")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Dabrafenib for treatment of BRAF-mutant melanoma
Authors Kainthla R, Kim K, Falchook G
Received 15 August 2013
Accepted for publication 9 October 2013
Published 31 December 2013 Volume 2014:7 Pages 21—29
DOI https://doi.org/10.2147/PGPM.S37220
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Radhika Kainthla,1 Kevin B Kim,2 Gerald S Falchook3
1Department of Internal Medicine, Baylor College of Medicine, 2Department of Melanoma Medical Oncology, 3Department of Investigational Cancer Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Abstract: Melanoma has the highest mortality of all the skin cancer subtypes. Historically, chemotherapy and immunologic therapies have yielded only modest results in the treatment of metastatic melanoma. The discovery of prevalent V600 BRAF mutations driving proliferation makes this oncogenic protein an ideal target for therapy. Dabrafenib, a reversible inhibitor of mutant BRAF kinase, improved response rates and median progression-free survival in patients with V600E BRAF-mutant metastatic melanoma, including those with brain metastases. With a well-tolerated toxicity profile, dabrafenib is effective as a monotherapy; however, resistance eventually develops in almost all patients. As a result, current research is exploring the role of combination therapies with dabrafenib to overcome resistance.
Keywords: dabrafenib, metastatic melanoma, V600E BRAF mutation
Introduction to melanoma and personalized medicine
Skin cancer is the most common cancer in the US, and melanoma has the highest mortality rate of all the skin cancer subtypes.1 The incidence and prevalence of cutaneous melanoma have increased over the last 30 years, and one in 50 Americans will be diagnosed with melanoma at some point in their lifetime.2 Although more than 80% of patients have localized disease at the time of diagnosis and a 5-year survival of more than 90%, metastatic melanoma continues to carry a poor prognosis, with a median overall survival of 9–11 months and one-year and 5-year survivals of about 33% and 15%, respectively.2,3
Until recently, standard treatments for metastatic melanoma have yielded only modest response rates and significant toxicities. Dacarbazine, an alkylating agent, was one of the first chemotherapies approved for metastatic melanoma, with a response rate of about 20% and a median response duration of 5–6 months.4 However, multiple studies have failed to demonstrate a survival benefit with dacarbazine.4 Similarly, high-dose interleukin (IL)-2 has a response rate of about 6%–16%, and responders have a progression-free survival of 13.1 months.5,6 For those with brain metastasis, the response rate with IL-2 is only 5%.7 Addition of the peptide vaccine gp-100 to high-dose IL-2 therapy slightly improved response rates and progression-free survival, but unfortunately the significant toxicity profile associated with high-dose IL-2, which includes capillary leak syndrome, arrhythmias, hypotension, and neurologic changes, makes the treatment difficult to tolerate in many patients.5,6 In spite of its modest response rates, significant toxicity profile, and lack of overall survival advantage, IL-2 continues to be a treatment option for metastatic melanoma because of the prolonged median progression-free survival of several years in patients who experience a complete response.5 Temozolomide, an oral alkylating agent that crosses the blood–brain barrier and has the same chemical active species that causes cell death as dacarbazine, has a response rate of approximately 7% and a median progression-free survival of about 1.2 months in patients with brain metastasis and no prior treatment.8 In addition to single-agent therapies, combination chemotherapy regimens have been explored but have also yielded relatively modest response rates.9 Strategies that combine cytotoxic chemotherapies with immune-modulating agents, such as biochemotherapy, have also been investigated.10,11 One biochemotherapy regimen combining cisplatin, vinblastine, dacarbazine, interferon, and IL-2 has demonstrated modest improvement in progression-free survival but not overall survival in Phase III trials.11,12 Although some combination regimens have slightly improved response rates, none have demonstrated improved overall survival when compared with dacarbazine monotherapy, and many regimens are associated with toxicities that are poorly tolerated by patients.4,10,11
Recent research efforts have explored the potential role of targeted therapy for metastatic melanoma. Understanding the driver mutations which contribute to the uncontrolled proliferation of cancer cells has been crucial for the development of drugs that specifically target the underlying cellular defect. In metastatic melanoma, oncogenic mutations in multiple cellular pathways have been identified, including BRAF and NRAS mutations in the mitogen-activated protein kinase (MAPK) pathway, p53 mutations, and PTEN mutations.13–15 Inhibitors have been developed that target specific proteins, such as BRAF and MEK, in unregulated proliferation cascades. Likewise, immune-modifying agents, such as anti-CTLA-4 and anti-PD-1 antibodies, have been developed to control melanoma growth by activating cytotoxic T-lymphocytes.16,17 Ipilimumab, an anti-CTLA-4 antibody, has a response rate of 10.9%, with a complete response rate of 1.5% and a median progression-free survival of 2.86 months.18 Importantly, ipilimumab was the first drug to demonstrate improved overall survival in patients with metastatic melanoma, with one-year and 2-year survival rates of 45.6% and 23.5%, respectively.18 Although severe immune-related adverse events are observed in about 10%–15% of patients, algorithmic management of the adverse events can significantly mitigate these serious toxicities.18 Additionally, combining ipilimumab with nivolumab, an anti-PD-1 antibody, in a Phase I study of patients with metastatic melanoma led to an objective response in 53% of patients, with all responding patients demonstrating tumor reductions of 80% or more. Although grade 3 and 4 adverse events occurred in 53% of patients, most side effects were reversible and manageable.19 Further, lambrolizumab, an anti-PD-1 antibody, had a response rate of about 38%, with a median progression-free survival of over 7 months and a fairly well-tolerated toxicity profile.20 The goal of molecular-targeted research has been to identify relevant molecular aberrations in individual patients and use this knowledge to guide treatment decisions.
Introduction to BRAF
The MAPK cascade plays an important role in cellular proliferation and differentiation.21,22 Mutations in the MAPK signaling pathway have been identified in patients with malignant melanoma.13–15,22 Growth factors activate a small GTP binding protein (RAS) on cell membranes, which triggers intracellular signaling (Figure 1).21 Downstream activation of RAF, a serine/threonine kinase protein, leads to phosphorylation of MEK proteins and eventual activation of the protein kinase ERK, which translocates into the nucleus and stimulates progrowth signals.21,22 Unregulated activation of the MAPK pathway can therefore lead to malignancy.21–23
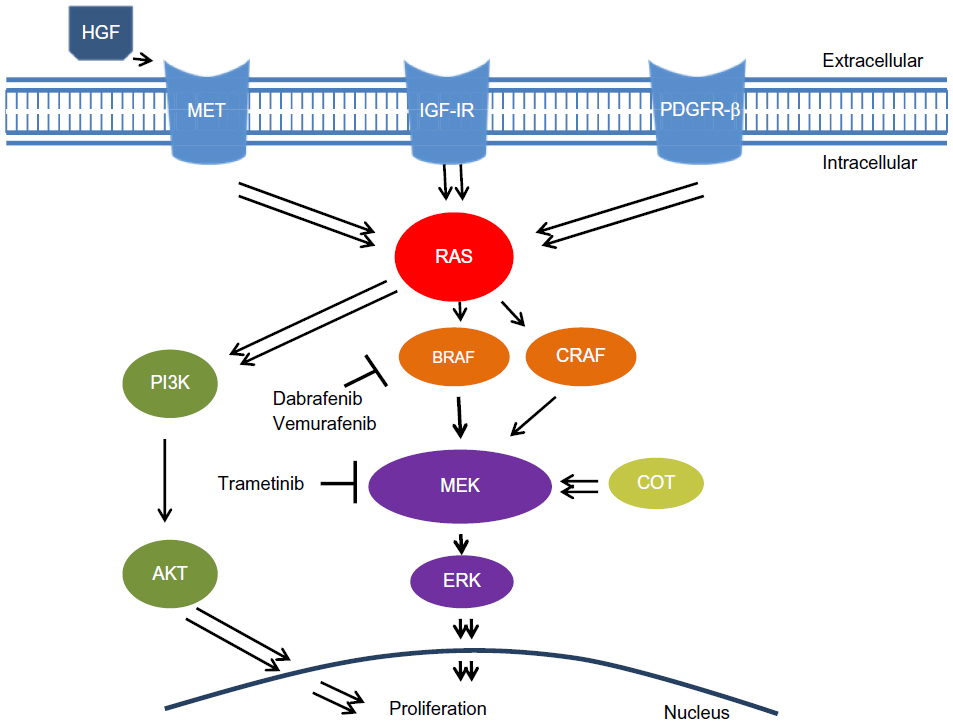 | Figure 1 Redundancy of the MAPK signaling cascade and targeted inhibitors. Single arrows signify direct pathways. Double arrows reflect a culmination of multiple steps in the signaling cascade. |
The RAF protein kinases have been extensively studied for their role in oncogenesis. Specifically, BRAF mutations have been identified in many malignancies, including cutaneous melanoma, colorectal cancer, and papillary thyroid carcinoma.24–27 In each tumor type, the presence of a BRAF mutation has been associated with a more aggressive disease course and worse overall prognosis.24–26
BRAF mutational status in malignant melanoma
BRAF mutations have been identified in 50%–60% of all metastatic melanomas, and 80%–90% of all BRAF mutations consist of an exchange of glutamine for valine at amino acid 600 (V600E).13,26,27 This alteration locks the kinase into a 500-fold more active conformation than wild-type BRAF and leads to oncogenesis via unregulated MAPK signaling.28 Substitution of lysine for valine (V600K) is another transformation observed in about 20% of BRAF mutations in metastatic melanoma.26 Historically, the prognosis of BRAF-mutant melanoma has been worse than melanomas with wild-type BRAF because untreated patients have a median overall survival of 11.1 months versus 46.1 months, respectively.26
The frequency of BRAF mutations in metastatic melanoma has driven the development of agents to disrupt the pathway signaling associated with these activating oncogenic mutations. Vemurafenib, an inhibitor that is ten times more selective for mutant BRAF than wild-type, induces potent cell cycle arrest, inhibits proliferation, and initiates apoptosis exclusively in V600E-mutant cells in a variety of experimental in vitro systems.29,30 Xenograft studies similarly demonstrated sensitivity and activity against melanomas with V600E BRAF mutations.30
Meanwhile, dabrafenib was developed separately as an ATP-competitive, reversible inhibitor of mutant BRAF kinase. Like vemurafenib, dabrafenib decreases phosphorylated ERK and causes cell cycle arrest.31 In preclinical studies, dabrafenib was almost 20 times more selective at inhibiting V600E BRAF-mutants than wild-type BRAF in multiple cancer cell lines.31 Additionally, dabrafenib has inhibitory effects on cell lines containing other activating BRAF mutations, including Val600Lys (V600K) and Val600Asp (V600D).31 Dabrafenib achieved a half maximal inhibitory concentration (IC50) with 0.6 nM, 0.5 nM, 1.9 nM, and 12 nM in V600E, V600K, V600D, and wild-type BRAF cell lines, respectively.31
Dabrafenib targeting mutated BRAF in metastatic melanoma
Clinical trials
The first-in-human Phase I trial of dabrafenib showed promising results. The treatment was well-tolerated with no maximum tolerated dose identified despite dose escalation that achieved pharmacokinetic concentrations well above the levels predicted to have adequate target inhibition.32 A recommended Phase II dose (RP2D) of 150 mg by mouth twice a day was selected for future studies.32 Patients with V600E BRAF-mutant melanoma were more responsive to treatment, with a confirmed response rate of 57% compared with 37% in patients with V600K mutations.32 Patients with either V600E or V600K had similar median progression-free survival of 5.5 and 5.6 months, respectively (Table 1).32 The most serious side effects with a grade 2 or higher at the RP2D were well differentiated cutaneous squamous cell carcinoma (7%), fever (6%), and fatigue (4%).32 Pyrexia was an unusual dose-limiting toxicity observed; however, almost all cases could be managed with antipyretics, low-dose steroids, or dose reduction. No patient required discontinuation of treatment secondary to side effects, and no deaths occurred from the treatment.32
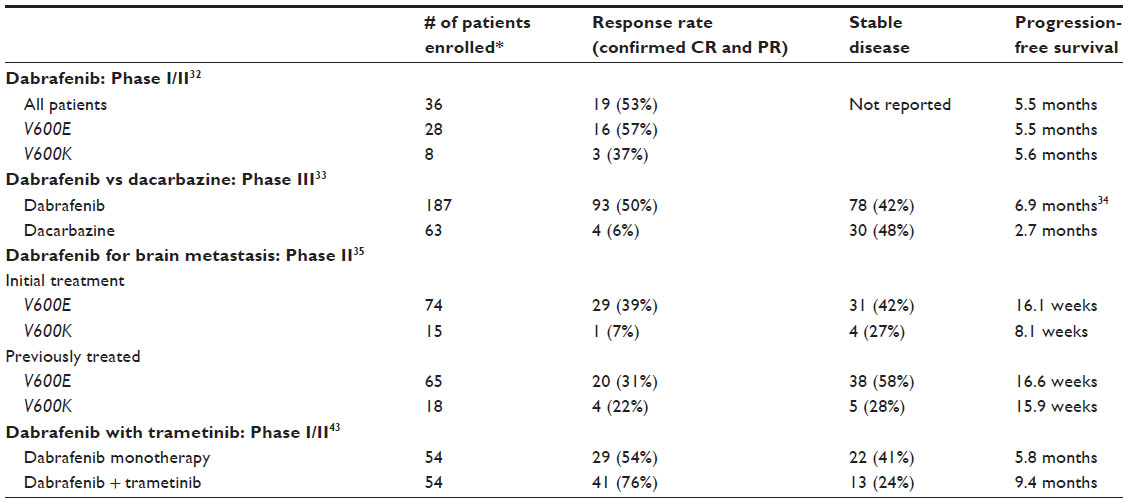 | Table 1 Comparison of endpoints among dabrafenib clinical trials |
Following the promising preliminary results of the Phase I trial, a randomized Phase III trial compared dabrafenib with dacarbazine in patients with V600E BRAF-mutant metastatic melanoma.33 Eligible patients had excellent performance status, with an Eastern Cooperative Oncology Group score of 0 (fully active and able to carry out all performance without restrictions) or 1 (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature) and no other prior treatments except high-dose IL-2.33 All patients with active central nervous system metastases were excluded.33 The confirmed response rate for dabrafenib was 50% compared with 6% for dacarbazine per the study’s independent review committee (Table 1).33 In those treated with dabrafenib, 47% had a partial response while 3% showed a complete response.33 The median progression-free survival in patients receiving dabrafenib was 6.9 months compared with 2.7 months in patients who received dacarbazine therapy.34 The most common side effects with dabrafenib were dermatologic, and included hyperkeratosis (39%) and squamous cell carcinoma (10%). Other common side effects observed were pyrexia (32%), headache (35%), and arthralgia (35%).34 The occurrence of pyrexia of at least grade 2 was higher in the Phase III study than in the Phase I study (11% versus 6%, respectively) but was manageable in most cases.32,33 Grade 3 and 4 adverse events were uncommon, and 28% of patients needed dose reductions secondary to toxicity.33,34 Overall, dabrafenib has markedly higher response rates and progression-free survival along with a well-tolerated toxicity profile compared with dacarbazine in patients with V600E BRAF-mutated metastatic melanoma.33
BRAF-mutant melanoma with brain metastasis
A small subset of ten patients with untreated brain metastases were enrolled at the RP2D in the first-in-human Phase I trial of dabrafenib.32 Nine patients responded to treatment, with four having complete resolution of brain lesions and a median progression-free survival of 4.2 months.32
Because of these encouraging preliminary results, a multicenter, open-label, Phase II trial compared dabrafenib treatment in V600E and V600K BRAF-mutant melanoma with metastases to the brain in patients with and without previous local treatment, including brain surgery, whole-brain radiation, or stereotactic radiosurgery (Table 1).35 In V600E mutants with no prior treatment, 39% had an intracranial response, with 3% having a complete reponse.35 Median progression-free survival was 16.1 weeks.35 In participants with V600E-BRAF and disease progression following prior local treatment, 31% demonstrated a partial response to dabrafenib, with a median progression-free survival of 16.6 weeks.35
Conversely, V600K BRAF mutants were less responsive, with an intracranial partial response of 7% and 22% in previously untreated and treated patients, respectively (Table 1).35 None had a complete response. Median progression-free survival in those with no prior treatment was 8.1 weeks versus 15.9 weeks in previously treated participants.35 The most common side effects in all groups were pyrexia (7% in untreated and 12% in previously treated patients) and cutaneous squamous cell carcinoma or keratoacanthoma (6% in previously untreated patients and 7% in those with prior local treatment).35 The results suggest that dabrafenib is a possible treatment option in patients with V600E or V600K BRAF-mutant melanoma metastatic to the brain regardless of history of prior local treatment.
Dabrafenib resistance and alternative combination strategies
Although dabrafenib has a high specificity and response rate in patients with V600E BRAF-mutated melanoma, resistance develops in almost all patients.31,33 Disease progression is observed in about 50% of patients on monotherapy within 6 months of treatment initiation.33,36 Multiple acquired mechanisms of resistance to BRAF inhibition have been investigated. Using sustained exposure to a specific V600E BRAF inhibitor, previously susceptible cell lines developed strong resistance.37,38 In vitro and in vivo analyses of melanoma cell lines and tumor biopsies with acquired resistance demonstrated intact V600E BRAF with no secondary mutation to account for the evasion of inhibition.37–39 Monitoring of MEK and ERK activation revealed distinct mechanisms of resistance, with elevated downstream phosphorylation in the setting of BRAF inhibition suggesting alternative MAPK pathway activation (Figure 1).37,40 Although multiple mechanisms appear to account for continued downstream signaling, utilization of different RAF isoforms, ie, ARAF or CRAF, to circumvent BRAF inhibition has been identified.38,40 Specifically, development of activating mutations of N-RAS kinase proteins, which tend to phosphorylate CRAF instead of BRAF, continue uncontrolled MAPK signaling.37,39,41 Another mechanism of resistance is acquisition of alternative splicing of BRAF (p61 BRAF), resulting in dimerization of RAF kinase and continued downstream ERK phosphorylation in the presence of RAF inhibitors.42 In addition, RAF-independent mechanisms have been identified with increased COT, a different serine/threonine MAP kinase, driving persistent MAPK cascade activation in the presence of BRAF inhibition.40 Alternatively, upregulation of receptor tyrosine kinases, such as platelet-derived growth factor receptor-β and insulin-like growth factor-1 receptor, confirms acquired MAPK-independent resistance.37,38 Ultimately, the mechanism of acquired BRAF inhibitor resistance appears to be complex, with multiple diverse pathways circumventing inhibition to cause disease progression after initial treatment response. Understanding resistance allows for combination treatment strategies to not only increase median progression-free survival but also potentially also improve complete response rates.
Combined BRAF and MEK inhibition
With evidence of persistent MAPK signaling through continued downstream MEK phosphorylation despite BRAF inhibition in vitro and in vivo, dabrafenib has been combined with trametinib, a MEK inhibitor, in both preclinical studies and clinical trials.39,43 In in vitro studies, cells having acquired resistance to dabrafenib with continued ERK phosphorylation in the presence of the BRAF inhibitor demonstrated restoration of inhibition similar to sensitive parental cell lines when treated with both dabrafenib and trametinib.39 These promising preclinical observations provided the rationale for a Phase I trial and a randomized Phase II clinical trial to determine the effects of this combination on response rates and median progression-free survivial.43 Enrolled participants had metastatic melanoma with confirmed V600E or V600K BRAF mutations but no prior treatment. Those with treated brain metastases and stable brain lesions for greater than 3 months were also eligible for enrollment.43 The recommended Phase II dose was 150 mg dabrafenib twice daily and 2 mg trametinib once daily, which are the recommended monotherapy doses for each agent.43
The response rate for combination therapy was improved at 76% compared with dabrafenib monotherapy, which was 54% (Table 1).43 The cohort treated with the recommended Phase II combination dose had a partial response rate of 67% and a complete response rate of 9%.43 Similar to previous studies, those treated with dabrafenib monotherapy had a partial response rate of 50% and complete response rate of 4%.33,43 The median progression-free survival in the group receiving combination dabrafenib and trametinib was 9.4 months, which was significantly better than the 5.8 months observed in those receiving dabrafenib monotherapy.43 Combination therapy was also generally tolerated well. The most frequent side effect of pyrexia, which was worse on combination therapy than on monotherapy (71% versus 26%), was the most common reason for dose reduction.43 Neutropenia was the most frequent grade 3 or 4 (11%) adverse effect with combination treatment.43 Surprisingly, the incidence of cutaneous squamous cell carcinoma was decreased on combination therapy compared with monotherapy using dabrafenib (7% versus 19%), possibly because MEK inhibition by trametinib attenuates dabrafenib-induced paradoxical activation of the MAPK pathway in normal keratinocytes.43 However, the dual therapy cohort experienced MEK inhibitor-associated toxic effects not seen with dabrafenib monotherapy, including decreased ejection fraction (9%) and chorioretinopathy (2%), although none were grade 3 or higher.43 Overall, combining dabrafenib with the MEK inhibitor trametinib led to improved response rates and median progression-free survival compared with treatment using dabrafenib alone and had a well tolerated toxicity profile.43
Combined BRAF, MEK, and PI3K/mTOR inhibition
BRAF and MEK inhibition vertically target two distinct proteins in the MAPK pathway; however, activation of alternative proliferative pathways can continue to drive oncogenesis (Figure 1). Mutations in both the MAPK and phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathways can coexist, with PI3K/mTOR activating mutations contributing to unregulated proliferation.44,45 An interaction between the MAPK and PI3K/mTOR signaling cascades has also been established, with increased AKT and mTOR phosphorylation occurring in the setting of BRAF inhibition.46,47 An in vitro study showed decreased cell proliferation in dabrafenib-resistant cells treated with both dabrafenib and trametinib; however, S6P, a protein activated by both the MAPK and PI3K/mTOR pathways, continued to be phosphorylated downstream.39 The addition of a dual PI3K/mTOR inhibitor to either dabrafenib or trametinib led to decreased S6P activation when compared with combination dabrafenib and trametinib therapy.39 The combination of dabrafenib with the PI3K/mTOR inhibitor decreased cell proliferation in both parental and resistant cell lines and offers a potential alternative treatment strategy in patients with acquired dabrafenib resistance.39 Clinical trials using a combination of dabrafenib, trametinib, and a PI3K/mTOR inhibitor are planned. Few overlapping toxicities are expected, given the individual mechanisms of action associated with each drug.
Combined dabrafenib and immune modulator strategies
BRAF activation induces downstream cytokine production that suppresses the immune system; therefore, dabrafenib-induced inhibition of the MAPK pathway could enable the immune system to play a vital role in clearing tumor cells and preventing recurrence.48,49 However, many immune cells, such as cytotoxic T-lymphocytes, use the MAPK pathway to function, and some MAPK pathway inhibitors negatively affect systemic immunity.50,51 To determine the effect of dabrafenib on the immune system, the peripheral blood of patients with metastatic melanoma treated with dabrafenib was analyzed using cytokine and immunologic assays along with flow cytometric analysis.49 Dabrafenib did not exhibit immunosuppression, likely secondary to its specificity for tumor cells containing V600 BRAF mutations, and spared the wild-type BRAF present in immune cells.49 In another study, biopsies obtained from patients before and after dabrafenib treatment were examined to determine the effect on tumor-infiltrating lymphocytes.52 Post-treatment tumor samples generally had high concentrations of both intratumoral and peritumoral CD4+ and CD8+ cells compared with samples before exposure to dabrafenib.52 Tumors with increased intratumoral CD8+ cells correlated with decreased tumor size and increased tumor necrosis.52 The initial increase in CD8+ cells found immediately post-treatment was reduced in biopsies taken from tumors following disease progression.52 These results suggest that dabrafenib combined with an immune stimulator, such as IL-2 or an anti-CTLA-4 antibody, could work synergistically. A recent Phase I study combining vemurafenib with ipilimumab was limited by grade 3 hepatotoxicity that was reversible with discontinuation of the drugs or use of glucocorticoids.53 Therefore, such combinations should be explored with caution in the future. Additional clinical trials are needed to assess the efficacy and safety of combining dabrafenib with immune modulators.
Combination BRAF and HGF/MET inhibition
In addition to acquired intracellular mechanisms of resistance, the tumor microenvironment can confer resistance to BRAF inhibition in cells containing V600E BRAF mutations.54 In vitro studies demonstrated that tumor cells initially sensitive to RAF inhibitors can become resistant when cultured with stromal cells that simulate the tumor microenvironment.54
Investigation of fibroblast-conditioned media using antibody array-based analysis identified hepatocyte growth factor (HGF) as the factor inducing resistance.54 When added to media containing a BRAF inhibitor, recombinant HGF caused growth of BRAF-inhibited cell lines and correlated with increased MET expression, whereas in other cell lines, HGF exposure resulted in undetectable MET expression with continued growth supression.54,55 HGF is a ligand to the receptor tyrosine kinase MET, which has been implicated in progression of melanoma (Figure 1).56 MET can initiate the MAPK cascade and bypass BRAF inhibition via phosphorylation of CRAF.56 MET also activates the PI3K-AKT pathway.54 In vitro studies demonstrated that dual inhibition of RAF and either HGF or MET resulted in reversal of resistance in cells with V600E BRAF mutations.54 Similarly, small molecules that inhibit MET eliminated resistance attributed to HGF secretion from surrounding fibroblast cells.54 These findings suggest that combining HGF/MET-specific inhibitors with BRAF inhibitors could possibly reduce some of the resistance conferred through the tumor microenvironment and ultimately improve both response rates and progression-free survival.
Place of dabrafenib in therapy
As a monotherapy, dabrafenib has been demonstrated in clinical trials to be an effective targeted agent in the treatment of metastatic melanoma with V600 BRAF mutations, especially in V600E and V600K mutants. Along with high response rates and a well tolerated toxicity profile, dabrafenib demonstrates durable activity against brain metastases. Taken by mouth twice daily, dabrafenib can be conveniently administered as an outpatient treatment. In May 2013, the US Food and Drug Administration approved dabrafenib for the treatment of unresectable or metastatic melanoma with V600E BRAF mutations.57
Although dabrafenib is effective as a single-agent treatment, resistance eventually develops in most patients. Preliminary studies examining combination strategies suggest enhanced response rates when dabrafenib is combined with a variety of inhibitors targeting proteins, not only in the MAPK cascade but also in alternative pathways conferring redundancy to oncogenesis. To improve overall response rates and survival outcome, more studies are needed to understand the interplay among the tumor microenvironment, systemic immune system, and intracellular signaling driving tumor progression. With recent approval from the US Food and Drug Administration, the role of dabrafenib as a single agent has been established, and combination therapy strategies to overcome resistance will be explored in future and ongoing clinical trials (Table 2).
 | Table 2 Active and recruiting clinical trials involving dabrafenib use in patients with melanoma |
Disclosure
KBK is a member of the GlaxoSmithKline (GSK) advisory board and has received research funding from GSK. GSF has received research funding and travel reimbursement from GSK. RK has no conflicts of interest to declare.
References
American Cancer Society. Cancer Facts and Figures 2013. Atlanta, GA: American Cancer Society; 2013. | |
Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). Bethesda, MD: National Cancer Institute. Available from: http://seer.cancer.gov/csr/1975_2009_pops09/. Accessed October 9, 2013. | |
Balch CM, Gershenwald JE, Soong S-J, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–6206. | |
Serrone L, Zeuli M, Sega FM, Cognetti F. Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res. 2000;19(1):21–34. | |
Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–2116. | |
Schwartzentruber DJ, Lawson DH, Richards JM, et al. Gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. | |
Guirguis LM, Yang JC, White DE, et al. Safety and efficacy of high-dose interleukin-2 therapy in patients with brain metastases. J Immunother. 2002;25(1):82–87. | |
Agarwala SS, Kirkwood JM, Gore M, et al. Temozolomide for the treatment of brain metastases associated with metastatic melanoma: a phase II study. J Clin Oncol. 2004;22(11):2101–2107. | |
Seigler HF, Lucas VS, Pickett NJ, Huang AT. DTIC, CCNU, bleomycin, and vincristine in metastatic melanoma. Cancer. 1980;46(11):2346–2348. | |
Punt CJA, van Herpen CML, Jansen RLH, Vreugdenhil G, Muller EW, de Mulder PH. Chemoimmunotherapy with bleomycin, vincristine, lomustine, dacarbazine (BOLD) plus interferon α for metastatic melanoma: a multicentre phase II study. Br J Cancer. 1997;76(2):266–269. | |
Atkins MB, Hsu J, Lee S, et al. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): a trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2008;26(35):5748–5754. | |
Eton O, Legha SS, Bedikian AY, et al. Sequential biochemotherapy for metastatic melanoma: results from a Phase III randomized trial. J Clin Oncol. 2002;20(8):2045–2052. | |
Shinozaki M, Fujimoto A, Morton DL, Hoon DS. Incidence of BRAF oncogene mutation and clinical relevance for primary cutaneous melanomas. Clin Cancer Res. 2004;10(5):1753–1757. | |
Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263. | |
Goel VK, Lazar AJF, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006;126(1):154–160. | |
Hersh EM, O’Day SJ, Powderly J, et al. A Phase II multicenter study of ipilimumab with or without dacarbazine in chemotherapy-naïve patients with advanced melanoma. Invest New Drugs. 2001;29(3):489–498. | |
Brahmer JR, Drake CG, Wollner I, et al. Phase I study of the single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. | |
Hamid O, Robert C, Daud D, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | |
Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9(9):726–735. | |
Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signaling pathways in cancer. Oncogene. 2007;26(22):3279–3290. | |
Fang JY, Richardson BC. The MAPK signaling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322–327. | |
Kalady MF, Dejulius KL, Sanchez JA, et al. BRAF mutations in colorectal cancer are associated with distinct clinical characteristics and worse prognosis. Dis Colon Rectum. 2012;55(2):128–133. | |
Frasca F, Nucera C, Pellegriti G, et al. BRAF (V600E) mutation and the biology of papillary thyroid cancer. Endocr Relat Cancer. 2008;15(2):191–205. | |
Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–1246. | |
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | |
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. | |
Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105(8):3041–3046. | |
Lee JT, Li L, Brafford PA, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010;23(6):820–827. | |
Laquerre S, Arnone M, Moss K, et al. A selective Raf kinase inhibitor induces cell death and tumor regression of human cancer cell lines encoding B-RafV600E mutation. Mol Cancer Ther. 2009;12 Suppl 1:Abstr B88. | |
Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastaese, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–1901. | |
Hauschild A, Grob J, Dimidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, Phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | |
Hauschild A, Grob J, Dimidov LV, et al. An update on BREAK-3, a Phase III, randomized trial: dabrafenib (DAB) versus dacarbazine (DTIC) in patients with BRAF V600E-positive mutation metastatic melanoma (MM). Poster 9013 presented at the 49th annual meeting of the American Society of Clinical Oncology, Chicago, IL, May 31 to June 4, 2013. | |
Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet. 2012;13(11):1087–1095. | |
Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011;364(8):772–774. | |
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. | |
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. | |
Greger JG, Eastman SD, Zhang V, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11(4):909–920. | |
Johnnessen CM, Boehm JS, Kim SY. COT/MAP3K8 drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. | |
Dumaz N, Hayward R, Martin J, et al. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006;66(19):9483–9491. | |
Poulikakos PI, Persaud Y, Janakirman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. | |
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | |
Janku F, Lee JJ, Tsimberidou AM, et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One. 2011;6(7):e22769. | |
Yuan TL, Cantley LC. PI3K pathway alterations in cancer: Variations on a theme. Oncogene. 2008;27(41):5497–5510. | |
Mendoza MC, Er EE, Blenis J. The RAS-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011; 36(6):320–328. | |
Sanchez-Hernandez I, Baquero P, Calleros L, Chiloeches A. Dual inhibition of (V600E) BRAF and the PI3K/AKT/mTOR pathway cooperates to induce apoptosis in melanoma cells through a MEK-independent mechanism. Cancer Lett. 2012;314(2):244–255. | |
Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer – immune evasion in human melanoma cells. J Exp Med. 2006;203(7):1651–1656. | |
Hong DS, Vence L, Falchook GS, et al. BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clin Cancer Res. 2012;18(8):2326–3715. | |
Weichsel R, Dix C, Woolridge L, et al. Profound inhibition of antigen-specific T-cell effector functions by dasatinib. Clin Cancer Res. 2008;14(8):2484–2491. | |
Zhao W, Gu YH, Song R, Qu BQ, Xu Q. Sorafenib inhibits activation of human peripheral blood T cells by targeting LCK phosphorylation. Leukemia. 2008;22(6):1226–1233. | |
Wilmott JS, Long GV, Howie JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–1394. | |
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–1366. | |
Straussman R, Morikawa T, Shee K, et al. Tumor micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | |
Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. | |
Puri N, Ahmed S, Janamanchi V, et al. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13(7):2246–2253. | |
US Food and Drug Administration. Dabrafenib. Available from: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm354477.htm. Accessed October 9, 2013. | |
National Institutes of Health. Dabrafenib. Available from: http://www.clinicaltrials.gov/ct2/results?term=dabrafenib&search=search. Accessed November 7, 2013. | |
Girotti MR, Marais R. Déjà vu: EGF receptors drive resistance to BRAF inhibitors. Cancer Discov. 2013;3(5):487–490. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.