")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 12
Cytotoxicity Of Chalcone Of Eugenia aquea Burm F. Leaves Against T47D Breast Cancer Cell Lines And Its Prediction As An Estrogen Receptor Antagonist Based On Pharmacophore-Molecular Dynamics Simulation
Authors Muchtaridi M , Yusuf M , Syahidah HN, Subarnas A, Zamri A , Bryant SD, Langer T
Received 26 May 2019
Accepted for publication 22 October 2019
Published 6 November 2019 Volume 2019:12 Pages 33—43
DOI https://doi.org/10.2147/AABC.S217205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Muchtaridi Muchtaridi,1 Muhammad Yusuf,2 Hasna Nur Syahidah,1 Anas Subarnas,1 Adel Zamri,3 Sharon D Bryant,4 Thierry Langer5
1Department of Pharmaceutical Analysis and Medicinal Chemistry, Faculty of Pharmacy, Universitas Padjadjaran, Bandung, West Java, Indonesia; 2Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Bandung, West Java, Indonesia; 3Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Riau, Pekanbaru, Riau 26293, Indonesia; 4Inte:Ligand GmbH, Mariahilferstrasse, Vienna 1070, Austria; 5Department of Pharmaceutical Chemistry, Faculty of Life Sciences, University of Vienna, Vienna A-1090, Austria
Correspondence: Muchtaridi Muchtaridi Tel/fax +62-22-7796200
Email [email protected]
Background: The 2ʹ,4ʹ-dihydroxy-6-methoxy-3,5-3-dimethylchalcone (ChalcEA) isolated from Eugenia aquea Burm f. leaves has potential anticancer activity against human breast-adenocarcinoma cell lines (MCF-7) with an IC50 value of 250 μM. However, its apoptotic activity on the T47D breast cancer cell lines which is involving caspase-3 has not been investigated.
Materials and methods: Therefore, this study aims to evaluate the cytotoxicity of ChalcEA on the T47D cell lines using the 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST) method and to predict its possible antagonistic activity on the human estrogen receptor alpha (hERα) using pharmacophore and molecular dynamics (MD) methods. The in vitro test of 10 synthesized ChalcEA derivatives was also performed as an insight into the further development of its structure as an anticancer agent.
Results: It is shown that ChalcEA has an IC50 of 142.58 ± 4.6 μM against the hERα-overexpressed T47D breast cancer cell lines, indicating its possible mechanism of anticancer activity as an antagonist of hERα. Pharmacophore study showed that ChalcEA shares similar features with the known hERα antagonist, 4-hydroxytamoxifen (4-OHT), which has hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), ring aromaticity (RA), and hydrophobicity (Hy) features. Molecular docking showed that ChalcEA formed hydrogen bonds with Glu353 and Arg394, and hydrophobic interactions in a similar manner with 4-OHT. Moreover, MD simulations showed that ChalcEA destabilized the conformation of His524, a remarkable behavior of a known hERa antagonist, including 4-OHT. Furthermore, the 10 best chalcone derivatives resulted from pharmacophore- and docking-based screening, were tested against the T47D cell lines. None of the derivatives have better activity than ChalcEA. It is suggested that the functional groups at the B-ring of ChalcEA are interesting to be further optimized in the next studies.
Conclusion: ChalcEA might act as an antagonist toward hERα, thus warranting further investigation as a potential anticancer agent.
Keywords: chalcone, estrogen receptor, pharmacophore, molecular docking, molecular dynamics
Introduction
Breast cancer is a leading cause of cancer deaths and is the most malignant form of cancer in women today.1,2 Statistics from 2005 show that Indonesia has 0.41 rate ratio of mortality to incidence (MR:IR), meaning that 41% of patients died from 100% of the cases.3 Previous studies suggested that breast cancer was highly affected by ovarian hormones.4,5 Circulation of estrogens has been associated with the increase of the risk for breast cancer in women. It is evidenced that estrogen is a key determinant of risk for breast cancer.6,7 Estradiol is a natural steroidal compound that acts as an agonist of estrogen. Also, there are various non-steroidal compounds, including tamoxifen, that acts as an agonist or an antagonist of estrogen.8 The non-steroidal compounds have been reviewed to have various activities as agonists or antagonists, depending on the organ system or genes studied.9,10
ERα is a ligand-activated transcription factor that has an important function in many tissues and plays a critical role in the aetiology of breast cancer.11,12 Since ERα is a major target for the treatment and prevention of breast cancer, many anti-breast cancer compounds have been designed to bind ERα to produce different pharmacological profiles. Hence, the discovery of natural product compounds that could potentially act as antagonists of estrogen in breast tissue would be useful.13,14
In rational drug design, the discovery of new biologically active compounds and leads from the diverse chemical scaffolds have been conducted to optimize the lead compounds from natural products.15 There are many chemotherapeutic agents for the treatment of cancer, including breast cancer. However, the efficacy of these anticancer agents is still below expectations, also including their undesirable side-effects.16 This situation has prompted efforts to discover new anticancer agents to overcome the current problems, especially exploring compounds derived from natural products. So far, numerous types of natural compounds with high anticancer activity have been discovered and further investigated.17
In an effort to discover anticancer agents from the natural product, we isolated a chalcone compound from the leaves of Eugenia aquea which have been identified as a 2ʹ,4ʹ-dihydroxy-6-methoxy-3,5-3-dimethylchalcone (ChalcEA). This compound inhibited the proliferation of breast cancer cell line MCF-7, with an IC50 of 74.5 µg/mL (250 μM), and promoted apoptosis via the activation of PARP.18 However, its apoptotic activity on the T47D breast cancer cell lines which is involving caspase-3 has not been investigated. Pharmacophore models produced from structure-based and ligand-based approaches are used to understand the key interaction features of homologs of an active compound and to understand the interaction of a target-binding site with the bioactive ligand.19 Previously, we have identified the pharmacophoric features of 4-OHT, which composed of 1) hydrophobic features from the butenyl group and aromatic rings; 2) positive ionizable features from the tertiary amine group; and 3) hydrogen bond features from the phenoxy and hydroxyl oxygens with the Glu353 and Arg394.19 ChalcEA has similar pharmacophoric features with 4-OHT.
For this reason, we predicted that the toxicity of ChalcEA to the breast cancer cell line was due to its binding to the hERα. Therefore, in this study, the cytotoxicity of ChalcEA on the T47D cell lines was evaluated using the 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST) method. The possible antagonistic activity of ChalcEA to the hERα receptor was further investigated by molecular docking, pharmacophore modelling, molecular dynamics simulation, and MM/GBSA binding energy calculation. In addition, the cytotoxicity of several synthetic ChalcEA derivatives was also tested. The result should be useful as an insight into the further development of ChalcEA activity on the cancer cells with selective estrogen receptor modulator (SERM)-like properties.20
Materials And Methods
Materials
Plant Materials
Sample collection: samples were collected from Manoko experiment station, Research Institute for Spices and Medicinal Plants, Indonesian Centre for Estate Crops Research and Development, Lembang, West Java, Indonesia. The isolation procedure was explained by Subarnas et al (2015) in the previous report.18
Chemicals
Chemicals for isolation: ethyl alcohol (EtOH) as an extraction solvent, ethyl acetate (EtOAc), n-hexane, acetone, chloroform, and methanol (MeOH) (Merck) for isolation, and chalcone derivatives from Zamri and coworkers. For assay: cell counting kit (CCK)-8 (Dojindo Lab., Tokyo Japan), foetal bovine serum (FBS) (Gibco), dimethyl sulfoxide (DMSO) (Sigma), Leibovitz L-1, Roswell Park Memorial Institute medium (RPMI) 1640 (Invitrogen), and human ductal breast epithelial tumour cell line T47D cell line were purchased from American Type Culture Collection (Manassas, Virginia).
Equipment
Enzyme-linked immunosorbent assay (ELISA) (Boehringer), Spectrometers (Shimadzu), microplate 96-well plate, and microplate reader, centrifuge, and incubator (Memmert).
Software And Hardware
Software: Gaussian03,21 LigandScout 4.1.,22 Autodock Vina,23 Accelrys Discovery Studio Visualizer 3.5,24 AMBER 14,25 and AmberTools 1.6.25 Hardware: A Linux PC powered by octacores Intel Xeon 2.4 GHz processor, GPU NVIDIA GTX 970, and 2 GB of RAM.
Methods
Isolation Of ChalcEA
The isolation process was guided by an application of thin-layer chromatography of a reference compound ChalcEA that was used in a previous study.18 All fractions were screened with thin-layer chromatography methods and compared with the reference standard. Only the fractions that contained the targeted compound were used for purification. Purification and isolation of the targeted compound were conducted at Universitas Padjadjaran, performed by column chromatography and preparative thin-layer chromatography methods.
Assay
Cytotoxicity tests on T47D human breast cancer cells were done according to the WST (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) method22 using CCK-8 reagent.26 CCK-8 was employed as a measure of cell viability. The mechanism of CCK-8 is based on the transformation of a water-soluble WST-8 to a water-soluble formazan (yellow color) by dehydrogenases in the presence of an electron carrier. The completed procedures of this method are published by Han et al.27
Cell Culture
The T47D cells were grown in a Roswell Park Memorial Institute (RPMI) medium supplemented with 2 mg/mL insulin, 1 mM sodium pyruvate, 1 mM non-essential amino acids, 4 mM glutamine, 10% FCS, and antibiotics (penicillin–streptomycin). One week before the experiment, the cells were transferred to phenol red-free medium supplemented with 5% C-SFCS.
Cytotoxicity Tests
Subcultured cells were provided every 3–4 days using a trypsin 0.25%–EDTA 0.02% solution (Gibco BRL). Cell viability was measured by WST assay using CCK-8 reagent. Briefly, cells were grown in 96-well plates for 48 hrs at a density of 2 × 104 cells/well in the growth medium. After 24 hrs, ChalcEA was put at over a range concentration from 1.95, 7.81, 31.25, 125, and 500 μg/mL in 5% DMSO. After 48 hrs incubation, the culture medium was removed from the cells and replaced with WST-Medium and dissolved CCK-8 (DOJINDO). Then, 10 μL of 1 N HCl was added to each well and the absorbance was measured at a wavelength of 450 nm with the reference wavelength at 620 nm. Absorbance was measured at 450 nm, then the absorbance measured at the reference wavelength (620 nm) was subtracted to obtain the absorbance of samples. Subsequently, the value of absorbance was used to count the percentage of cell proliferation inhibition (%CPI), known as % inhibition.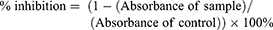
The value of % inhibition was used to determine graphic inhibition, after which the linear equation was made from the graphic inhibition. The value of the half-maximal inhibitory concentration (IC50) was calculated using this formula.
Molecular Docking Simulation
The X-ray crystallography of ERα complexed with 4-OHT as positive control system was derived from Protein Data Bank (PDB ID: 3ERT). The ligand structures and macromolecule were separated by Discovery Studio Visualizer 3.5. 3D structures of ChalcEA and its derivatives were optimized by Ligandscout 4.1 Advanced and Hyperchem. All ligands and 3ERT were prepared for molecular docking simulation using AutoDockTools (ADT) 1.5.6. The molecular docking methods were carried out according to a previous study.19 The receptor and ligands were protonated. The solvation parameters and default Kollman charges28 were added to the protein atoms. Gasteiger charges were allocated to each ligand atom.29 A grid box size was 40 x 40 x 40 with spaced by 0.375 Å. This grid box centered on active site of ERα (x = 30.282, y = −1.913, and z = 24.207).
Molecular docking simulations were done by AutoDock 4.2. The Lamarckian Genetic Algorithm (LGA) parameters were: 100 runs, the mutation rate of 0.02, the population size of 150, elitism of 1, 5,000,000 energy evaluations, and a crossover rate of 0.80.30 Root-mean-square deviation (RMSD) tolerance was set 1 Å to cluster the docking conformation results. The docking results were visualized by Accelrys Discovery Studio Visualizer 3.5.
Pharmacophore Mapping
The polling algorithm and the best energy option, based on a CHARMm forcefield embedded in LigandScout 4.1, were performed in order to set a conformational for each molecule.22 The best confirmation of molecules generated was screened to map against the pharmacophore model using the “best fit” option. LigandScout 4.1 was performed to derive the 3D chemical feature-based pharmacophores from the structural data of complex protein structure from PDB (3ERT) using the default settings. The training set from DUD was used for validation.
Structure-Based 3D-Pharmacophore Modeling
The 3D-pharmacophore model was generated from the X-ray structure of ERα in complex with 4-OHT (PDB code: 3ERT) using Ligandscout 4.1.22 Validation of the 3D-interaction pharmacophore models was done for its capability to differentiate true active compounds from decoys by screening a set of 625 known active and 20,772 decoy compounds obtained from the enhanced Database of Useful Decoys (DUDe: http://dude.docking.org).31 The dataset from DUDe was transformed into 3D multi-conformational databases for virtual screening using the idbgen algorithm in LigandScout 4.1, which computes conformations and annotates each conformation with pharmacophore features. All of the ChalcEA derivatives were screened virtually using the validated 3D-SB pharmacophore model and the LigandScout 4.1 VS algorithm iscreen, with a maximum of two omitted features to identify and rank ligands in the library that could fit the geometry and features of the 3D-model. The pharmacophore fit score measured the fit of features of each hit compound to the pharmacophore model features and was used to rank the hit molecules retrieved by the pharmacophore model.
Molecular Dynamics Simulation
The crystal structure of hERα in complex with 4-OHT (antagonist, PDB ID 3ERT) was used in this study. The structure of 3ERT was manually corrected using Accelrys Discovery Studio 3.532 and AmberTools25 was used to add the hydrogen atoms to the protein structure.
The 3D structure of ChalcEA was modeled by Accelrys Discovery Studio 3.5.32 The parameterization of ChalcEA was done AM1-BCC method in the Antechamber module33 of AMBER 14. The initial structure of ChalcEA in complex with the protein was done by superimposing it onto the crystal structure of 4-OHT complex with hERα.
The minimization and molecular dynamics simulation were performed by using the PMEMD module of AMBER 14. Sterical clashes in the initial structure of ChalcEA complex were removed by stepwise minimization using the steepest descent and conjugate gradient algorithm. The AMBER force field ff14SB was used. Since there was no disulfide bridge formed, the notation of CYS remained unchanged. The MD system was solvated in the TIP3P water box and neutralized by the counterions. A periodic boundary condition (PBC) was applied with the cutoff of the non-bonded interaction of 9 Å. The MD system was gradually heated to a physiological temperature, i.e., 310 K in the NVT ensemble using the Langevin thermostat with a frequency of collision of 1.0 ps−1 and a harmonic restraint of 5 kcal mol−1 Å−2 on the backbone protein. Furthermore, the system was equilibrated for 3 ns in the NPT ensemble with the gradual decrease of the restraint. Finally, a 20 ns production run was performed under the NPT ensemble with the SHAKE algorithm enabled.
Result And Discussion
Cytotoxicity Of ChalcEA On The T47D Human Breast Cancer Cell Lines
The activity of ChalcEA and its derivatives was evaluated against T47D human breast cancer cell lines. The cells were exposed to 10 concentrations of the samples (1.95, 7.81, 15.63, 31.25, 65, 125, 250, 500, 625, 750, 875 μg/mL) for 24–48 hrs. These range concentrations were adjusted based on the previous study.34 The cytotoxicity of ChalcEA was indicated by its IC50 value, calculated from the ratio of absorbance from the presence of formazan, the product of the cell metabolism of WST (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt).
Figure 1 shows a dose-dependent T47D cell death after the treatment of ChalcEA. The calculated IC50 value against T47D cells was 42.49 μg/mL (142.58±4.6 µM), lower than that of MCF-7 (250 μM) reported before.35 This result indicates that the inhibitory activity of ChalcEA against T47D cell lines was stronger than MCF7 cell lines. In other words, the T47D cells were more susceptible than MCF-7 to the ChalcEA due to the different characteristics of each type of cancer cells. Different from the MCF-7, the apoptosis in T47D cells involves caspase-3 activation.36,37 It is known that caspase-3 plays an important role in apoptotic pathways through the cleavage of a variety of key cellular proteins.38
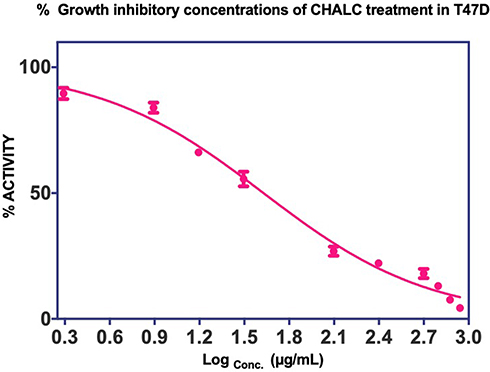 |
Figure 1 Cell proliferation inhibition profiles of ChalcEA in T47D cell lines. |
Nevertheless, the mode of action of ChalcEA in breast cancer cells has not been clear. In this study, ChalcEA was predicted to bind to hERα due to the dominant presence of this receptor in breast cancer cells. A hERα full antagonist, 4-OHT, was used as a control in this study.39,40 To support our prediction, we implemented pharmacophore mapping, molecular docking simulation, and molecular dynamics simulation to evaluate the possibility of ChalcEA mode of actions as a hERα antagonist.
Structure-Based Pharmacophore Of ChalcEA With 4-OHT
The crystal structure of hERα in complex with 4-OHT was selected as a reference for structure-based pharmacophore study.41 LigandScout 4.1 was employed to perform the pharmacophore modeling, including its comparative analysis with the docking result. The conformations of ChalcEA were optimized using all possible combination of features and interfeatures distance. Figure 2A shows the seven pharmacophore features derived from the complex of 4-OHT with hERα (IC50: 2nM): one positive ionizable (PI), one hydrogen-bond donor (HBD), one hydrogen-bond acceptor (HBA), one hydrophobic moiety (Hy), and three aromatic rings (RA). As compared with 4-OHT, ChalcEA missed three features: one RA, one HBD, and one PI on the tail of 4-OHT. In Figure 2B, the superimposition between the docking pose of ChalcEA and the bound-conformation of 4-OHT suggested that the two aromatic rings (A and B) and 4-OH group of ChalcEA were well mapped with those of 4-OHT, except the hydrophobic tail.
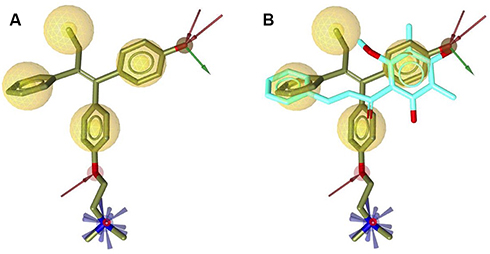 |
Figure 2 (A) Pharmacophore features of 4-OHT (yellow-colored sticks) in hERα (3ERT) and (B) in comparison with the docking pose of ChalcEA (cyan-colored sticks). |
Interactions Of ChalcEA With hERα From Docking
The binding mode of ChalcEA on the antagonist-form of hERα42 was predicted using AutoDock 4.2.43 Initially, the co-crystallized ligand, 4-OHT, was re-docked into the ligand-binding site of hERα, resulted in a docking score (free energy of binding) of −9.7 kcal/mol. Figure 3 showed a successful control docking, which indicated low RMSD value between the docking pose and the crystal structure (1.12 Å).
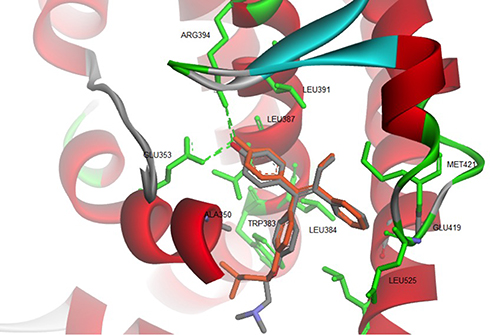 |
Figure 3 Re-docking of co-crystallized 4-OHT into the ligand-binding site of hERα using AutoDock 4.2 (crystal and docking poses are colored in grey and orange, respectively). |
The docking pose of 4-OHT formed hydrogen bonds and hydrophobic interactions with the hERα. Residues involved in the hydrogen bond formations are Glu353 and Arg394. Whereas Leu346, Thr347, Ala350, Trp383, Leu384, Leu391, Met421, and Leu525 are participating in the hydrophobic interactions.
The computed free energy of binding of the ChalcEA in hERα was −8.23 kcal/mol, higher than that of 4-OHT (−9.7 kcal/mol). Similar to the structure-based pharmacophore result, the superimposition between docking pose of 4-OHT and ChalcEA also suggested that the less affinity of ChalcEA was due to the absence of hydrophobic tail (Figure 4).
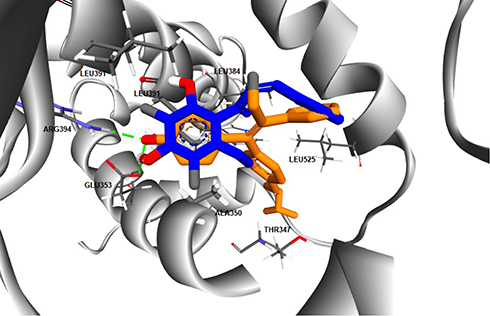 |
Figure 4 The superimposition between the docking poses of ChalcEA and 4-OHT (blue and orange-colored sticks, respectively) inside the ligand-binding site of hERα. |
Previously, we have observed that the hydrophobic tail of 4-OHT plays an important role in its antagonistic activity. In a 20 ns of MD simulation, the hydrophobic tail interfered the hydrogen bond network between helix-3 and helix-11 of hERα in agonist-form.34 One of the known molecular mechanisms of hERα antagonist is through the disruption of zipper-like hydrogen bond network among Glu419, His524, and Lys531.34,39,44 Moreover, the hydrophobic tail of 4-OHT also in contact with the flexible loop, namely 534-loop, in between helix-11 and helix-12. From a 158 ns of MD simulation, the conformation of this loop was different in apo-, agonist-, and antagonist forms.45 Therefore, the missing 4-OHT’s hydrophobic tail features in the structure of ChalcEA might be a reason behind the lower affinity and IC50 of this compound.
Molecular Dynamics Simulation Of ChalcEA
To further study the binding of ChalcEA inside the ligand-binding site of hERα, two sets of 20 ns of MD simulations were performed, namely 3ERT-ChalcEA and 3ERT-4OHT. The RMSD profile of hERα in the presence of ChalcEA was higher than that of 4-OHT, indicating its antagonistic activity by changing the conformation of the receptor (Figure 5). Moreover, the similar action of ChalcEA with the 4-OHT is also suggested by the identical residual RMSF profile between the two MD systems (Figure 6). A high fluctuation at the C-terminal of two systems was expected, due to the flexibility of the helix 11–12 regions in the antagonist-form of hERα.45
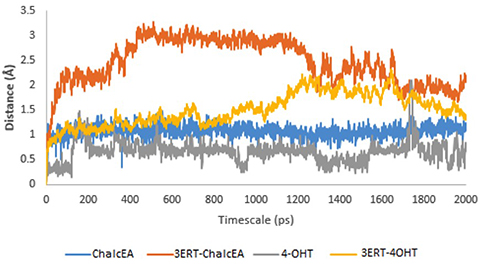 |
Figure 5 RMSD profile of 3ERT-ChalcEA and 3ERT-4OHT during 20 ns of MD simulation. |
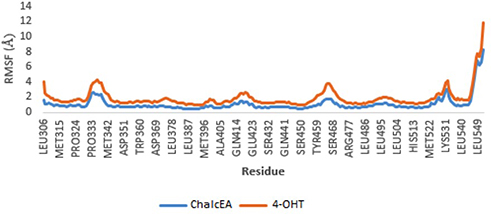 |
Figure 6 RMSF profile of 3ERT-ChalcEA and 3ERT-4OHT systems throughout 20 ns of MD simulation. |
MD simulations showed no hydrogen bond with His524 was formed. The stabilization of His524 is important for the agonistic activity of hERα ligand. It is required to maintain the hydrogen bond network around the ligand-binding site.34 For this reason, it is predicted that ChalcEA is an antagonist, not an agonist. To support our prediction, the rotation of His524 throughout 20 ns of simulations was monitored. Figure 7 showed that the side chain of His524 in both MD systems was rotated, implying that ChalcEA acted similarly with the 4-OHT inside the ligand-binding site of hERα.
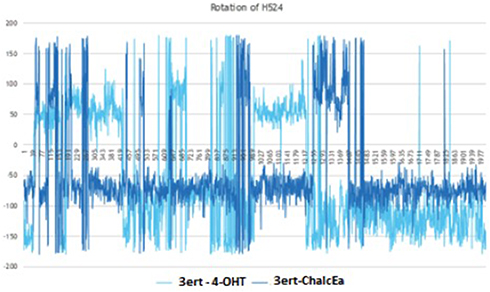 |
Figure 7 The plot of dihedral analysis of His524’s side-chain rotation during 20 ns of simulation. |
From the trajectory of MD simulations, the MM/GBSA interaction energy of ChalcEA and 4-OHT was −38.56 kcal/mol and −51.24 kcal/mol, respectively, as shown in Table 1. This result corresponds with the hypothesis of the absence of hydrophobic tail as the source of ChalcEA’s lower affinity as compared to 4-OHT.
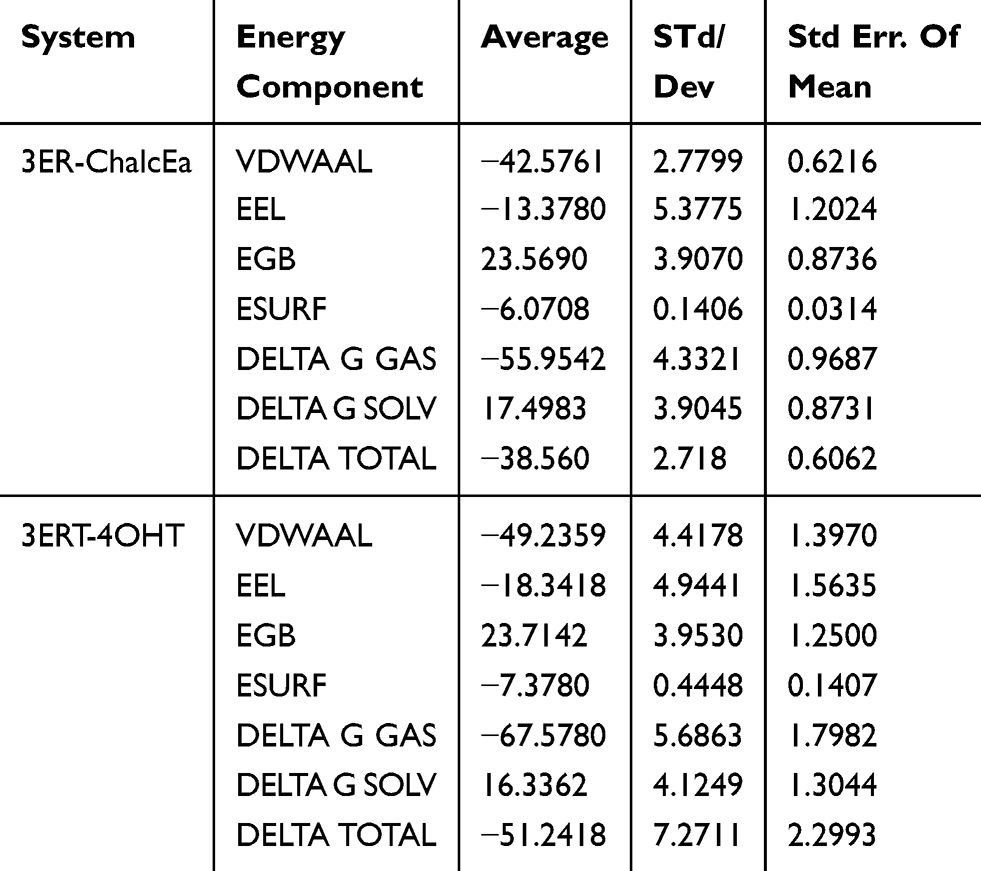 |
Table 1 Van Der Waals (VDW) Analysis Of The ChalcEA And 4-OHT System In Both Antagonist Receptor Forms Throughout 20 ns Of MD Simulation |
Pharmacophore-Based Screening
The pharmacophore model that is built by LigandScout 4.1 software (Vienna, Austria) was validated using the ROC curve. Validation of the pharmacophore screening method in Ligandscout 4.1 was performed using a test set consisting of a decoy set and an active set. The active set is a set of compounds or ligands that are known to be active on certain receptors with IC50 values known from previous research results. Decoy sets are a set of ligands that have structures similar to the active set but are inactive against the receptor. As shown in Figure 8, the total number of compounds used in the validation process is 21,397, comprising 625 active compounds and 20,772 decoy compounds. These compounds are obtained from http://dude.docking.org/.46 From the screening results, 17,796 best hits were obtained. EF100% and AUC100% values obtained were 1.2 and 0.94, respectively. Based on this result, the method has been validated due to the high specificity determined by the AUC value of 94%. The X-axis of the ROC curve is the rate of the active compound and the Y-axis is the rate of the decoy compounds. The sensitivity (Se) and specificity (Sp) are plotted in ROC curves, and it is noted that Spsubset = ((Decoystotal − Decoysselected)/Decoystotal) and Sesubset = (Ligandsselected/Ligandstotal).47
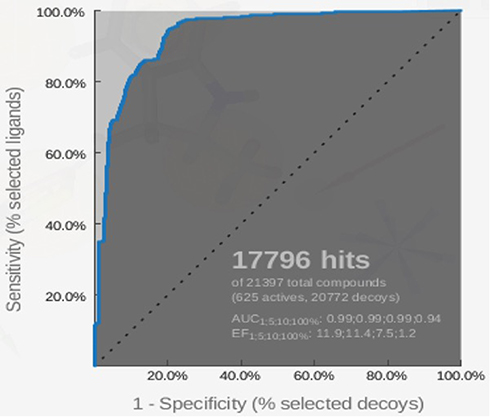 |
Figure 8 ROC curve for validation of the pharmacophore model by using the 625-compound active set and the 20,722-compound decoy set. |
Virtual Screening
Virtual pharmacophore screening was performed on 113 chalcone derivatives. The best 40 compounds were selected, which were further simulated by molecular docking against hER-α receptors. There are 37 compounds that have fit pharmacophore with tamoxifen more than 50% and 3 other compounds were less than 50% (Table 2).
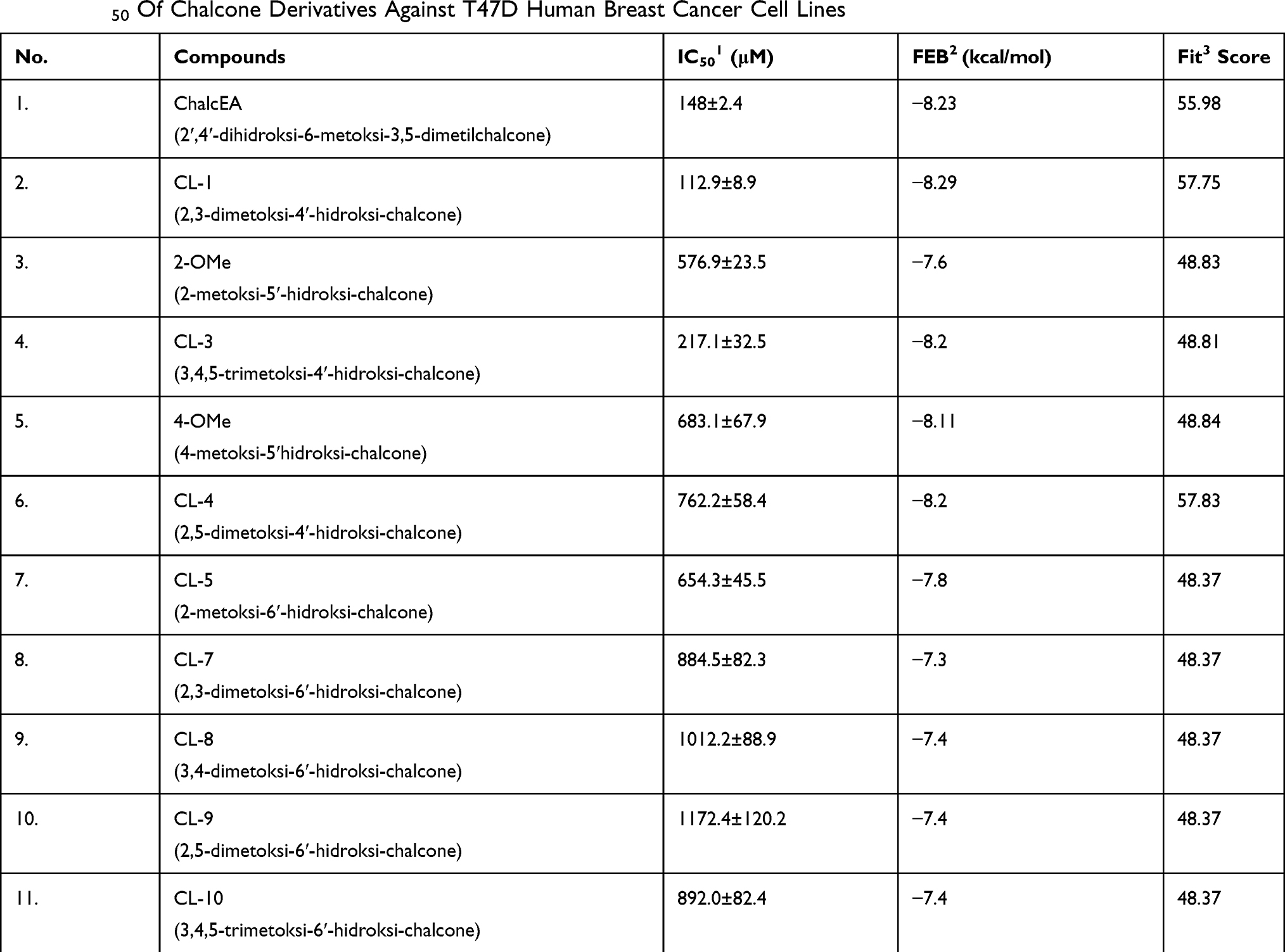 |
Table 2 IC50 Of Chalcone Derivatives Against T47D Human Breast Cancer Cell Lines |
In Vitro Test Of The Best Compounds
The chalcone derivatives (113 structures) from the ChEMBL database48 were screened using molecular docking and pharmacophore fit. The activity of 10 synthesizable compounds of the top 20 hit compounds was evaluated by in vitro assay against T47D human breast cancer cell lines. These compounds were synthesized by Zamri and coworkers.49
Based on Table 2, the 4-OH group on ring A on chalcone skeleton plays an important role that might affect inhibitory activity. As shown in Table 2, ChalcEA, CL-4, and CL-1 have a good fit score. However, the substituents of the methoxy on ring B decreased their inhibitory activity due to the change of hydrophobicity of ring B.
ChalcEA has the same hydrophobic aromatic with 4-OHT as indicated by the good overlay between their structures. It is observed that the hydroxyl group at position no. 4 contributed to the binding affinity of CL-1, CL-3, and CL-4. This functional group is similar with that of 4-OHT to form interaction with Arg394 and Glu393. Hydrogen bond interaction was formed between His524 and the hydroxyl group of CL1, CL4, and CL8. However, CL5 and CL10 did not form a hydrogen bond interaction with the residue.
ChalcEA had free energy binding of −8.23 kcal/mol. Table 2 showed that only two chalcone derivatives (4-OMe and CL4) had low free energy binding with −8.11 kcal/mol and −8.29 kcal/mol, respectively. The ligand of 4-OMe formed hydrogen bonds with Glu353 and Arg394, while CL4 only formed a hydrogen bond with Arg394. The increasing number of hydrogen bond resulted in a lower free energy of binding of 4-OMe than CL4. Thus, 4-OMe has a better affinity compared to CL4 (Figure 9).
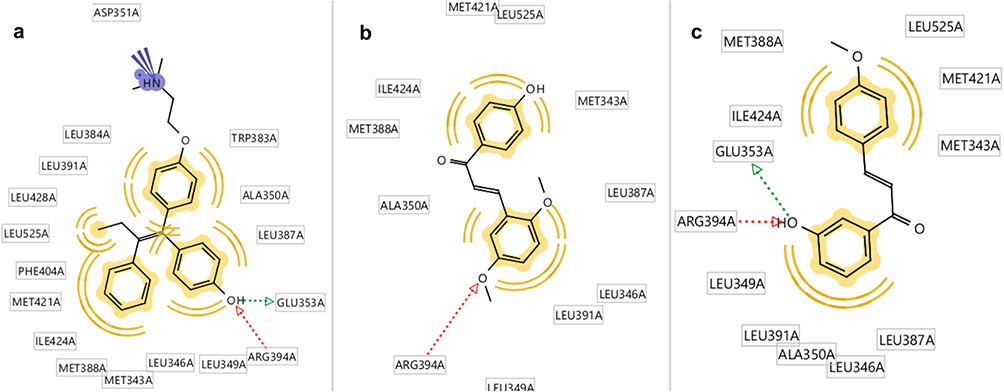 |
Figure 9 Molecular docking results of chalcone derivatives against human estrogen receptor alpha (HERα). (A) Hydroxyl group of 4-OGT formed hydrogen bonds with GLU353 and Arg394 of HERα, (B) while CL4 was play role as hydrogen bond acceptor against Arg394, and (C) 4-OMe also formed hydrogen bonds with GLU353 and Arg394 of HERα. |
Conclusion
ChalcEA showed proliferation inhibition activity of T-47D cell lines, with an IC50 142.58±4.6 µM. The possible anticancer action of ChalcEA was due to the antagonistic activity to the estrogen receptor alpha. This suggestion was based on the similarity of pharmacophoric features between ChalcEA and 4-OHT. Furthermore, its mechanism as ERα antagonist was predicted by MD simulation. RMSD, RMSF, and hydrogen bond analysis throughout 20 ns MD simulation showed that ChalcEA had a similar effect toward loop-534 with a disruption of the hydrogen bond network between Glu419, His524, and Lys531. Pharmacophore analysis showed that ChalcEA missed the hydrophobic and negative ionizable features that present in the tail chain of 4-OHT. It can be concluded that ChalcEA might act as an antagonist toward hERα, thus deserving further investigation as a potential anticancer agent from natural products.
Acknowledgments
We gratefully acknowledge the Rector of Universitas Padjadjaran and the Minister of Research and Higher Education for funding this project through the Grant of Research and Publication 2016–2018 and Academic Leadership Grants 2019 no. 1373b/UN6.O/LT/2019 from Universitas Padjadjaran. We also acknowledge the support of IntelLigand Company for the use of their facilities.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–386. doi:10.1002/ijc.29210
3. Ghoncheh M, Pournamdar Z, Salehiniya H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac J Cancer Prev. 2016;17(S3):43–46. doi:10.7314/APJCP.2016.17.S3.43
4. Zhu HH, Hu CH, Strickland P. Perspectives of breast cancer etiology: synergistic interaction between smoking and exogenous hormone use. Chin J Cancer. 2011;30(7):433–441.
5. Boonyaratanakornkit V, Pateetin P. The role of ovarian sex steroids in metabolic homeostasis, obesity, and postmenopausal breast cancer: molecular mechanisms and therapeutic implications. Biomed Res Int. 2015;2015:140196. doi:10.1155/2015/140196
6. Arslan AA, Shore RE, Afanasyeva Y, Koenig KL, Toniolo P, Zeleniuch-Jacquotte A. Circulating estrogen metabolites and risk for breast cancer in premenopausal women. Cancer Epidemiol Biomarkers Prev. 2009;18(8):2273–2279. doi:10.1158/1055-9965.EPI-09-0312
7. Ziegler RG, Fuhrman BJ, Moore SC, Matthews CE. Epidemiologic studies of estrogen metabolism and breast cancer. Steroids. 2015;99(Pt A):67–75. doi:10.1016/j.steroids.2015.02.015
8. Farooq A. Structural and functional diversity of estrogen receptor ligands. Curr Top Med Chem. 2015;15(14):1372–1384. doi:10.2174/1568026615666150413154841
9. Martinkovich S, Shah D, Planey SL, Arnott JA. Selective estrogen receptor modulators: tissue specificity and clinical utility. Clin Interv Aging. 2014;9:1437–1452. doi:10.2147/CIA.S66690
10. Lewis DK, Johnson AB, Stohlgren S, Harms A, Sohrabji F. Effects of estrogen receptor agonists on regulation of the inflammatory response in astrocytes from young adult and middle-aged female rats. J Neuroimmunol. 2008;195(1–2):47–59. doi:10.1016/j.jneuroim.2008.01.006
11. Zhou Z, Qiao JX, Shetty A, et al. Regulation of estrogen receptor signaling in breast carcinogenesis and breast cancer therapy. Cell Mol Life Sci. 2014;71(8):1549.
12. Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics. 2006;7(8):497–508. doi:10.2174/138920206779315737
13. Oseni T, Patel R, Pyle J, Jordan VC. Selective estrogen receptor modulators and phytoestrogens. Planta Med. 2008;74(13):1656–1665. doi:10.1055/s-0028-1088304
14. Marino M, Galluzzo P. Are flavonoids agonists or antagonists of the natural hormone 17beta-estradiol? IUBMB Life. 2008;60(4):241–244. doi:10.1002/iub.34
15. Sliwoski G, Kothiwale S, Meiler J, Lowe EW
16. Kayl AE, Meyers CA. Side-effects of chemotherapy and quality of life in ovarian and breast cancer patients. Curr Opin Obstet Gynecol. 2006;18(1):24–28. doi:10.1097/01.gco.0000192996.20040.24
17. Newman D. Screening and identification of novel biologically active natural compounds. F1000Research. 2017;6:783. doi:10.12688/f1000research
18. Subarnas A, Diantini A, Abdulah R, et al. Apoptosis induced in MCF-7 human breast cancer cells by 2ʹ, 4ʹ-dihydroxy-6-methoxy-3, 5-dimethylchalcone isolated from eugenia aquea burm f. leaves. Oncol Lett. 2015;9(5):2303–2306. doi:10.3892/ol.2015.2981
19. Muchtaridi M, Syahidah H, Subarnas A, Yusuf M, Bryant S, Langer T. Molecular docking and 3D-pharmacophore modeling to study the interactions of chalcone derivatives with estrogen receptor alpha. Pharmaceuticals. 2017;10(4):81. doi:10.3390/ph10040081
20. Miller CP. SERMs: evolutionary chemistry, revolutionary biology. Curr Pharm Des. 2002;8(23):2089–2111. doi:10.2174/1381612023393404
21. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 03. Wallingford, CT: Gaussian, Inc; 2003.
22. Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45(1):160–169. doi:10.1021/ci049885e
23. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
24. Accelrys. Discovery Studio Modeling Environment, Release 3.5. San Diego: Accelrys Software Inc; 2007.
25. Case DA, Cheatham TE
26. Yin LM, Wei Y, Wang WQ, Wang Y, Xu YD, Yang YQ. Simultaneous application of BrdU and WST-1 measurements for detection of the proliferation and viability of airway smooth muscle cells. Biol Res. 2014;47:75. doi:10.1186/0717-6287-47-75
27. Han SB, Shin YJ, Hyon JY, Wee WR. Cytotoxicity of voriconazole on cultured human corneal endothelial cells. Antimicrob Agents Chemother. 2011;55(10):4519–4523. doi:10.1128/AAC.00569-11
28. Scott J, Weiner SJ, Kollman PA, et al. A new force field for molecular mechanical simulation of nucleic acids and proteins. J Amer Chem Soc. 1984;106(3):765–784. doi:10.1021/ja00315a051
29. Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity – a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. doi:10.1016/0040-4020(80)80168-2
30. Ikram NK, Durrant JD, Muchtaridi M, et al. A virtual screening approach for identifying plants with anti H5N1 neuraminidase activity. J Chem Inf Model. 2015;55(2):308–316. doi:10.1021/ci500405g
31. Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–6594. doi:10.1021/jm300687e
32. Accelrys. Discovery Studio Modeling Environment, Release 2.5.5. San Diego: Accelrys Software Inc; 2007.
33. Duan Y, Wu C, Chowdhury S, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Compu Chem. 2003;24(16):1999–2012. doi:10.1002/jcc.10349
34. Muchtaridi M, Yusuf M, Diantini A, et al. Potential activity of fevicordin-A from phaleria macrocarpa (Scheff) boerl. Seeds as estrogen receptor antagonist based on cytotoxicity and molecular modelling studies. Int J Mol Sci. 2014;15(5):7225. doi:10.3390/ijms15057225
35. Subarnas A. Antiproliferative activity of primates-consumed plants against MCF-7 human breast cancer cell lines. E3 J Med Res. 2012;1(4):038–043.
36. Yang S, Zhou Q, Yang X. Caspase-3 status is a determinant of the differential responses to genistein between MDA-MB-231 and MCF-7 breast cancer cells. Biochim Biophys Acta Mol Cell Res. 2007;1773(6):903–911. doi:10.1016/j.bbamcr.2007.03.021
37. Mooney LM, Al-Sakkaf KA, Brown BL, Dobson PRM. Apoptotic mechanisms in T47D and MCF-7 human breast cancer cells. Br J Cancer. 2002;87(8):909–917. doi:10.1038/sj.bjc.6600541
38. Devarajan E, Sahin AA, Chen JS, et al. Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene. 2002;21(57):8843–8851.
39. Celik L, Lund JD, Schiott B. Conformational dynamics of the estrogen receptor alpha: molecular dynamics simulations of the influence of binding site structure on protein dynamics. Biochemistry. 2007;46(7):1743–1758.
40. Hu R, Hilakivi-Clarke L, Clarke R. Molecular mechanisms of tamoxifen-associated endometrial cancer (Review). Oncol Lett. 2015;9(4):1495–1501. doi:10.3892/ol.2015.2962
41. Eiler S, Gangloff M, Duclaud S, Moras D, Ruff M. Overexpression, purification, and crystal structure of native ER alpha LBD. Protein Expr Purif. 2001;22(2):165–173. doi:10.1006/prep.2001.1409
42. Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95(7):927–937. doi:10.1016/S0092-8674(00)81717-1
43. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and autodocktools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi:10.1002/jcc.v30:16
44. Musfiroh I, Muchtaridi M, Muhtadi A, et al. Cytotoxicity studies of xanthorrhizol and its mechanism using molecular docking simulation and pharmacophore modelling. J Appl Pharm Sci. 2013;3(06):007–015.
45. Celik L, Lund JDD, Schiøtt B. Conformational Dynamics of the estrogen receptor α: molecular dynamics simulations of the influence of binding site structure on protein dynamics. Biochemistry. 2007;46(7):1743–1758. doi:10.1021/bi061656t
46. Mysinger MM, Carchia M, Irwin JJ, Shoichet BK, Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–6594. doi:10.1021/jm300687e
47. Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49(23):6789–6801. doi:10.1021/jm0608356
48. Gaulton A, Bellis LJ, Bento AP, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(Database issue):D1100–D1107.
49. Zamri A, Teruna H, Ikhtiarudin I. The influences of power variations on selectivity of synthesis reaction of 2ʹ-hydroxychalcone analogue under microwave irradiation. Molekul. 2016;11(2):299–307. doi:10.20884/1.jm.2016.11.2.220
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.