.
Keywords: intratumoural T cell infiltration, nanoparticles accumulation, tumor vascular barriers, nitric oxide, tumor-associated platelets
Introduction
Cancer immunotherapy has become a powerful clinical strategy for cancer treatment that has achieved remarkable breakthroughs.1–3 However, despite recent successes, some cancer patients are resistant to immunotherapy, which may be due to the inability of T cells to infiltrate the tumors. Many groups have demonstrated that the number of CD8+ and CD4+ T cells in tumors was closely associated with a good clinical outcome.4–6 Tumors induce vascular barriers which inhibit T-cell homing, thereby weakening the efficacy of immunotherapy.7–9 Moreover, the tumor vascular barriers also impair the delivery efficiency of antitumor drugs, leading to ineffective antitumor therapy.10–12 New strategies aimed at breaking tumor vascular barriers with enhanced intratumoral T cells infiltration and improved drug accumulation are urgently needed to advance the field of anticancer therapy.
Besides maintaining hemostasis, platelets play a crucial role in promoting cancer progression, particularly in maintaining tumor vascular integrity.13,14 Tumor blood vessels always show vascular damage and decreased endothelial connections, which is called an enhanced permeability and retention (EPR) effect. Nanoparticles penetrate the tumor microenvironment through EPR effect.15–18 However, the activated platelets repair broken blood vessels by adhering to the vascular endothelium, thus restraining vascular permeability, and hindering the diffusion of immune cells and medicines into solid tumors. In addition, it has been reported that the depletion of platelets in tumor-bearing mice can cause loss of vascular integrity, leading to massive bleeding in the tumors.19,20 Platelet depletion has been shown to promote the intratumoral accumulation of chemotherapeutic drugs, thereby enhancing the anti-tumor effect.21,22 Considering the vital role of platelets in maintaining tumor vascular barriers, we hypothesized that a specific blockade of tumor-associated platelets may represent a new method to destabilize tumor vasculature, leading to increased endothelial permeability and efficient infiltration of immune cells and drugs. Although several interesting works have shown that the systemic platelet depletion significantly improved tumor vascular permeability and enhances the efficacy of cancer therapy.23–25 However, systemic depletion of platelets in cancer patients is not a therapeutic option due to severe side effects on hemostasis.
As an endogenous platelet inhibitor, nitric oxide (NO) suppresses multiple platelet functions, including preventing its aggregation and adhesion to the vascular endothelium, thereby preventing thrombus and atherosclerosis.26–29 Recently, it has been validated that NO molecules hold great potential in tumor therapy due to its unparalleled killing effect on cancer cells at high concentrations.30,31 Small molecule NO donors include S-nitrosothiols (SNOs), metal-NO-complexes, sydonomines, diazeniumdiolates (NONOates), and NO-drug hybrids.32 However, the application of these NO-release strategies still limited by nonbiodegradable components, uncontrollable release and insufficient tumor perfusion, which leads to the leakage of NO into normal tissues and restricted killing effect of NO.33,34 Therefore, an ideal NO release therapeutic system should have good biocompatibility and release high levels of NO locally and controllably under specific stimuli, which can be conveniently applied to tumor sites, such as ultrasound (US), laser and magnetism.
Herein, we designed an ultrasound-responsive NO release nanosystem, SNO-HSA-PTX, which was constructed by loading an anticancer drug (paclitaxel, PTX) into a NO donor-modified albumin shell (Figure 1A). We have demonstrated the SNO-HSA-PTX stimulated by US irradiation achieved the on-demand release of NO for the inhibition of tumor-associated platelets activation. As shown in Figure 1B, when the SNO-HSA-PTX nanoparticles were administered intravenously into the mice, they reached into tumor regions via the leaky tumor vasculature. The US irradiation was performed to trigger the NO release from SNO-HSA-PTX, which locally inhibited tumor-associated platelets activation. The absence of platelets in tumors induced openings in the tumor vascular barriers, which promoted the accumulation of SNO-HSA-PTX nanoparticles to the tumor sites. Meanwhile, the damaged vascular barriers allowed oxygen-carrying hemoglobin to infiltrate tumor regions, alleviating hypoxia of the tumor microenvironment and reverse tumor immunosuppression. In addition, a significant amount of NO was fast released from SNO-HSA-PTX under US irradiation to reach a high concentration, directly killing cancer cells as NO gas therapy. PTX also exerted the chemotherapeutic effect and further induced tumor cell apoptosis.35 Finally, the intratumoral infiltration of T cells was augmented, together with chemotherapy and NO therapy, which greatly inhibited tumor growth. We for the first time to reveal the role of NO in opening the tumor vascular barrier through the platelet pathway. Systemic administration of SNO-HSA-PTX to mice with US irradiation resulted in enhanced tumor vascular leakage, accompanied by increased drug accumulation, tumor hypoxia alleviation and T cells infiltration, ultimately leading to tumor suppression.
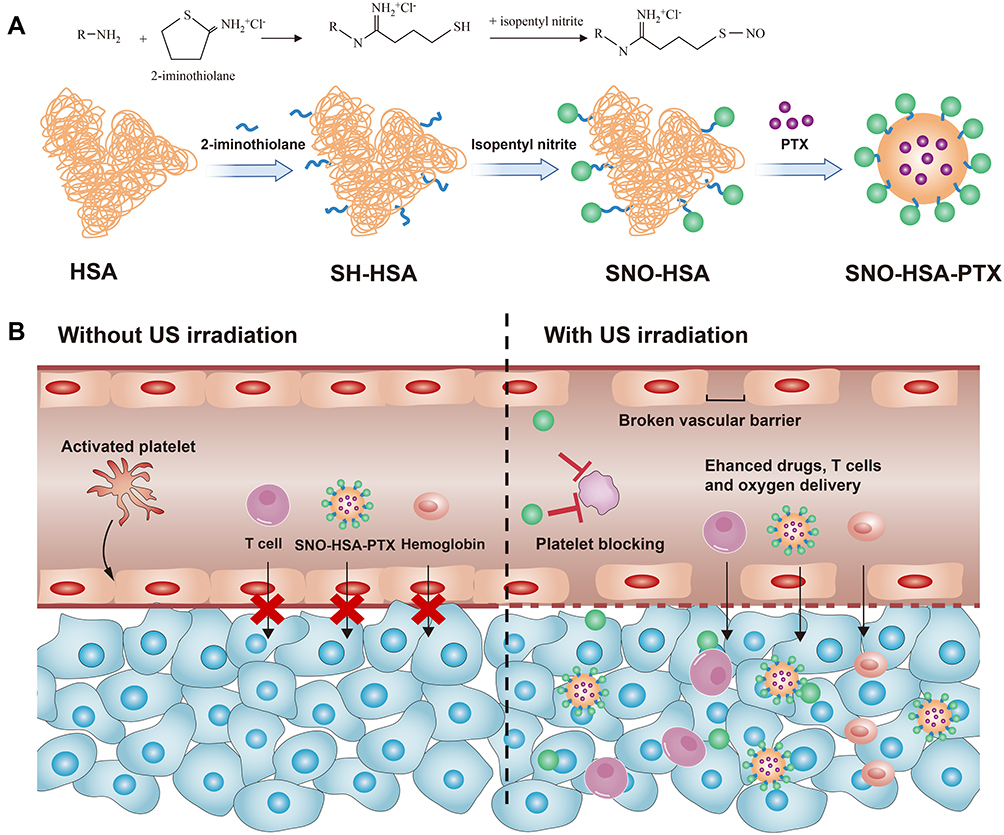 |
Figure 1 Schematic illustration of improved efficacy of US-triggered NO release nanosystem mediated platelet blockade. (A) Schematic diagram of preparation of SNO-HSA-PTX nanoparticles. (B) The NO released by SNO-HSA-PTX under US stimulation inhibited platelet activation thus opening the tumor vascular barrier, leading to enhanced T cells infiltration, drug accumulation and tumor hypoxia relief. |
Experimental Section
Materials
Human serum albumin solution (HSA) was purchased from Octapharma. 2-iminothiolane, DTPA and 2-mercaptoethanol were provided by Aladdin Industrial Co. (Shanghai, China). Paclitaxel (PTX) was purchased from Dalian Meilun Biotechnology (Dalian, China). Isopentyl nitrite and IR780 were brought from J&K Scientific Co. (Beijing, China). BCA kits, NO probe and Griess kits were obtained from Beyotime Biotechnology (Shanghai, China). CFSE were obtained from Sigma Chemical Co. (Shanghai, China). The CCK-8, DAPI and DIL were purchased from Dojindo Molecular Technologies (Rockville, USA). All other reagents were used directly as supplied without any further purification.
Animal Model
All animal procedures were approved by Laboratory Animal Management Committee at Nanjing Tech University (SYXK(Shu) 2016–0031). All animal procedures were performed according to the guidelines of the Administration Committee of Experimental Animals in Jiangsu Province and the Ethics Committee of Nanjing Tech University. BALB/c female mice were brought from Qinglongshan Laboratory Animal Center (Yangzhou, China). Mice were housed in groups of six maintaining free water and food supply. The 4T1 cells (1 × 107/mL, 50 μL) were subcutaneous injected into the lower flanks of the mice. The tumor was taken off when its volume reached 150 mm3 and processed into small pieces of about 4 mm3. Then, these pieces were implanted subcutaneously into BALB/c mice to establish the subcutaneous tumor models.
Synthesis of SNO-HSA
S-nitrosated human serum albumin (SNO-HSA) was obtained by modifying the S-Nitrosothiol groups on the surface of HSA. Briefly, 400 mg of HSA (200 mg/mL, 2 mL) was reacted with 3 mM of 2-iminothiolane dissolved in 20 mL of phosphate buffer for 1 h. The reaction product was concentrated by an ultrafiltration cup to obtain S-thiolated HSA (SH-HSA). The concentration of SH-HSA was adjusted to 0.2 mM and then 15 mM of isopentyl nitrite was added to react at 37°C for 3 h. The reaction solution was concentrated and purified to obtain the final product SNO-HSA through an ultrafiltration cup. The concentration of nitric oxide (NO) and protein in SNO-HSA was measured by Griess Reagent and BCA kits, respectively.
Preparation and Characterization of SNO-HSA-PTX Nanoparticles
SNO-HSA-PTX nanoparticles were prepared by encapsulating hydrophobic drugs paclitaxel (PTX) in the hydrophobic cavities of proteins. Briefly, 2-mercaptoethanol was added to HSA or SNO-HSA solution (2 mg/mL, 100 mL) to open the hydrophobic regions of proteins. PTX dissolved in ethyl alcohol (10 mg/mL, 2 mL) was dropped uniformly and slowly into the above HSA or SNO-HSA solution to form HSA-PTX and SNO-HSA-PTX nanoparticles. In addition, 2 mg of IR780 was added to PTX ethyl alcohol to form IR780-labeled SNO-HSA-PTX. The obtained nanoparticle dispersions were enriched by ultrafiltration. SNO-HSA-PTX nanoparticles were freeze-dried and stored at −20 4 °C. The particle size of SNO-HSA-PTX was monitored by Malvern Zetasizer (Nano ZS, Malvern, UK). The shape of SNO-HSA-PTX was observed under a transmission electron microscopy (TEM, JEM-2100, Japan). The PTX concentration was quantified using high-performance liquid chromatography (HPLC).
Drug loading of PTX = (actual PTX content in nanoparticles/weight of nanoparticles) × 100%
Encapsulation efficiency of PTX = (actual drug loading of nanoparticles/theoretical drug loading of nanoparticles) × 100%
US-Triggered Drug Release
The effect of US irradiation on PTX release was studied. The SNO-HSA-PTX solution (20 mg/mL) was dealt with US irradiation (1 MHz, 2 W/cm2) for different times (1, 2, 3, 4, 5 min), and the NO generation was measured by Griess Reagent. Similar, different concentration of SNO-HSA-PTX (2.5, 5, 10, 15, 20 mg/mL) was dealt with US irradiation (1 MHz, 2 W/cm2, 5 min), and the NO generation was measured by Griess Reagent. The effect of US irradiation on PTX release was also studied. The SNO-HSA-PTX solution (PTX: 2 mg/mL) was dealt with US irradiation (1 MHz, 2 W/cm2) for 5 min. The SNO-HSA-PTX solution with or without US irradiation was put into a dialysis bag to study its release behavior in 0.2% (w/v) Tween 80-PBS.
Platelet Preparation
Whole blood was quickly collected from the retro-orbital sinus of BALB/c mice into tubes with sodium citrate (Blood: sodium citrate=9:1). Then, the anticoagulated blood was centrifuged at 200×g for 8 min to obtain the supernatant, which is platelet-rich plasma (PRP). Centrifuge PRP at 1000 ×g for 8 min and discard the supernatant to obtain white platelet pellet. Finally, the platelet suspension was obtained by resuspending the platelet pellet with modified Tyrode’s buffer for future use. Tyrode’s buffer was composed of 137 mM NaCl, 2 mM KCl, 12 mM NaHCO3, 0.3 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, and 5 mM HEPES.
Platelet Aggregation Assay
SNO-HSA-PTX, HSA-PTX or saline was added into platelet suspensions followed by US irradiation (1 MHz, 2 W/cm2) for 5 min (platelets: 4×107/mL, PTX: 2 μg/mL and NO: 2 μM). After that, 5 μL of thrombin (200 U/mL) was added to activate the platelet aggregation. The ultraviolet absorption of platelet suspensions was measured at 650 nm before and after thrombin addition. Changes in absorbance reflect platelet aggregation. The saline group served as the positive control. The supernatant was collected for ATP detection according to the luminescence level.
The platelets were labeled with CFSE (5(6)-Carboxyfluorescein diacetate, succinimidyl ester) to directly show platelet aggregation. After treating platelets as described above, the platelet suspensions were dropped onto the glass slide to capture the aggregated platelets by the fluorescence microscope.
Platelet Adhesion Assay
SNO-HSA-PTX, HSA-PTX or saline was added into CFSE-labeled platelet suspensions followed by US irradiation (1 MHz, 2 W/cm2) for 5 min (platelets: 4×107/mL, PTX: 2 μg/mL, NO: 2 μM). Then, the processed platelet suspensions were added to the collagen I-coated 96-well plates and incubated for 1 h at 37°C. After that, the collagen I-coated 96-well plates were washed three times with buffer. The adhesion of platelets was observed under the microscope. The number of the platelets in the field of view was counted to characterize platelet adhesion. The adhesion of saline-treated platelets was served as a positive control.
Cell Culture
The mouse breast cancer cell lines 4T1 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA).
Intracellular NO Generation
DAF-FM DA (Ex/Em =495/515 nm) was a typical NO indicator to characterize the intracellular NO production. The 4T1 cells were seeded in 24-well plates 24 h in advance. The cells were incubated with DAF-FM DA (5 μM) for 20 min then washed by PBS. Next, HSA-PTX or SNO-HSA-PTX (PTX: 2 μg/mL, NO: 2 μM) was added to the cells and incubated for 2 h. After incubation, the extracellular nanoparticles were removed, the cells were dealt with US irradiation (1 MHz, 2 W/cm2) for 5 min. The fluorescence was captured by confocal laser scanning microscopy (CLSM) and a fluorescence microplate. The cells after the above treatments were collected for intracellular NO analysis by the flow cytometry analysis (ACEA Biosciences).
In vitro Cytotoxicity
4T1 cells were prepared in 96-well plates. HSA-PTX and SNO-HSA-PTX were added to the cells and incubated for 2 h and then washed to remove extracellular nanoparticles. Cells were exposed to US irradiation (1 MHz, 2 W/cm2) for 5 min per well followed by the addition of a mixture of CCK-8 (10 μL) and fresh medium (100 μL). After incubation for another 2 h, the cell viability was analyzed by measuring the absorbance at 450 nm by an Infinite F50 microplate reader (Tecan Life Sciences, Mannedorf, Switzerland).
In vivo Pharmacokinetics Study
Healthy SD rats were randomly divided into 3 groups (n= 3). PTX, HSA-PTX and SNO-HSA-PTX were administered via the tail vein at the dose of 20 mg PTX/kg, respectively. At predetermined time intervals (0.5, 1, 2, 4, 8, 12, 24, 48 and 72 h), blood samples were collected with heparin and centrifuged to obtain the plasma. The concentration of PTX in plasma was determined by HPLC. Drug and statistics (DAS) software (version 2.1.1, Mathematical Pharmacology Professional Committee, China) was utilized to calculate the pharmacokinetics parameters.
In vivo Imaging and Biodistribution
To study the biodistribution of SNO-HSA-PTX, the NIR fluorescent dye IR780 was applied to label SNO-HSA-PTX. Briefly, 200 μL of IR780-labeled SNO-HSA-PTX (IR780: 80 μg/mL) was injected into mice when the tumor size reached approximately 150–200 mm3. Then, these groups received US irradiation (1 MHz, 2 W/cm2, 5 min) at 3 h after injection. At a different time after US irradiation (0, 4, 24, 48 h), the fluorescence images of IR780 (Ex/Em = 740/810 nm) in mice were obtained using the Maestro system (Cri Inc., Woburn, MA, USA). IR780 fluorescence data were analyzed using IVIS Living imaging software (PerkinElmer).
4T1 tumor-bearing mice were injected with HSA-PTX and SNO-HSA-PTX via the tail veins (PTX: 2 mg/mL, NO: 2 mM, 200 μL), and then received US irradiation (1 MHz, 2 W/cm2, 5 min) at 3 h after injection. The tumor tissues were collected and weighted at 0, 6, 24 and 48 h after US irradiation. The PTX in tumors was extracted with dichloromethane. Then, the extracted PTX was further concentrated by evaporating the dichloromethane using a nitrogen blower, and then dissolved in acetonitrile for HPLC detection.
In vivo NO Generation
4T1 tumor-bearing mice were injected with SNO-HSA-PTX via the tail veins, and then received US irradiation (1 MHz, 2 W/cm2, 5 min) at 3 h after injection. The tumors were removed for anti-nitrotyrosine antibody staining 24-h post-injection to observe the NO generation in vivo. Also, tumor slices were stained with CD62P to investigate the activation of platelets.
In vivo Tumor Vessel Permeability Assay
A typical Evans blue assay was used to evaluate the tumor vascular permeability. 4T1 tumor-bearing mice were injected with SNO-HSA-PTX via the tail veins, and then received US irradiation (1 MHz, 2 W/cm2, 5 min) at 3 h after injection. After 3 h, the mice were intravenously injected with Evans blue (40 mg/kg). After another 5 h, the tumors were collected and weighed. Then, the tumors were homogenized with formamide to extract Evans blue, and the ultraviolet absorption of Evans blue was detected at 620 nm. The fluorescence of Evans blue was observed using a fluorescence microscope by frozen sections of tumors.
In vivo Blood Perfusion
4T1 tumor-bearing mice were injected with SNO-HSA-PTX via the tail veins, and then received US irradiation (1 MHz, 2 W/cm2, 5 min) at 3 h after injection. The changes in tumor blood flow were observed by laser speckle contrast imaging at different time after US irradiation. One day later, the mice were sacrificed to collect tumors for H&E staining. Then, these tumors were homogenized in saline and centrifuged to obtain supernatant. The hemoglobin concentration was measured using a hemoglobin assay kit (Jiancheng, Nanjing). Also, tumor slices were stained with anti-mouse HIF-1α antibody for HIF-1α-based hypoxia detection.
In vivo Analysis of T Cells
For the evaluation of T cell infiltration, 4T1 tumor-bearing mice were injected with SNO-HSA-PTX via the tail veins, and then received US irradiation (1 MHz, 2 W/cm2, 5 min) at 3 h after injection. Two days after the treatment, the mice were sacrificed, and the tumors were collected for FACS analyses. Briefly, tumor tissues were cut into small pieces and digested in digestion solution at 37°C for 30 min. The digested tissues were filtered to obtain single-cell suspensions. For CD8+ T cells infiltration and regulatory T cells (Tregs) evaluation, cells were stained with anti-CD3-FITC, anti-CD8-PE, anti-CD4-APC, and anti-FoxP3-PE-CY7 antibodies (BD Pharmingen). For intracellular Foxp3 staining, the cells were further fixed and permeabilized using a Foxp3/Transcription Factor Buffer Set (BD Pharmingen). The immunofluorescence staining of CD8 was also performed and captured by fluorescence microscopy.
In vivo Antitumor Efficacy
When the tumor size reached approximately 80 mm3, 4T1 tumor-bearing mice were randomly assigned into six groups (n = 6). The mice were intravenously injected with 200 μL of saline (groups 1 and 2), HSA-PTX (PTX: 2 mg/mL; groups 3 and 4), and SNO-HSA-PTX (NO: 2 mM and PTX: 2 mg/mL; groups 5 and 6). Groups 2, 4, and 6 additionally received US irradiation (1 MHz, 2 W/cm2, 5 min) 3 h post-injection. The treatments were given on mice on days 1, 3, and 5. The tumor size and body weight were recorded every 2 days, and tumor volume was calculated as width2 × length/2. On day 15, the tumors were collected and weighted. To evaluate the therapeutic efficacy of the different treated groups, the tumor-bearing mice were treated as describedand tumors were removed on day 2 for H&E staining and TUNEL staining. The images were acquired by a digital microscope (Nikon, Japan).
Biosafety of SNO-HSA-PTX in vivo
The blood biochemical analyses of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) were performed to indicate the biosafety of SNO-HSA-PTX. After treatment with saline and SNO-HSA-PTX for 1 day, the whole blood was collected for analysis. In addition, tail blooding test was used to evaluate the effect of US-triggered release NO nanoparticles on the hemostatic function of platelets. The mice were intravenously injected with saline or SNO-HSA-PTX (PTX: 2 mg/mL, NO: 2 mM, 200 μL) and were then exposed to US irradiation (1 MHz, 2 W/cm2, 5 min) at 3 h after injection. Twelve hours after administration, the tail tips were truncated and the tails were soaked immediately in the saline at 37 °C. The tail bleeding time of mice was recorded.
Statistical Analysis
Multiple comparisons were analyzed by ANOVA or Tukey’s post hoc tests. All data were presented as the mean ± SD or the mean ± SEM. Significance was expressed as *p < 0.05, **p < 0.01, and ***p < 0.001.
Results and Discussion
Preparation and Characterization of SNO-HSA-PTX
A previous study reported that the characteristic absorption peak of S-NO exists at 340 nm.36 As shown in Figure 2A, there was an obvious absorption peak at 340 nm in SNO-HSA compared with HSA, indicating the successful synthesis of SNO-HSA. There was 6.32±1.19 mol of SNO per mol of SNO-HSA. The SNO-HSA-PTX nanoparticles were constructed by encapsulating PTX into the hydrophobic cavity of SNO-HSA through hydrophobic interaction. The average size of SNO-HSA-PTX was ~139±3.22 nm obtained by dynamic light scattering (DLS) (Figure 2B). As shown in the representative TEM images in Figure 2C, the synthesized SNO-HSA-PTX nanoparticles were regular spherical and uniform in size with an average diameter of about 136±4.13 nm. The concentration of PTX was detected by HPLC, and the load capacity of PTX in SNO-HSA-PTX nanoparticles was about 10.5±0.89%. The encapsulation efficiency of PTX in SNO-HSA-PTX was approximately 78.66±2.12%. The load capacity of SNO in SNO-HSA-PTX nanoparticles was about 0.68±0.04%. It was observed that SNO-HSA-PTX nanoparticles exhibited good stability in physiological media (Figure S1). These results indicated the successful preparation of SNO-HSA-PTX.
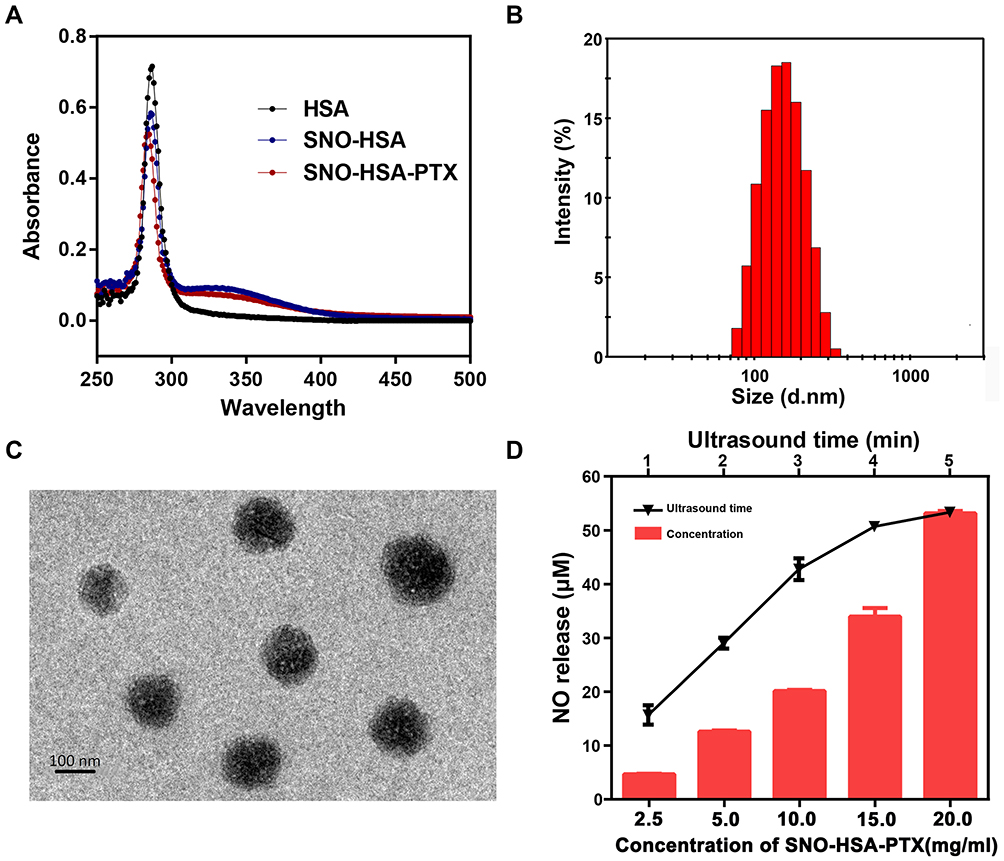 |
Figure 2 Characterization of SNO-HSA-PTX. (A) UV-vis absorption spectra HAS, SNO-HSA and SNO-HAS-PTX. (B) Particle size distribution of SNO-HSA-PTX. (C) The TEM images of SNO-HSA-PTX. (D) Concentration and US time-dependent changes of NO generated from SNO-HSA-PTX. |
In order to evaluate the ultrasound (US) stimuli-responsive NO release behavior of SNO-HSA-PTX, the accumulative release of NO was measured by a typical Griess assay. NO can react with Griess assay to form diazo compounds, which can be measured by 540 nm ultraviolet light. The NO released from SNO-HSA-PTX increased with the prolongation of US time (Figure S2). As shown in Figure 2D, treating SNO-HSA-PTX with US irradiation resulted in an obvious release of NO. We also observed that the NO release rates of SNO-HSA-PTX after US irradiation were increased and reached the maximum at 5 min. Therefore, we determined the optimal time of US irradiation was 5 min. The evaluation of the NO generation revealed that SNO-HSA-PTX displayed a clear US responsive NO release profile. The US response release of NO from SNO-HSA-PTX may be attributed to the instability of the S-NO bonds under ultrasonic energy, resulting in the breaking of the S-NO bond thus releasing NO. In addition to triggering the release of NO, US stimulation also showed an acceleration effect on the release of PTX (Figure S3). We inferred that this may be due to the energy of US irradiation destroying the nanostructure of SNO-HSA-PTX, causing the leakage of PTX thus leading to a faster release of PTX.
The Inhibitory Ability of SNO-HSA-PTX on Platelets
Platelets act as the crucial modulator in regulating angiogenesis and maintaining vasculature function in the tumor microenvironment.37 The tumor site is called a non-healing wound, allowing platelets to accumulate at the tumor site to maintain the integrity of the tumor blood vessel wall, thus limiting the delivery of drugs to the tumor site. As an endogenous inhibitor of platelets, NO can prevent platelets from activation, inhibiting platelet adhesion and aggregation. In order to study whether SNO-HSA-PTX has the effect of enhancing tumor vascular permeability, we first study the anti-platelet aggregation effect of NO released by SNO-HSA-PTX. Platelets labeled with CFSE were incubated with HSA-PTX or SNO-HSA-PTX, and US irradiation was used to trigger the generation of NO.
As shown in Figure 3A, it can be clearly observed that platelets treated with SNO-HSA-PTX+US irradiation still maintain their individual shape, suggesting that the platelets were inactive. While the platelets in the other treatment groups aggregated and stuck into clumps. Then, the platelet aggregation rate of the platelets with different treatments was detected (Figure 3B). The platelet aggregation induced by thrombin served as a positive control. Compared with the HSA-PTX (91.67±6.14%), HSA-PTX+US (89.87±8.53%) and SNO-HSA-PTX (81.68±6.08%) groups, the platelet aggregation of the SNO-HSA-PTX with US irradiation pretreatment group was minimal (30.24±6.18%). These results indicated that the NO produced by SNO-HSA-PTX in response to US irradiation can effectively prevent platelet aggregation.
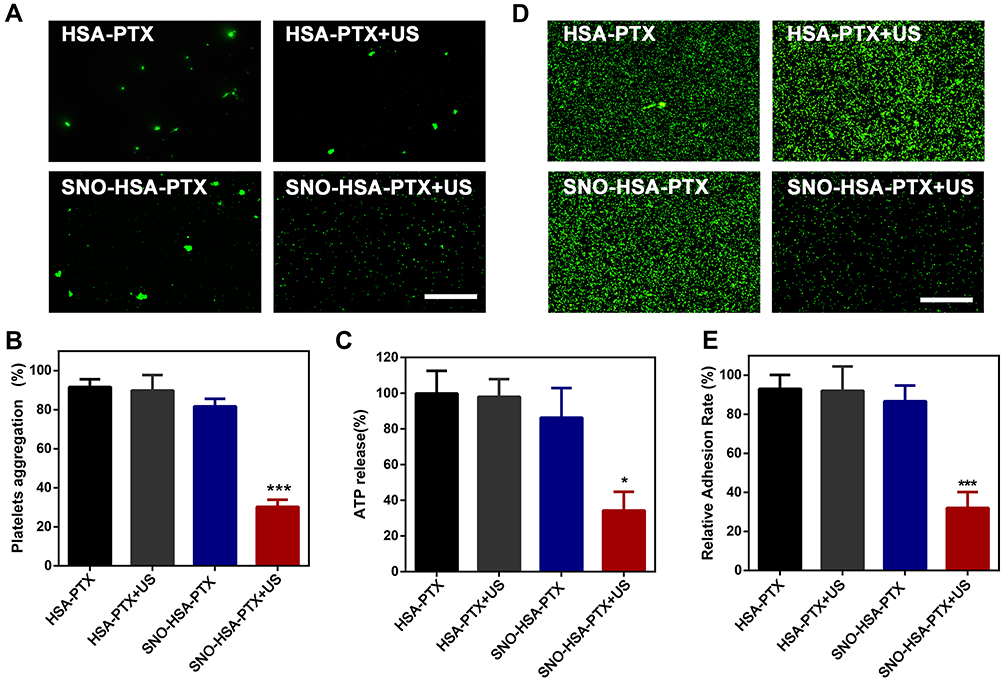 |
Figure 3 The inhibitory ability of SNO-HSA-PTX on platelets. (A) Aggregation images of CFSE-labeled platelets treated with HSA-PTX, HSA-PTX+US, SNO-HSA-PTX or HSA-PTX+US (1 MHz, 2 W/cm2, 5 min). Scale bar = 50 μm. (B) The aggregation rates of platelets with different treatments induced by thrombin. ***p < 0.001. (C) ATP release from platelets activated by thrombin. *p < 0.05. (D) Representative image of platelet adhesion. Scale bar: 50 μm. (E) Quantification of platelet adhesion after treatment with HSA-PTX, HSA-PTX+US, SNO-HSA-PTX or HSA-PTX+US (1 MHz, 2 W/cm2, 5 min). ***p < 0.001. |
Activated platelets can release ATP. The release of ATP from platelets is an important indicator of platelet activation.38 Next, we analyzed the ATP levels in the supernatant obtained by centrifuging the platelets suspension after different treatments (Figure 3C). The ATP released from platelets under thrombin stimulation was used as a positive control. SNO-HSA-PTX with US irradiation significantly reduced the ATP levels, while other treatments without NO generation did not reduce ATP levels, indicating that the released NO possessed the capability of inhibiting platelet activation.
The platelet adhesion was examined to assess the ability of US-triggered release from SNO-HSA-PTX in preventing platelets from adhering to the vascular endothelium. As shown in Figure 3D and E. SNO-HSA-PTX with US irradiation significantly reduced the adherent platelets to collagen-coated plates, as reflected by the decreased quantification of platelet adhesion rate. Together, these results indicated that SNO-HSA-PTX under US irradiation possessed the ability to inhibit platelet aggregation, activation and adhesion.
Intracellular NO Generation and NO-Assisted Cytotoxicity
To confirm the US responsive NO generation in 4T1 cells, the released NO was stained with the DAF-FM DA fluorescence probe. As revealed in Figure 4A, only after exposure to US irradiation, SNO-HSA-PTX groups showed significant green fluorescence, and the quantitative flow cytometry analysis also demonstrated the large amount of NO generation (Figure 4B), indicating the on-demand US responsive NO release. Simultaneously, the US responsive NO release of SNO-HSA-PTX was further validated by quantitative of DAF-FM fluorescence (Figure 4C). Taken together, the SNO-HSA-PTX with US irradiation displayed a high level of NO generation and would offer the opportunity for more effective anti-tumor NO therapy.
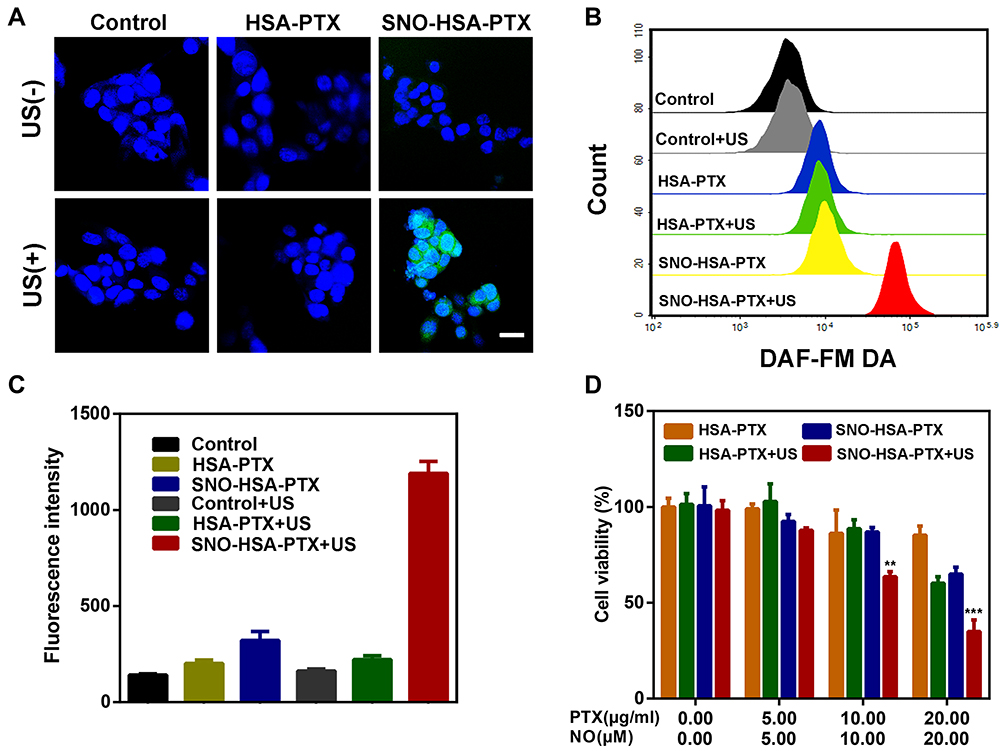 |
Figure 4 Intracellular NO generation and cell cytotoxicity. (A) Representative CLSM images of intracellular NO production in 4T1 cells stained with DAF-FM DA (green) and DAPI (blue). Scale bars: 20 μm. (B) NO generation detected by the flow cytometry. (C) Quantification of NO production by fluorescence microplate reader. (D) The cell viability of 4T1 cells treated with HSA-PTX or SNO-HSA-PTX nanoparticles with or without US irradiation (1.0 MHz, 2 W/cm2, 5 min). **p < 0.01, ***p < 0.001. |
Next, we assessed whether the US-triggered NO release was sufficient to achieve NO-mediated apoptosis, as the high concentration of NO possesses a killing effect on tumor cells. The CCK-8 kit was employed to investigate the cytotoxicity of US-triggered NO release nanoparticles on 4T1 cells. As shown in Figure 4D, with the increased concentration of the loaded drug, the cytotoxicity of SNO-HSA-PTX with US irradiation group was significantly higher than that of HSA-PTX with US irradiation group, which could be speculated by the knowledge that the NO released from SNO-HSA-PTX was considered as the therapeutic accelerator. The carrier of SNO-HSA-PTX or HSA-PTX did not cause any significant toxicity to cells at the same concentration of nanoparticles, indicating the safety of nanoparticles in circulation (Figure S4).
In vivo Pharmacokinetics
The pharmacokinetics of PTX, HSA-PTX and SNO-HSA-PTX were investigated by i.v. injection at a dose of 20 mg PTX/kg. The time-dependent plasma concentration profiles were co-plotted in Figure S5 and pharmacokinetic parameters were summarized in Table S1. PTX was cleared from the circulatory system very quickly, while the clearance (CL) of HSA-PTX and SNO-HSA-PTX was much slower, which probably related to the reticuloendothelial system (RES) escape and decreased leakiness. Compared with PTX, the blood circulation of HSA-PTX/SNO-HSA-PTX was prolonged with 7.85/8.71 times longer half-life (t1/2), 1.93/2.05 times larger area under the curve (AUC), and 4.70/4.88 times higher mean retention time (MRT) respectively. The prolonged PTX presence in the circulation system might be attributed to the protein shell, which contributed to the enhanced accumulation of nanoparticles in tumor tissue and effective anticancer activity in vivo. In addition, there was no difference between HSA-PTX and SNO-HSA-PTX in pharmacokinetic parameters, probably owing to the similar carrier structures and relatively high stability in physiological condition.
Enhancement of Tumor Vascular Permeability by SNO-HSA-PTX
Previous studies have identified platelets as the key regulators of tumor vascular integrity. Therefore, strategies to selectively inhibit platelet activation at tumor sites to increase the accumulation of drugs in solid tumors represent an important goal of cancer therapy. In addition, previous papers have reported that NO influence nanoparticles distribution.39–41 The near-infrared dye IR780 was loaded into the nanosystems to monitor the accumulation of SNO-HSA-PTX to the tumor site after US irradiation. As shown in Figure 5A, there was no significant difference in the accumulation of HSA-PTX and SNO-HSA-PTX in the absence of US irradiation (0 h). However, with increased time, the difference of intratumoral accumulation between the SNO-HSA-PTX group and the HSA-PTX group gradually widened. The quantified data reveal that the accumulation of SNO-HSA-PTX in the tumor site was at levels about 1.5 times higher than that of HSA-PTX as reflected in Figure 5B. At 48 h after US irradiation, mice were euthanatized, and the major organs and tumors were dissected for ex vivo biodistribution analyses (Figure S6). Strong fluorescence signals and relatively high fluorescence intensity were found in the tumors of mice treated with SNO-HSA-PTX, while weak fluorescence was observed in HSA-PTX treated mice. We speculated that this probably due to the NO triggered by US irradiation enhanced the tumor vascular permeability through inhibiting platelets and subsequent breaking the tumor vascular barrier, thereby increasing the accumulation of nanoparticles in the tumor site.
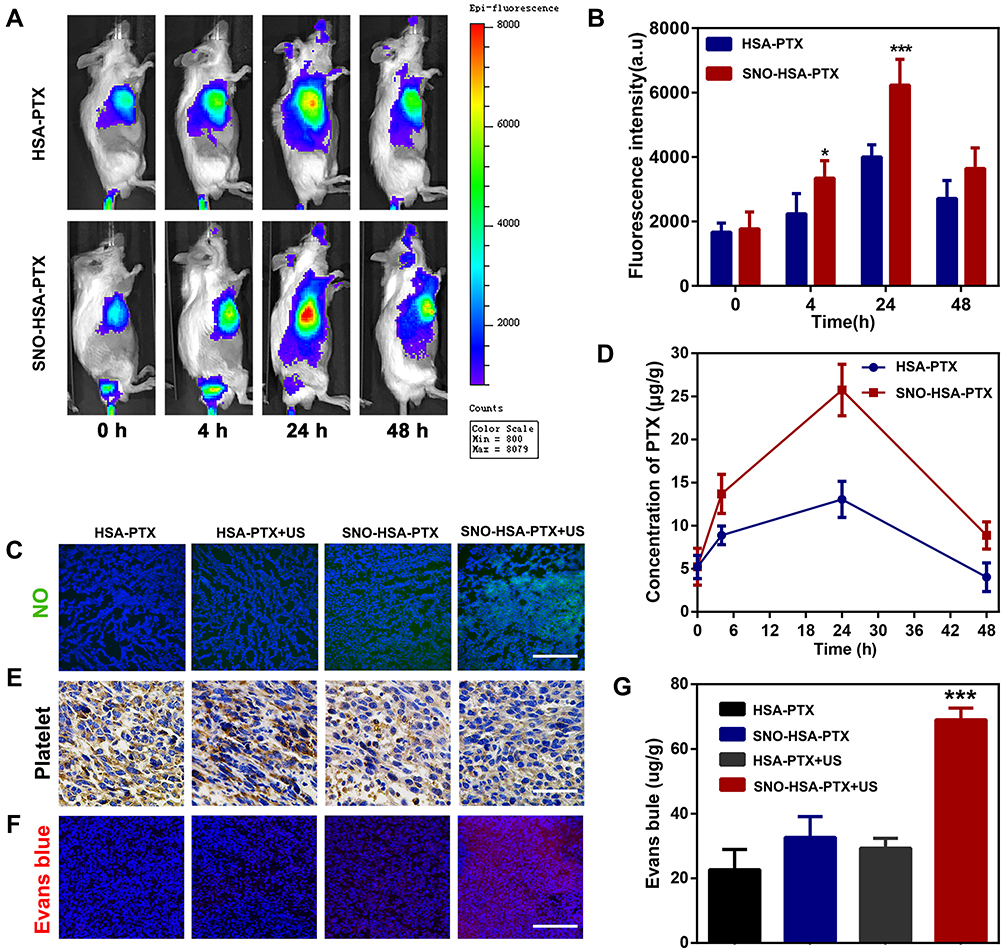 |
Figure 5 Effect of SNO-HSA-PTX treatment on tumor targeting and tumor vascular permeability in vivo. (A) The in vivo distribution of IR780-labeled HSA-PTX and IR780-labeled HSA-PTX at different time points after US irradiation (1.0 MHz, 2 W/cm2, 5 min) in 4T1 tumor-bearing mice. (B) Quantitative analysis of IR780 fluorescence intensity. ***p < 0.001. (C) Immunofluorescence images of tumors slices stained with anti-nitrotyrosine from mice with different treatments. Scale bar: 200 μm. ***p < 0.001. (D) The content of PTX in tumor tissues after treatments. (E) Immunohistochemistry images of tumor sections stained with anti-CD62P antibodies. Scale bar: 50 μm. (F) Representative fluorescence images of Evans blue (red) in tumor frozen sections (DAPI; blue). Scale bar: 100 μm. (G) Quantitative determination of Evans blue in tumors by spectrophotometry at 620 nm. ***p < 0.001. |
In order to verify our speculation, it is very important to confirm NO release and platelet inhibition in vivo. To verify NO generation in vivo, the tumor sections were stained by anti-nitrotyrosine antibody, which is a standard marker indicating NO released in vivo.42 As shown in Figure 5C, compared with the other groups, tumors treated with SNO-HSA-PTX+US displayed a significant increase in nitrotyrosine signals, which indicated the effective NO generation within the tumors. The intratumoural accumulation of PTX was assessed after US irradiation. Quantitative analysis displayed that the PTX content in the tumors treated with SNO-HSA-PTX+US was nearly twice that of the HSA-PTX+US treatment group (Figure 5D). Platelet surface CD62P is the “gold standard” marker of platelet activation.43 In order to investigate whether released NO possessed the ability to reduce platelet activation in vivo, we supplemented immunohistochemistry assay for CD62P in tumor tissue after treatment with HSA-PTX, HSA-PTX+US, SNO-HSA-PTX or SNO-HSA-PTX+US. As shown in Figure 5E, platelet-positive area was reduced in the SNO-HSA-PTX+US treatment, suggesting that the released NO can effectively reduce the number of intratumor platelets. In view of the important role of platelets in maintaining the tumor vasculature integrity, our SNO-HSA-PTX nanosystem combined with US irradiation may possess the capacity of disrupting vascular barriers.
Next, the typical Evans blue assay was used to characterize the integrity of the tumor vascular barriers in mice with different treatments. Three hours after US irradiation, 4% Evans blue was injected via the tail veins and the tumors were collected 5 h later. As shown in Figure 5F, the SNO-HSA-PTX+US treatment showed strong red fluorescence, indicating higher Evans blue delivery to solid tumors compared with other treatments. Extensive extravasation of Evans blue was detected in the SNO-HSA-PTX with the US irradiation group (Figure 5G). The content of Evans blue in tumors of SNO-HSA-PTX with US irradiation was at least twice that of the other groups, indicating increased vascular leakage and impaired vascular barrier caused by US-triggered NO.
Enhancement of Tumor Hemorrhage by SNO-HSA-PTX
It has been shown that the blockade of platelets in tumor-bearing mice caused damage to the vascular barrier, thereby facilitating the delivery of drugs to the tumor site. Interestingly, we found severe intratumor hemorrhage from the mice treated with SNO-HSA-PTX+US. As shown in Figure 6A, increased blood cells infiltration was observed by H&E staining in the tumors of mice treated with SNO-HSA-PTX+US, reflected as the significant increase in hemoglobin content (Figure 6B), while no obvious effect of the other treatments was observed, indicating an increase in leakage of the tumor vessels caused by US-triggered NO. To visualize the change of tumor blood flow upon administration of SNO-HSA-PTX, the mice were treated with saline or SNO-HSA-PTX followed by US irradiation. As shown in the laser speckle contrast imaging of the tumors (Figure 6C), only a small amount of blood flow was seen in the tumors before applying US irradiation. However, in mice treated with SNO-HSA-PTX+US, the blood flow increased from 338.5±14.85 to 640±52.33 in 4 hours (Figure 6D). No obvious enhancement of blood flow was found in the controls. These results confirmed that the US-triggered release of NO in SNO-HSA-PTX destabilized tumor vascular integrity in tumors, leading to profound tumor hemorrhage.
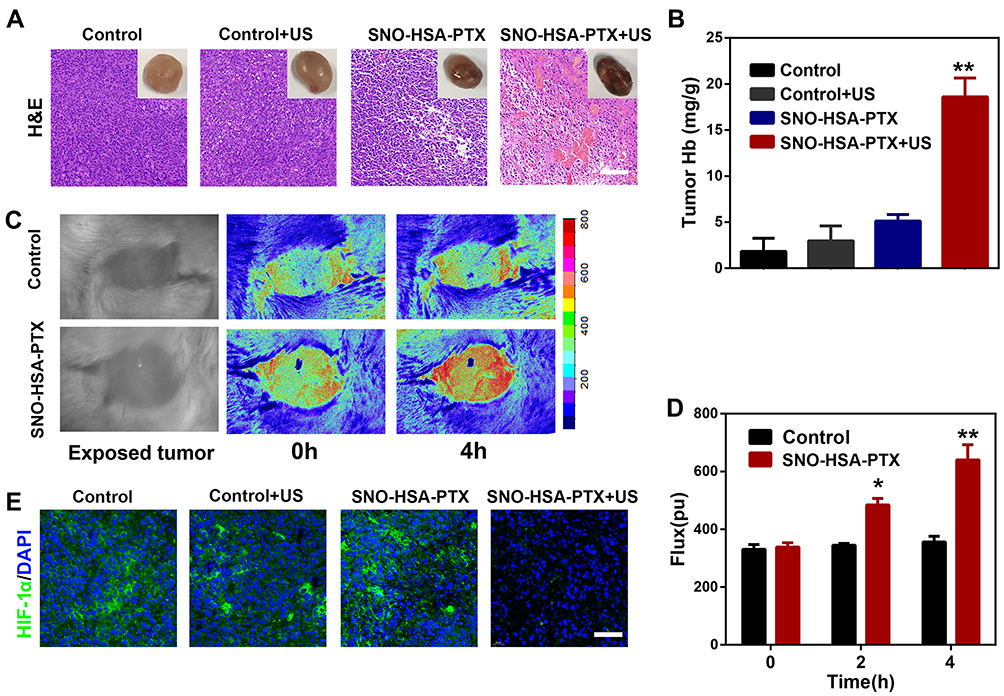 |
Figure 6 SNO-HSA-PTX combined with US irradiation enhanced tumor hemorrhage and relieved tumor hypoxia. (A) Red blood cells infiltration in tumors as visualized by H&E staining 24 h after the treatments of saline, saline+US, SNO-HSA-PTX, or SNO-HSA-PTX+US. The upper right corner was the photo of the representative tumor. Scale bar: 100 μm. (B) The content of hemoglobin inside the tumors detected by the hemoglobin assay kits. **p < 0.01. (C) Laser speckle contrast imaging of blood flow signals in 4T1 tumor-bearing mice after intravenous injection of saline or SNO-HSA-PTX with US irradiation. (D) Flux of blood flow measured by Doppler flow imaging. *p < 0.05, **p < 0.01. (E) HIF-1α staining of tumor sections harvested from mice treated with saline, saline+US, SNO-HSA-PTX or SNO-HSA-PTX+US. Scale bar: 50 μm. |
It is well known that hemoglobin has the function of carrying oxygen. The increase in hemoglobin content in the tumors would likely endow SNO-HSA-PTX with the ability to increase oxygen perfusion to relieve tumor hypoxia. To validate this hypothesis, the tumors after treated with saline, saline+US, SNO-HSA-PTX and SNO-HSA-PTX+US were collected for HIF-1α immunofluorescence staining. As shown in Figure 6E, the hypoxic tumor microenvironment led to high expression of HIF-1α in the tumors of the control group, while the expression of HIF-1α level was significantly downregulated in SNO-HSA-PTX+US treatment. Those results demonstrated the damage of the tumor endothelial integrity leads to the enhanced red blood cells infiltration, thereby increasing the oxygen perfusion and relieving tumor hypoxia.
Enhancement of Intratumoral T Cells Infiltration by SNO-HSA-PTX
It has been previously shown that the NO triggered by US irradiation from SNO-HSA-PTX damaged the tumor vascular barrier, leading to increased drug accumulation and blood perfusion. It can be questioned whether the intratumoral infiltration of lymphocytes could be augmented in the same manner.44 The levels of CD8+ T cells in tumors were further analyzed by immunofluorescence staining. As shown in Figure 7A and B, only slightly increased the CD8+ T cells infiltration in the tumor treated with HSA-PTX. The CD8+ and CD4+ T cells infiltration exhibited a significant increase in SNO-HSA-PTX+US treatment, suggesting that the US-triggered NO generation would significantly improve the intratumoral T cells infiltration. The quantification of CD4+ T cells and CD8+ T cells in tumors were further investigated using flow cytometry assay (Figure 7C). The proportions of CD8+ T cells gating on CD3+ cells from SNO-HSA-PTX with US irradiation treatment were remarkably increased compared with those from the control group (approximately 30.9% vs 14.6%). The percentage of CD8+ T cells in tumors of SNO-HSA-PTX+US group accounted for approximately 3-fold higher than that from saline-treated mice (Figure 7D), indicating that the specific release of NO induced by US irradiation contributed to increased CD8+ T cells infiltration in tumors. In recent years, extensive evidences have uncovered that the hypoxic tumor microenvironment could induce the differentiation of T cells into Tregs in tumors, thereby maintaining the immunosuppression tumor microenvironment.45,46 Considering the ability of our US-triggered NO release nanoparticles on relieving tumor hypoxia, we thus inferred that SNO-HSA-PTX combined with US may have the effect of inhibiting Tregs in tumors. As shown in Figure 7E, the ratio of Foxp3+ Tregs gating on CD4+ T cells in SNO-HSA-PTX+US treatment was lower than that in the other treatments, implying that SNO-HSA-PTX+US treatment may decrease the distribution of Tregs at tumor sites. Also, the ratio of the CD8+ T cell/Tregs within the tumor was significantly improved in SNO-HSA-PTX+US treatment as shown in Figure 7F. In general, SNO-HSA-PTX with US irradiation treatment increased the number of anti-tumor CD8+ and CD4+ T cells in the tumors, while reducing the proportion of tumor-promoting Tregs. This NO-based nanosystem can increase the homing of T cells by breaking the vascular barrier, and at the same time reverse tumor immunosuppression by improving the tumor hypoxic microenvironment.
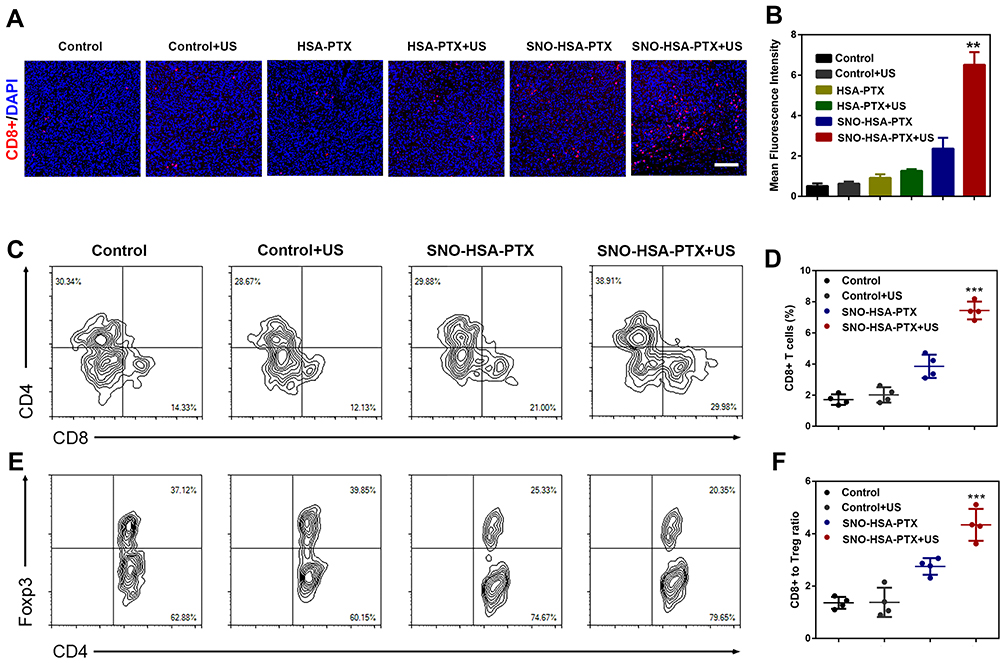 |
Figure 7 SNO-HSA-PTX combined with US irradiation enhanced intratumoral T cells infiltration. (A) Representative fluorescent images of CD8+ T cells infiltration after SNO-HSA-PTX treatment with or without US irradiation. (CD8+ T cells: red color; DAPI: blue color). Scale bar: 100 μm. (B) Statistical analysis of CD8+T cells positive fluorescence in tumor slices calculated by Image J software. **p < 0.01. (C) Representative flow cytometry analysis images of CD4+ T cells and CD8+ T cells gating on CD3+ cells. (D) Quantification of CD8+ T cells percentages in tumors treated with saline, saline+US, SNO-HSA-PTX or SNO-HSA-PTX+US. ***p < 0.001. (E) Representative flow cytometry analysis images of Foxp3+ T cells gating on CD4+ T cells. (F) Ratios of CD8+ T cells versus Tregs (Foxp3+ T cells) in the tumors after treatment. ***p < 0.001. |
In vivo Antitumor Efficacy
To investigate the antiplatelet-assisted therapeutic strategy based on our US-triggered NO release platform in tumor growth, the therapeutic efficacy of SNO-HSA-PTX was evaluated in 4T1 tumor-bearing mice. As shown in Figure 8A, US irradiation had no obvious effect on tumor growth. In contrast, SNO-HSA-PTX+US treatment showed a significant inhibitory effect on tumor growth when compared to HSA-PTX, HSA-PTX+US or SNO-HSA-PTX treatments, which also be reflected from the tumor weight and the tumor photos (Figure 8B and C). As shown in the H&E staining images of tumors (Figure 8D), the contracted nuclei were found in tumors of SNO-HSA-PTX+US treatment, indicating cell apoptosis. The cell death was accompanied by obvious intratumoral hemorrhage in SNO-HSA-PTX+US treatment. US-triggered NO release of SNO-HSA-PTX promoted tumor apoptosis, which was also proved by the increased positive TUNEL assay (Figure 8E). The excellent efficacy of SNO-HSA-PTX with US irradiation on inhibiting tumor growth may be attributed to the toxic effect of NO, combined with enhanced drug accumulation and T cells infiltration caused by the destruction of the tumor vascular barrier.
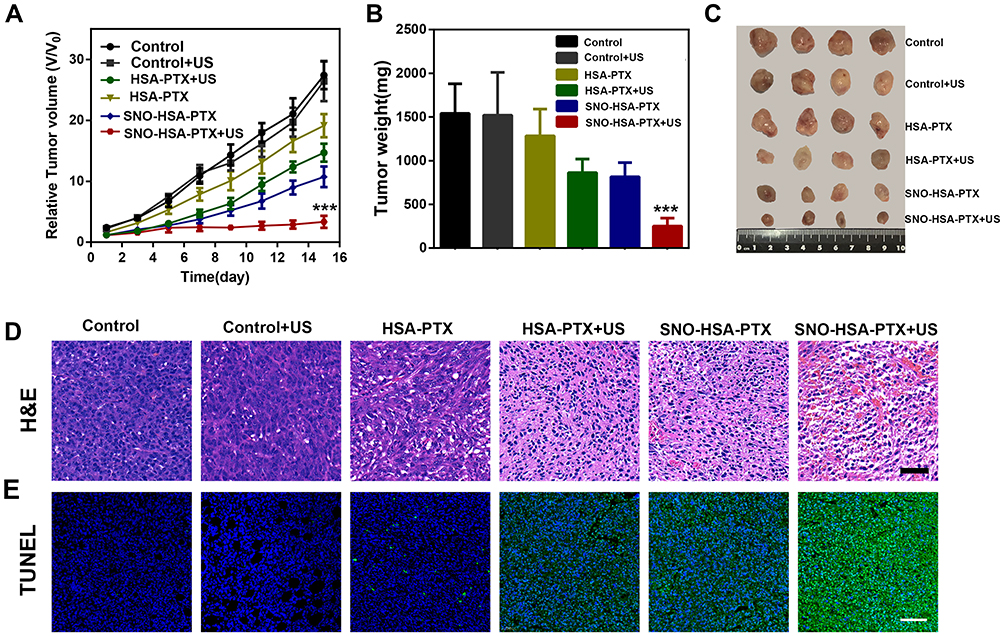 |
Figure 8 In vivo antitumor efficacy of SNO-HSA-PTX combined with US irradiation in 4T1 tumor-bearing mice. (A) The relative tumor growth curves of 4T1 tumor-bearing mice after various treatments for 15 days. ***p < 0.001. (B) The average tumor weight in each treatment at day 15. ***p < 0.001. (C) The photo images of tumors from different treatment groups at day 15. (D) Images of H&E staining of tumor tissues after different treatments. Scale bar: 50 μm. (E) Representative immunofluorescent images of tumor apoptosis stained by TUNEL. Scale bar: 50 μm. |
Biosafety Evaluation of SNO-HSA-PTX
The blood biochemical analyses of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) were performed to indicate the biosafety of SNO-HSA-PTX. There were no obvious changes in the levels of AST, ALT, ALP and CREA in the SNO-HSA-PTX treated group, which indicated that SNO-HSA-PTX was safe for the liver and kidney (Figure S7). Considering the inhibitory effect of our US-triggered NO release nanoparticles on platelets at tumor sites, we evaluated the effect of SNO-HSA-PTX+US on the normal hemostatic function of platelets (Figure S8). Compared with the saline group, the SNO-HSA-PTX+US treatment had no significant effect on the bleeding time of mice, indicating that the tumor-specific NO release did not inhibit systemic platelet functions. All these results demonstrated that the safety of SNO-HSA-PTX for cancer treatment in vivo.
Conclusion
In summary, we designed a US-responsive NO release nanosystem, SNO-HSA-PTX, which was constructed by loading an anticancer drug (paclitaxel, PTX) into a NO donor-modified albumin shell. We have demonstrated the SNO-HSA-PTX stimulated by US irradiation achieved the on-demand release of NO for the local inhibition of tumor-associated platelets activation. The NO produced by SNO-HSA-PTX in response to US irradiation can effectively prevent platelet aggregation. The absence of platelets in tumors induced openings in the tumor vascular barriers, which promoted the accumulation of SNO-HSA-PTX nanoparticles to the tumor sites. Meanwhile, the damaged vascular barriers allowed oxygen-carrying hemoglobin to infiltrate tumor regions, alleviating hypoxia of the tumor microenvironment. In addition, the intratumoral infiltration of T cells was augmented, together with chemotherapy and NO therapy, which greatly inhibited tumor growth. Systemic administration of SNO-HSA-PTX to mice with US irradiation resulted in enhanced tumor vascular leakage, accompanied by increased drug accumulation, tumor hypoxia alleviation and T cells infiltration, ultimately leading to tumor suppression.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Nos. 81803439), Youth Foundation of Jiangsu province (Nos. BK20180699), Jiangsu Synergetic Innovation Center for Advanced Bio-Manufacture (Nos.XTD1820). The language of the manuscript has been carefully checked and revised by Professor Xueming Li.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sang W, Zhang Z, Dai Y, Chen X. Recent advances in nanomaterial-based synergistic combination cancer immunotherapy. Chem Soc Rev. 2019;48(14):3771–3810.
2. Mi Y, Hagan CT, Vincent BG, Wang AZ. Emerging nano-/microapproaches for cancer immunotherapy. Advan Sci. 2019;6:1801847. doi:10.1002/advs.201801847
3. Kruger S, Ilmer M, Kobold S, et al. Advances in cancer immunotherapy 2019 – latest trends. J Exp Clin Cancer Res. 2019;38(1):268. doi:10.1186/s13046-019-1266-0
4. Melero I, Rouzaut A, Motz GT, Coukos G. T-Cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4(5):522. doi:10.1158/2159-8290.CD-13-0985
5. Ino Y, Yamazaki-Itoh R, Shimada K, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108(4):914–923. doi:10.1038/bjc.2013.32
6. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves Anti-PD-1 immunotherapy. Cell. 2017;170(6):1109–1119.e10. doi:10.1016/j.cell.2017.08.027
7. Motz GT, Santoro SP, Wang L-P, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–615. doi:10.1038/nm.3541
8. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. 2015;33:55–63. doi:10.1016/j.coi.2015.01.011
9. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9(2):115. doi:10.1038/s41419-017-0061-0
10. Pan L, Liu J, He Q, Shi J. MSN-mediated sequential vascular-to-cell nuclear-targeted drug delivery for efficient tumor regression. Adv Mater. 2014;26(39):6742–6748. doi:10.1002/adma.201402752
11. Maeda H. The link between infection and cancer: tumor vasculature, free radicals, and drug delivery to tumors via the EPR effect. Cancer Sci. 2013;104(7):779–789. doi:10.1111/cas.12152
12. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25.
13. Bambace NM, Holmes CE. The platelet contribution to cancer progression. J Thrombosis Haemostasis. 2011;9(2):237–249. doi:10.1111/j.1538-7836.2010.04131.x
14. Ho-Tin-NoÉ B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thrombosis Haemostasis. 2011;9(s1):56–65. doi:10.1111/j.1538-7836.2011.04317.x
15. Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem. 2010;21(5):797–802. doi:10.1021/bc100070g
16. Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. doi:10.1016/j.addr.2010.03.011
17. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. doi:10.1016/j.addr.2010.04.009
18. Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602–2663. doi:10.1021/acs.chemrev.5b00346
19. Volz J, Mammadova-Bach E, Gil-Pulido J, et al. Inhibition of platelet GPVI induces intratumor hemorrhage and increases efficacy of chemotherapy in mice. Blood. 2019;133(25):2696–2706. doi:10.1182/blood.2018877043
20. Ho-Tin-Noé B, Boulaftali Y, Camerer E. Platelets and vascular integrity: how platelets prevent bleeding in inflammation. Blood. 2018;131(3):277–288. doi:10.1182/blood-2017-06-742676
21. Demers M, Wagner DD. Targeting platelet function to improve drug delivery. OncoImmunology. 2012;1(1):100–102. doi:10.4161/onci.1.1.17962
22. Ma Z, Liu S, Ke Y, et al. Biomimetic nano-NOS mediated local NO release for inhibiting cancer-associated platelet activation and disrupting tumor vascular barriers. Biomaterials. 2020;255:120141. doi:10.1016/j.biomaterials.2020.120141
23. Xu XR, Yousef GM, Ni H. Cancer and platelet crosstalk: opportunities and challenges for aspirin and other antiplatelet agents. Blood. 2018;131(16):1777–1789.
24. Lucotti S, Cerutti C, Soyer M, et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J Clin Invest. 2019;129(5):1845–1862. doi:10.1172/JCI121985
25. Elaskalani O, Berndt MC, Falasca M, Metharom P. Targeting Platelets for the Treatment of Cancer. Cancers. 2017;9(7):94. doi:10.3390/cancers9070094
26. Riddell DR, Graham A, Owen JS. Apolipoprotein E inhibits platelet aggregation through the L-arginine:nitric oxide pathway: implications for vascular disease. J Biol Chem. 1997;272(1):89–95. doi:10.1074/jbc.272.1.89
27. Gresele P, Momi S, Guglielmini G. Nitric oxide-enhancing or -releasing agents as antithrombotic drugs. Biochem Pharmacol. 2019;166:300–312. doi:10.1016/j.bcp.2019.05.030
28. Makhoul S, Walter E, Pagel O, et al. Effects of the NO/soluble guanylate cyclase/cGMP system on the functions of human platelets. Nitric Oxide. 2018;76:71–80. doi:10.1016/j.niox.2018.03.008
29. Qiu H, Qi P, Liu J, et al. Biomimetic engineering endothelium-like coating on cardiovascular stent through heparin and nitric oxide-generating compound synergistic modification strategy. Biomaterials. 2019;207:10–22. doi:10.1016/j.biomaterials.2019.03.033
30. Fan W, Lu N, Huang P, et al. Glucose-responsive sequential generation of hydrogen peroxide and nitric oxide for synergistic cancer starving-like/gas therapy. Angewandte Chemie Int Ed. 2017;56(5):1229–1233. doi:10.1002/anie.201610682
31. Zhang X, Guo Z, Liu J, et al. Near infrared light triggered nitric oxide releasing platform based on upconversion nanoparticles for synergistic therapy of cancer stem-like cells. Sci Bull. 2017;62(14):985–996. doi:10.1016/j.scib.2017.06.010
32. Qin L, Gao H. The application of nitric oxide delivery in nanoparticle-based tumor targeting drug delivery and treatment. Asian J Pharm Sci. 2019;14(4):380–390. doi:10.1016/j.ajps.2018.10.005
33. Yang T, Zelikin AN, Chandrawati R. Progress and promise of nitric oxide-releasing platforms. Advan Sci. 2018;5(6):1701043. doi:10.1002/advs.201701043
34. Fan W, Yung BC, Chen X. Stimuli-responsive NO release for on-demand gas-sensitized synergistic cancer therapy. Angewandte Chemie Int Ed. 2018;57(28):8383–8394. doi:10.1002/anie.201800594
35. Huang C-S, You X, Dai C, et al. Targeting super-enhancers via nanoparticle-facilitated BRD4 and CDK7 inhibitors synergistically suppresses pancreatic ductal adenocarcinoma. Advan Sci. 2020;7(7):1902926. doi:10.1002/advs.201902926
36. Xu Y, Liu J, Liu Z, et al. Blockade of platelets using tumor-specific NO-releasing nanoparticles prevents tumor metastasis and reverses tumor immunosuppression. ACS Nano. 2020;14(8):9780–9795. doi:10.1021/acsnano.0c01687
37. Doshi N, Orje JN, Molins B, Smith JW, Mitragotri S, Ruggeri ZM. Platelet mimetic particles for targeting thrombi in flowing blood. Adv Mater. 2012;24(28):3864–3869. doi:10.1002/adma.201200607
38. Soslau G, Parker J. The bioluminescent detection of platelet released ATP: collagen-induced release and potential errors. Thromb Res. 1992;66(1):15–21. doi:10.1016/0049-3848(92)90151-Y
39. Liu R, Xiao W, Hu C, Xie R, Gao H. Theranostic size-reducible and no donor conjugated gold nanocluster fabricated hyaluronic acid nanoparticle with optimal size for combinational treatment of breast cancer and lung metastasis. J Controlled Release. 2018;278:127–139. doi:10.1016/j.jconrel.2018.04.005
40. Hu C, Cun X, Ruan S, et al. Enzyme-triggered size shrink and laser-enhanced NO release nanoparticles for deep tumor penetration and combination therapy. Biomaterials. 2018;168:64–75. doi:10.1016/j.biomaterials.2018.03.046
41. Hu C, Yang X, Liu R, et al. Coadministration of iRGD with multistage responsive nanoparticles enhanced tumor targeting and penetration abilities for breast cancer therapy. ACS Appl Mater Interfaces. 2018;10(26):22571–22579. doi:10.1021/acsami.8b04847
42. Masri F. Role of nitric oxide and its metabolites as potential markers in lung cancer. Ann Thorac Med. 2010;5(3):123–127. doi:10.4103/1817-1737.65036
43. Burdess A, Michelsen AE, Brosstad F, Fox KAA, Newby DE, Nimmo AF. Platelet activation in patients with peripheral vascular disease: reproducibility and comparability of platelet markers. Thromb Res. 2012;129(1):50–55. doi:10.1016/j.thromres.2011.08.015
44. Yang X, Hu C, Tong F, et al. Tumor microenvironment-responsive dual drug dimer-loaded PEGylated bilirubin nanoparticles for improved drug delivery and enhanced immune-chemotherapy of breast cancer. Adv Funct Mater. 2019;29(32):1901896. doi:10.1002/adfm.201901896
45. Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Chapter 3 - Regulatory T cells in cancer. In: Vande Woude GF, Klein G, editors. Advances in Cancer Research. Academic Press; 2010:57–117.
46. Westendorf A, Skibbe K, Adamczyk A, et al. Hypoxia enhances immunosuppression by inhibiting CD4+ Effector T cell function and promoting treg activity. Cell Physiol Biochem. 2017;41(4):1271–1284. doi:10.1159/000464429
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.