")
Back to Journals » OncoTargets and Therapy » Volume 12
Cystatin SN Affects Cell Proliferation by Regulating the ERα/PI3K/AKT/ERα Loopback Pathway in Breast Cancer
Authors Liu Y, Ma H, Wang Y, Du X, Yao J
Received 11 October 2019
Accepted for publication 27 November 2019
Published 23 December 2019 Volume 2019:12 Pages 11359—11369
DOI https://doi.org/10.2147/OTT.S234328
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Yanfang Liu,* Hong Ma,* Ye Wang, Xinyang Du, Jing Yao
Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Yao
Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Jiefang Road 1277, Wuhan 430022, Hubei Province, People’s Republic of China
Tel +86-18986271157
Email [email protected]
Background: Cystatin SN (CST1) has been reported to act as an oncogene in cancers, but its underlying mechanism remains unclear.
Methods: We performed Western blotting analyses to observe protein expression and conducted transwell invasion, wound healing, and colony formation assays to assess cell invasion, migration, and proliferation, respectively. We also performed cell cycle analyses by flow cytometry to determine the role of CST1 in the cell cycle. In vivo experiments used subcutaneous tumor models in BALB/c-nu athymic female mice to evaluate the effect of CST1 on tumor growth.
Results: Western blotting analyses showed that CST1 was upregulated in ER+ breast cancer cells such as MCF7, T47D, and BT474. CST1 knockdown led to slower cell growth and inhibited the G1 to S phase transition in ER+ breast cancer cells. In vivo experiments showed that CST1 deletion inhibited tumor growth, and led to decreased expression of estrogen receptor α (ERα) and p-AKT. In vitro experiments showed that the over-expression of CST1 led to the upregulation of ERα, and inhibition of CST1 inhibited the expression of ERα. Western blotting analyses showed that CST1 regulated the activity of the PI3K/AKT signaling pathway in breast cancer cells. We confirmed that CST1 acted as an oncogene in ER+ breast cancer by regulating the ERα/PI3K/AKT/ERα loopback pathway.
Conclusion: CST1 acts as an oncogene in ER+ breast cancer, and CST1 contributes to cancer development by regulating the ERα/PI3K/AKT/ERα loopback pathway in ER+ breast cancer. Our findings indicate that CST1 could be a significant therapeutic target for ER+ breast cancer patients. Our discovery should inspire further studies on the role of CST1 in cancers.
Keywords: ERα, CST1, breast cancer, cancer, PI3K/AKT signaling pathway
Introduction
Breast cancer is common among women worldwide and is the leading cause of cancer deaths in women.1,2 About 70% of breast cancers are estrogen receptor-positive (ER+). Studies have shown that the estrogen receptor is essential for the development of luminal breast cancers types,3 and the activation and upregulation of estrogen receptor α (ERα) signaling promote tumorigenesis and tumor invasion in breast cancer.4 Treatment of ER+ breast cancer relies mostly on endocrine therapies,5 mainly aromatase inhibitors, selective estrogen receptor modulators, and selective estrogen receptor down-regulators.6 These endocrine agents prolong the survival of ER+ breast cancer patients;7 however, one-third of patients who initially benefit from endocrine therapy frequently relapse after long-term treatment;8,9 therefore, it is significant for us to find a target for the treatment of ER+ breast cancer patients.
Cystatin SN (CST1) is a secretory protein belonging to the type 2 cystatin family,10 which affects the cell cycle, cell senescence, tumor formation, and cancer metastasis.11–19 CST1 is highly expressed in non-small-cell lung cancer, gastric cancer, pancreatic cancer and colorectal cancer, where it is significantly related to poor outcome, recurrence, metastasis and poor survival.12,14,16,20–23 In gastric cancer, CST1 leads to cell proliferation by targeting the Wnt signaling pathway;21 in colorectal cancer, CST1 knockdown suppresses tumor growth by affecting the IL-6 signaling pathway;24 in pancreatic cancer, knocking down CST1 reduces p-AKT expression, inhibits colony formation, and inhibits tumor growth in vitro.16 The function of CST1 is widely studied in various cancer types, however, the role of CST1 in breast cancer remains unclear.
In this research, we focus on the role of CST1 in breast cancer, and found that CST1 is significantly upregulated in ER+ breast cancer cells. Studies demonstrate that the upregulation of ERα and activation of the PI3K/AKT signaling pathway promote cell proliferation, tumor recurrence and metastasis.4,25 In this research, we found that CST1 knockdown inhibits the expression of ERα and the PI3K/AKT signaling. We reveal that CST1 regulates the ERα/PI3K/AKT/ERα signaling pathway in ER+ breast cancer.
This study aimed to uncover the mechanism of CST1 in ER+ breast cancer, in particular, the regulation between CST1 and the ERα/PI3K/AKT/ERα loopback pathway. Our findings demonstrate that CST1 may be a potential therapeutic target in ER+ breast cancer.
Materials and Methods
Cell Culture and Reagents
Human breast cancer cell lines were purchased from the Chinese Academy of Science Cell Bank (Shanghai, China). Human breast cancer cell lines MCF7, T47D, BT474, and SKBR3 were maintained in DMEM (Gibco) mixed with 10% fetal bovine serum (FBS) (Gibco, USA). MDA-MB231, BT549, MDA-MB468, and normal mammary epithelial cells (HBL-100) were maintained in RPMI 1640 (Gibco, USA) mixed with 10% FBS (Gibco, USA). Cells were cultivated in 5% CO2 at 37°C, in a humidified atmosphere. Short interfering RNA (siRNA) was obtained from Ruibo (Guangzhou, China). We used Lipofectamine 2000 (Invitrogen, USA) to transfect siCST1-1 (5ʹ-GGTGAAATCCAGGTGTCAA-3ʹ), siCST1-2 (5ʹ-CAGAAGGTCCCTGGTGAAA-3ʹ) and negative control siRNA into cells according to the manufacturer’s instructions. We also used Lipofectamine 2000 (Invitrogen, USA) to transfect pOE3.1-CAHyg-CST1 into MCF7 to obtain CST1-overexpressing cell lines. Two shRNA hairpins LV3(H1/GFP&Puro)-shCST1-1 (5ʹ-GAAGAACAGTTGTGCTCTTT-3ʹ), LV3(H1/GFP&Puro)-shCST1-2 (5ʹ-CCAGGCCATTCGCACCAGCCA-3ʹ) were used to knock down CST1 constitutively in MCF7 to produce shCST1 cells.
Plasmids and Stable Cell Line Generation
LV3(H1/GFP&Puro)-CST1 plasmids were purchased from GenePharma. MCF7 cells with stable knockdown of CST1 were generated by transfecting LV3(H1/GFP&Puro)-CST1 along with Lipofectamine 2000, followed by selection with Puro (Gibco, USA).
Transwell Invasion Assay
We used the Boyden chamber assay to detect cell invasion. Briefly, 600 µL of a serum-containing (10%) medium was added to each well of 24-well plates, and the chamber was added to the medium in the plates. A suspension of 1×105 cells in 100 μL of a serum-free medium was added to the top chamber with a Matrigel-coated membrane (8 μM pole size, BD Biosciences, Shanghai, China) and incubated at 37°C for 48 hrs. Invading cells on the lower membrane surface were fixed with 75% ethanol and stained with crystal violet. Cells were counted and photographed under an optical microscope, and cell counts were expressed as the mean ± SD from five random fields per well.
Western Blot Analysis
Cells were harvested in cell lysis buffer that contained protease and phosphatase inhibitors (CST, USA), and heated for 5–10 mins at 100°C. Equal quantities of proteins samples were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gels at a constant voltage of 90 V. The gels were then transferred onto nitrocellulose membranes, which were blocked and incubated with primary antibodies followed by secondary antibody. Proteins were visualized using enhanced chemiluminescence reagents. The antibodies used were as follows: anti-CST1 (diluted 1:500; Abcam, UK), anti-ERα (diluted 1:1000; Abcam, UK), anti-p-ERα (phospho Ser167; diluted 1:1000; CST, USA), anti-PI3K p85 alpha (diluted 1:1000; Abcam, UK), anti-p-PI3K p85 alpha (phospho Y607; diluted 1:500; CST, USA), anti-AKT (diluted 1:1000; CST, USA), anti-p-AKT (phospho Thr308; diluted 1:1000; CST, USA), anti-GAPDH (diluted 1:2000; Abcam, UK), mouse secondary antibody (diluted 1:4000; Gibco, USA), and rabbit secondary antibody (diluted 1:4000; Gibco, USA). Results were quantified using the Image J program (NIH, USA).
Cell Proliferation
Cell proliferation was monitored using the colony formation assay. Briefly, 500 cells per well of each plate were plated in six-well plates and left to generate, before being fixed with 75% ethanol, stained with crystal violet, and photographed 14 days later.
Flow Cytometric Cell Cycle Analyses
Single-cell suspensions were prepared for cell cycle analysis. We centrifuged cells at 1200 rpm for 5 mins, repeated twice, and subsequently fixed them in 75% ethanol (−20°C) for 2 h. The cells were then stained with propidium iodide (diluted to 50 μg/mL with phosphate-buffered saline (PBS), Sigma) in the dark and 100 μg/mL of DNase-free RNase A (Sigma) and analyzed on a BD FACSCanto™ II (BD Biosciences, USA) 30 mins later.
Wound-Healing Assay
Cells were plated in six-well plates and scratched with sterile 200 µL tips when the cell density reached about 90%. At 48 h after scratching, the cells were photographed under an optical microscope. The images were analyzed to determine the position of the migrating cells at the wound edges.
RNA Isolation and Real-Time (RT)‐PCR
Total RNA was extracted from cells (HBL-100, MCF7, T47D, BT474, SKBR3, MDA-MB231, MDA-MB468, and BT549) using TRIzol® reagent (Invitrogen, USA) according to manufacturer’s instructions. RNA (1 µg) was used for cDNA synthesis with primers. The reaction conditions were 37°C for 15 mins, 85°C for 5 s. RT‐PCR was carried out on an Applied Biosystems 7500 RT-PCR system (Applied Biosystems, USA) following standard procedures: 40 cycles of amplification at 95°C for 30 s, 95°C for 5 s, 60°C for 30 s, 95 °C for 15 s, 60 °C for 60 s, and 95°C for 15 s. The primers used were as follows. CST1: forward, 5′-CCTGTGCCTTCCATGAACAGCC-3′, reverse, 5′-GGGTGGTGGCTGGTGCCAATG3′. GAPDH: forward, 5ʹ-CGGATTTGGTCGTATTGGG-3ʹ, reverse, 5ʹ-CTGGAAGATGGTGATGGGATT-3ʹ.
Mouse Xenograft Studies
All experiments were approved by the Experimental Animal Center of Huazhong University of Science and Technology (Permission Number: SYXK 2016–0057), and all animals were managed according to the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006). Five-week-old BALB/c-nu female mice were bought from SHU BEI LI (China). To stimulate the growth of MCF7-shNC and MCF7-shCST1 tumors, 17-estradiol pellets (0.72 mg, Innovative Research of America) were implanted into mice. A week later, 5× 106 MCF7-shNC/MCF7-shCST1 cells were suspended in 100 μL of a mixture of PBS and Matrigel (1:1) (Sigma, USA), and injected subcutaneously. We used a Vernier caliper to measure tumors, and evaluated tumor volume using the following formula: V= length × width2/2 mm3.
Immunohistochemistry (IHC) in Xenograft Tumors
Tumor tissues from tumor-implanted mice were immunostained with antibodies according to the manufacturers’ instructions. All the results were photographed. The antibodies used were: anti-CST1 (diluted 1:100; Abcam, UK), anti-Ki-67 (diluted 1:200; CST, USA), anti-ERα (diluted 1:100; Abcam, UK), anti-AKT (diluted 1:200; CST, USA), and anti-p-AKT (diluted 1:100; CST, USA).
Statistical Analysis
All statistical analyses were carried out using the GraphPad Prism 8.0 software package. A two-tailed Student’s t-test was used to evaluate the significance of the differences between two groups of data. Differences were considered statistically significant at *p<0.05; **p<0.01; ***p<0.001; and ****p < 0.0001.
Results
CST1 Was Upregulated in ER+ Breast Cancer Cells
To investigate the expression of CST1, we performed Western blotting analyses in normal mammary epithelial cells (HBL-100), ER+ breast cancer cell lines (MCF7, T47D, and BT474), and ER− breast cancer cell lines (SKBR3, MDA-MB-231, MDA-MB-468, and BT549). The results showed that CST1 was upregulated more significantly at the protein level in ER+ breast cancer cell lines than in normal cells or ER− breast cancer cell lines (Figure 1A). We then analyzed the mRNA level of CST1 in all three cell types by RT-PCR. The results showed that the mRNA level of CST1 was higher in ER+ breast cancer cells than in HBL-100 cells or ER− breast cancer cells (Figure 1B). In summary, CST1 was up-regulated in ER+ breast cancer cells.
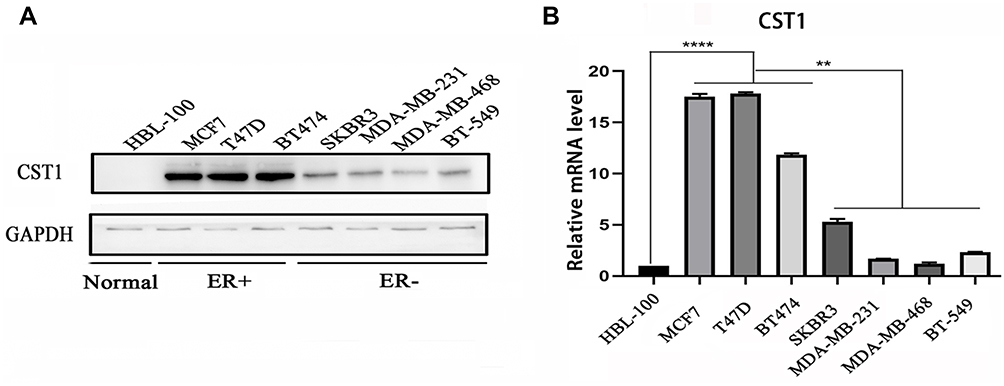 |
Figure 1 CST1 was overexpressed in ER+ breast cancer. (A) Western blotting analysis in normal mammary epithelial cells (HBL-100), ER+ breast cancer cell lines (MCF7, T47D, and BT474), and ER− breast cancer cell lines (SKBR3, MDA-MB-231, MDA-MB-468, and BT549) showing that CST1 was significantly overexpressed in ER+ breast cancer cells compared with normal mammary epithelial cells and ER− breast cancer cell lines. (B) RT-PCR to assess the mRNA levels of CST1 in HBL-100, ER+ breast cancer cell lines, and ER− breast cancer cell lines, showing that the mRNA level of CST1 was higher in ER+ breast cancer cells. Differences were considered statistically significant at **p<0.01; ****p<0.0001 (Student’s t-test). |
CST1 Functioned as an Oncogene in ER+ Breast Cancer
We knocked down CST1 in MCF7, T47D and BT474 ER+ breast cancer cells (Figure 2A). The colony formation assay showed that CST1 knockdown reduced the proliferation of these cells significantly (Figure 2B). Transwell invasion and wound-healing assays were performed on MCF7 to examine the extent of cell invasion and migration. We confirmed that the knockdown of CST1 inhibited cellular migration (Figure 2C). In the wound-healing assay, the gap between the scratched area was larger in the CST1 knockdown group than in the control group after 48 hrs (Figure 2D). To investigate the ability of CST1 to accelerate tumorigenesis in vivo, MCF7 cells stably depleted of CST1 were injected into athymic nude mice. Compared with the control group, the MCF7-deleted had smaller tumors (Figure 2E). IHC of xenograft tumor tissues revealed lower expression of Ki-67, CST1, ERα, p-AKT and AKT in CST1-depleted tumor tissues (Figure 2F). Taken together, these results indicate that CST1 had a strong function in ER+ breast cancer cells and possibly regulated the expression of ERα.
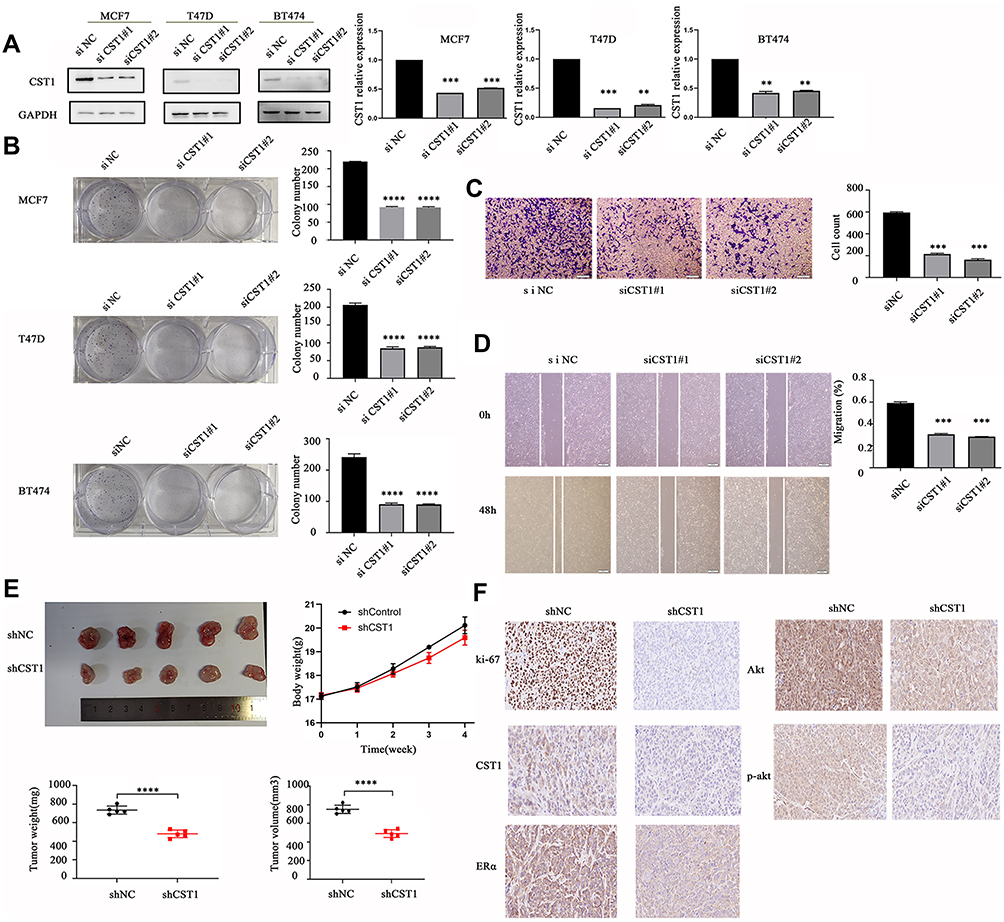 |
Figure 2 CST1 knockdown inhibited proliferation and cell migration in ER+ breast cancer. (A) Western blotting analysis of ER+ breast cancer cells (MCF7, T47D, and BT474) transfected with siCST1, verifying that CST1 was significantly knocked down. (B) Colony formation analysis to monitor cell proliferation. 500 cells per well from each plate were plated in six-well plates and left to generate. The cells were fixed with 75% ethanol, stained with crystal violet, and photographed 14 days later. The results showed a significant decrease in cell proliferation after knocking down CST1 in MCF7, T47D and BT474 cells. (C) Transwell invasion assay to assess cell migration in MCF7 breast cancer cells. We used the Boyden chamber assay to detect cell invasion. Briefly, in 24-well plates, 600 µL of a serum-containing (10%) medium per well was added, after which, the chamber was added to the medium in the plates. A suspension of 1×105 MCF7 cells in 100 μL of a serum-free medium was added to the top chamber with a Matrigel-coated membrane. Then, the cells were incubated at 37°C for 48 hrs. Invading cells on the lower membrane surface were fixed with 75% ethanol, and stained with crystal violet. The results showed a significant inhibition of cell invasion after knocking down CST1 in MCF7. (D) Wound-healing assay to assess cellular migration. MCF7 cells were plated in six-well plates and scratched with a sterile 200 µL tips when the cell density reached about 90%. At 48 hrs after scratching, the cells were photographed under an optical microscope. The images were analyzed to determine the position of the migrating cells at the wound edges. The results showed wider wound areas in CST1 knockdown MCF7 breast cancer cells. (E) Mouse xenograft studies were conducted to assess CST1’s ability to accelerate tumorigenesis in vivo. 5× 106 MCF7-shNC/MCF7-shCST1 cells were suspended in 100 μL of a mixture of PBS and Matrigel and injected subcutaneously into BALB/c-nu female mice. The body weight of the mice, the tumor weight and the tumor volume were measured and calculated. The results showed slower growth and lower volume of tumor in the CST1 stably depleted group compared with the control group. (F) IHC to assess the expression of Ki-67, CST1, ERα, AKT and p-AKT. Tumor tissues from tumor-implanted mice were immunostained with antibodies according to the manufacturers’ instructions. All results were photographed, and showed lower expression of Ki-67, CST1, ERα, AKT and p-AKT in the CST1-deleted group. Differences were considered statistically significant at **p<0.01, ***p<0.001, ****p<0.0001 (Student’s t-test). |
CST1 Affected the G1 to S Phase Transition in ER+ Breast Cancer Cells
Given that CST1 knockdown inhibited cell growth, we next assessed its effect on the cell cycle. Flow cytometry data revealed that the percentage of cells in the S phase decreased significantly in the siCST1-transfected group compared with the siNC-transfected cells; however, the opposite was observed for cells in the G1 phase, suggesting that the knockdown of CST1 inhibited cell proliferation by inhibiting the G1 to S phase transition (Figure 3). Together, these results suggest that the knockdown of CST1 inhibited cell growth by affecting the G1 to S phase transition in ER+ breast cancer cells.
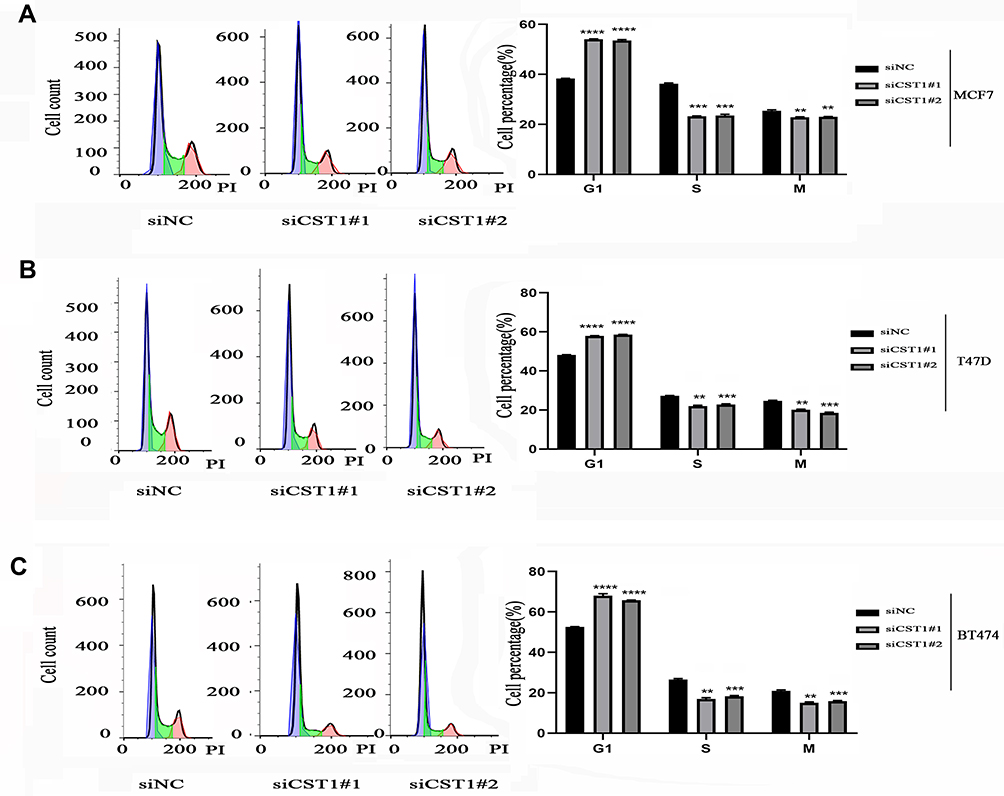 |
Figure 3 CST1 affected the G1 to S phase transition in ER+ breast cancer cells. Flow cytometry analysis was conducted to assess the effects of CST1 on the cell cycle. Single-cell suspensions were prepared. We centrifuged cells at 1200 rpm for 5 mins, repeated twice, then fixed them in 75% ethanol (−20°C) for 2 hrs. The cells were then stained with propidium iodide in the dark and 100 μg/mL of DNase-free RNase A, and analyzed on a BD FACSCanto™ II (BD Biosciences, USA) 30 mins later. (A) The results showed that CST1 knockdown inhibited the G1 to S phase transition in MCF7 breast cancer cells. (B) Cell cycle analyses showed that CST1 knockdown inhibited the G1 to S phase transition in T47D breast cancer cells. (C) Cell cycle analyses showed that CST1 knockdown inhibited the G1 to S phase transition in BT474 breast cancer cells. Differences were considered statistically significant at **p<0.01; ***p<0.001; ****p<0.0001 (Student’s t-test). |
CST1 Regulated ERα Expression in ER+ Breast Cancer
The IHC results showed that CST1-depleted tumor tissue had lower expression of ERα. To assess the ability of CST1 to regulate ERα expression, we knocked down and overexpressed CST1 in ER+ breast cancer cells. The results showed that knockdown of CST1 led to decreased expression of ERα (Figure 4A); correspondingly, overexpression of CST1 upregulated ERα expression (Figure 4B).
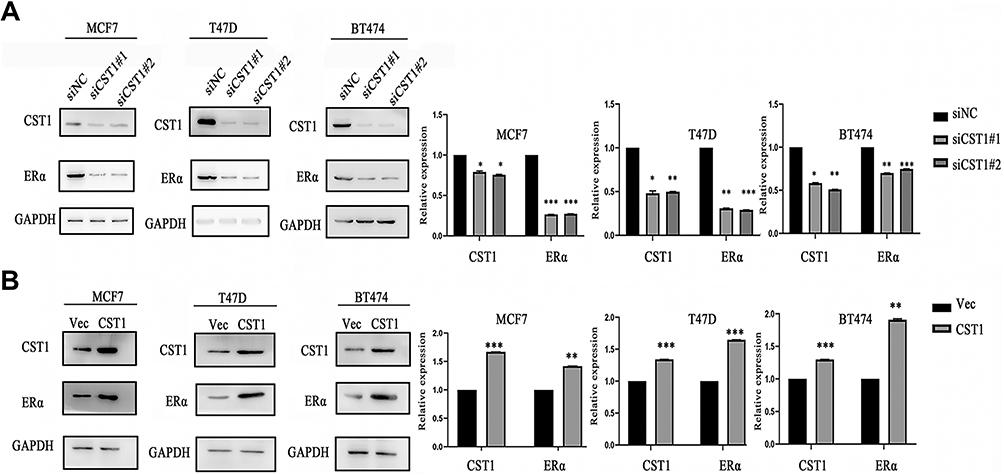 |
Figure 4 CST1 regulated the expression of ERα in ER+ breast cancer. (A) SiCST1 was transfected to MCF7, T47D, BT474 breast cancer cell lines, then we conducted Western blotting analysis to assess the change in ERα expression. CST1 knockdown led to decreased ERα expression in ER+ breast cancer cells. (B) Over-expressed CST1 plasmids were transfected to MCF7, T47D, and BT474 breast cancer cell lines, then Western blotting analysis was conducted to assess the expression of ERα. CST1 overexpression contributed to growing ERα expression in ER+ breast cancer cells. Differences were considered statistically significant at *p<0.05, **p<0.01, ***p<0.001 (Student’s t-test). |
CST1 Regulated the PI3K/AKT Signaling Pathway in Breast Cancer
Previous study of pancreatic cancer revealed that CST1 knockdown significantly reduced the expression of p-AKT,16 and our study showed that CST1 deletion inhibited the expression of p-AKT in tumor tissues, indicating a possible regulatory process between CST1 and the AKT-related signaling pathway. In breast cancer, the activation of many AKT-related signaling pathways contributed to the development of breast cancer, affecting cell proliferation and metastasis.26–32 Common AKT-related signaling pathways that affect the spread of cancer cells include the PI3K/AKT/mTOR signaling.31,32 We speculated that CST1 regulated PI3K/AKT signaling pathway in breast cancer. To avoid interference of hormone receptors, we conducted Western blotting analyses using three-negative breast cancer cells (estrogen receptor-negative, progesterone receptor-negative, and erbb2 negative; MDA-MB231, BT549 and MDA-MB468). The results showed that CST1 knockdown inhibited the expression of p-PI3K and p-AKT (Figure 5A). Consistent with this, the Western blotting analyses showed that CST1 overexpression led to increased expression of p-PI3K and p-AKT (Figure 5B), indicating that CST1 overexpression contributed to the activation of PI3K/AKT signaling pathway in ER− breast cancer cells. Together, CST1 led to the activation of PI3K/AKT signaling pathway.
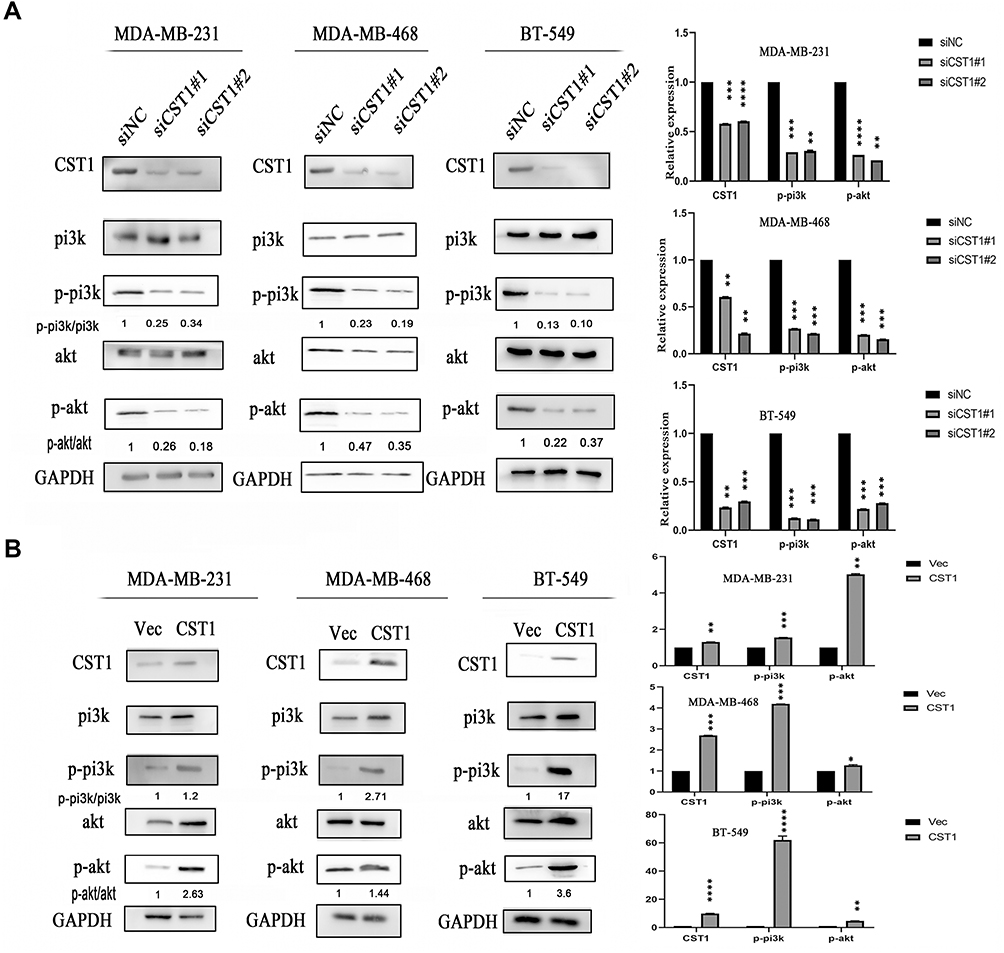 |
Figure 5 CST1 regulated PI3K/AKT signaling pathway in ER− breast cancer. (A) SiCST1 was transfected to ER− breast cancer cells (MDA-MB-231, MDA-MB-468, and BT-549), then we conducted Western blotting analysis to assess the change of PI3K/AKT signaling pathway. (B) The over-expressed CST1 plasmids were transfected to ER− breast cancer cells, then the Western blotting analysis was conducted to assess the expression of the PI3K/AKT signaling pathway. Differences were considered statistically significant at *p<0.05; **p<0.01; ***p<0.001; ****P<0.0001 (Student’s t-test). |
CST1 Regulated the ERα/PI3K/AKT/ERα Loopback Pathway in ER+ Breast Cancer
CST1 Regulated the ERα/PI3K/AKT Signaling Pathway in ER+ Breast Cancer Cells
Our results showed that CST1 regulated the expression of ERα, and also regulated the activity of the PI3K/AKT signaling pathway. Studies have shown that there is a crosstalk between ERα and the PI3K/AKT signaling pathway in ER+ breast cancer.33 Based on our experimental results and previous studies, we hypothesized that CST1 affected the proliferation of breast cancer cells in ER+ breast cancer by regulating the ERα/PI3K/AKT/ERα loopback signaling pathway. Treatment with E2 has been shown to activate ERα;3,34 therefore, to verify that CST1 regulated the ERα/PI3K/AKT pathway, we treated ER+ breast cancer cells with 10 nm of E2 and/or siCST1. Our results showed that siCST1 treatment led to the downregulation of ERα, p-PI3K, and p-AKT compared with the control group (Figure 6A). Compared with controls, E2 treatment increased the activity of the PI3K/AKT signaling pathway (Figure 6A), indicating that ERα was upstream of the PI3K/AKT signaling pathway. Transfection of siCST1 reduced ERα expression and decreased the activity of the PI3K/AKT signaling pathway (Figure 6A), demonstrating that CST1 regulated the ERα/PI3K/AKT signaling pathway. Hence, CST1 regulated the ERα/PI3K/AKT signaling pathway in both the absence and presence of E2 in ER+ breast cancer cells.
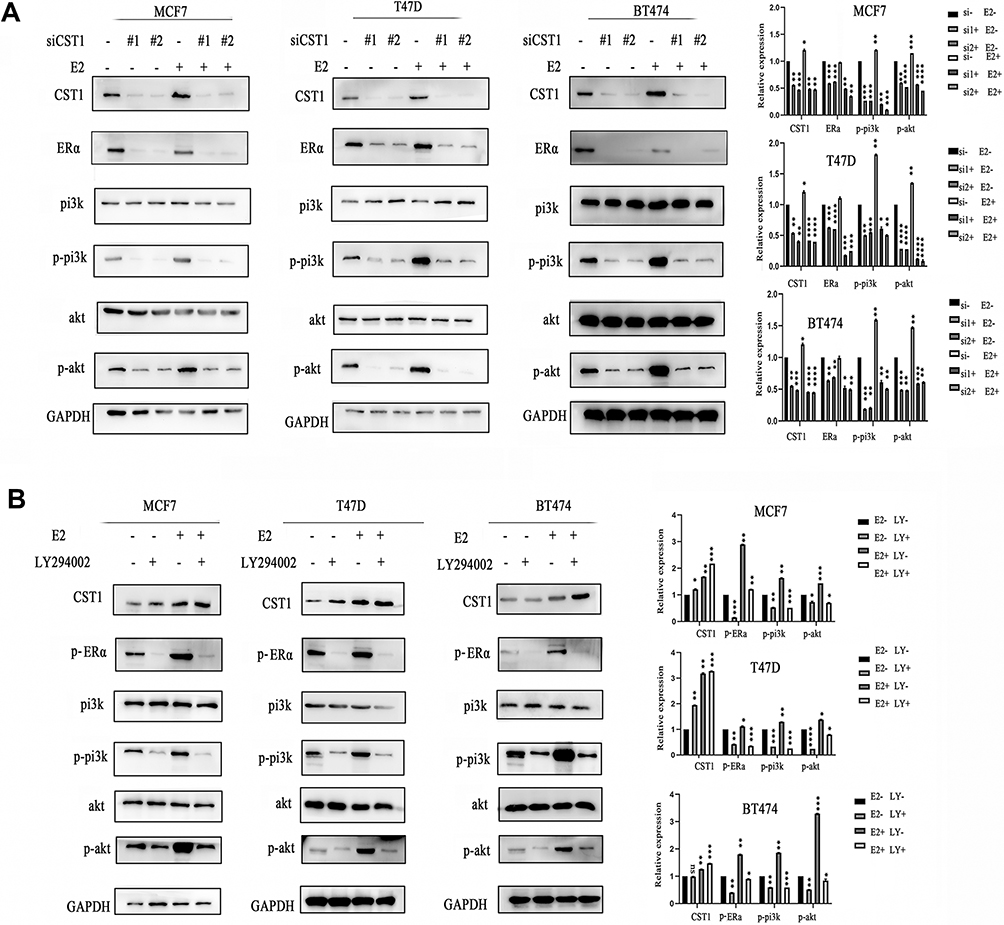 |
Figure 6 CST1 regulated the ERα/PI3K/AKT/ERα loopback pathway in ER+ breast cancer. (A) ER+ breast cancer cells were treated with 10 nm of E2 and/or siCST1, Western blotting analysis was conducted to assess the regulation by CST1 of the ERα/PI3K/AKT signaling pathway. The results showed that CST1 regulated the expression of the ERα/PI3K/AKT signaling pathway in ER+ breast cancer cells. (B) ER+ breast cancer cells (MCF7, T47D, and BT474) were treated with E2 and/or LY294002. Western blotting analysis showed a reciprocal regulation between ERα and the PI3K/AKT signaling pathway in ER+ breast cancer cells. Differences were considered statistically significant at *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (Student’s t-test). |
A Reciprocal Regulation Existed Between ERα and the PI3K/AKT Signaling Pathway
The above experiments confirmed that ERα regulated the PI3K/AKT signaling pathway in ER+ breast cancer. A previous study showed that a crosstalk exists between ERα and this pathway.33 To check for a reciprocal regulation, we treated ER+ breast cancer cells (MCF7, T47D, and BT474) with E2 and/or LY294002 (a selective PI3K inhibitor). The results showed that LY294002 exposure decreased the expression of p-ERα, p-PI3K and p-AKT in the presence or absence of E2, indicating that the PI3K/AKT signaling pathway regulated p-ERα expression (Figure 6B). These results confirm the crosstalk between ERα and the PI3K/AKT signaling pathway, and demonstrate that CST1 regulates the ERα/PI3K/AKT/ERα loopback pathway in ER+ breast cancer.
Discussion
CST1 is regarded as a tumor biomarker in various cancer types, and its high expression is significantly related to poor outcome, recurrence, metastasis and poor survival in cancer patients.14,16,21–23 Though CST1 is widely studied in cancers, however, the role of CST1 in ER+ breast cancer does not receive enough attention from researchers. In this research, we found that CST1 is significantly upregulated in ER+ breast cancer cells. Knocking down CST1 in ER+ breast cancer cells inhibit cell proliferation, cell migration and invasion, and inhibits the number of S phase cells. CST1 knockdown also inhibits tumor growth in vivo. Our research demonstrates that CST1 might serve as a potential therapeutic target in ER+ breast cancer.
In vivo experiments, CST1 depletion decreases the expression of ERα and p-AKT in tumor tissues. We speculate that CST1 regulates the expression of ERα and the AKT-related signaling pathways. Western blotting analysis confirms that the upregulation of CST1 leads to high expression of ERα, and inhibition of CST1 results in decreased ERα expression. Studies show that the activation of AKT-related signaling pathways contributes to the development of cancers.26–32 Common AKT-related signaling pathways that affect cell proliferation include the PI3K/AKT signaling pathway.31,32 Based on our experimental results and previous studies, we speculate that CST1 affecting cell proliferation in breast cancer is related to the PI3K/AKT signaling pathway. The speculation is verified in three-negative breast cancer cells. We confirm that CST1 regulates the activity of the PI3K/AKT signaling pathway.
Studies show that activation of ERα contributes to ERα-mediated transcriptional targets, and upregulation of the PI3K/AKT signaling pathway leads to cell proliferation, both of which are important reasons for cancer progression and metastasis in ER+ breast cancer patients.8,9,35–37 The crosstalk between ERα and the PI3K/AKT signaling pathway promotes tumor progression,33 and limits the efficacy of cancer therapy.25,35,36,38 In this study, we report that CST1 performs an oncogenic function in ER + breast cancer by regulating the ERα/PI3K/AKT/ERα loopback pathway (Figure 7).
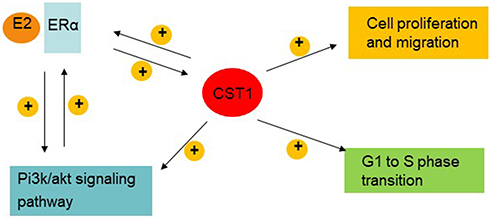 |
Figure 7 CST1 model affected cell proliferation by regulating the ERα/PI3K/AKT/ERα loopback pathway in breast cancer. A positive loopback regulation exists between ERα and the PI3K/AKT signaling pathway in ER+ breast cancer cells. Also, there is mutual positive regulation between ERα and CST1. CST1 positively regulates the PI3K/AKT signaling pathway; functions as an oncogene, contributing to cell growth and migration; and also affects the G1 to S phase transition in ER+ breast cancer cells. |
CST1 is a secretory protein,10 however, this work considered only intracellular CST1; it remains unknown whether extracellular CST1 would have the same effects. This should be the subject of our further research. In this research, we confirm that CST1 regulates the PI3K/AKT signaling pathway in breast cancer cells; however, the mechanism of CST1 regulating the PI3K/AKT signaling pathway, the interaction between CST and AKT, and the interaction domain, all of which are not studied in this research because of time constraints. Recently, we are focusing on the relationship between CST1 and ERα, but we do not find any interaction.
In conclusion, CST1 is upregulated and has an oncogenic role in ER+ breast cancer. CST1 contributes to cell proliferation by regulating the ERα/PI3K/AKT/ERα loopback pathway. Targeting CST1 might be a potentially effective way to treat ER+ breast cancer. Our results should form a basis for further studies on the role of CST1 in cancers.
Acknowledgment
This work was supported by the Provincial Natural Science Foundation of Hubei (No. 2015BCA270).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107
2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. doi:10.3322/caac.21349
3. Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378(9805):1812–1823. doi:10.1016/s0140-6736(11)61539-0
4. Strom A, Hartman J, Foster JS, et al. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci U S A. 2004;101(6):1566–1571. doi:10.1073/pnas.0308319100
5. Turner NC, Neven P, Loibl S, et al. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet. 2017;389(10087):2403–2414. doi:10.1016/s0140-6736(16)32419-9
6. Najim O, Seghers S, Sergoynne L, et al. The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: a systematic review and meta-analysis of randomized and non-randomized trials. Biochimica Biophysica Acta Rev Cancer. 2019:188315. doi:10.1016/j.bbcan.2019.188315
7. Cuzick J. The ATAC trial: the vanguard trial for use of aromatase inhibitors in early breast cancer. Expert Rev Anticancer Ther. 2007;7(8):1089–1094. doi:10.1586/14737140.7.8.1089
8. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nature Rev Cancer. 2009;9(9):631–643. doi:10.1038/nrc2713
9. Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–1717. doi:10.1016/s0140-6736(05)66544-0
10. Oh BM, Lee SJ, Cho HJ, et al. Cystatin SN inhibits auranofin-induced cell death by autophagic induction and ROS regulation via glutathione reductase activity in colorectal cancer. Cell Death Dis. 2017;8(3):e2682. doi:10.1038/cddis.2017.100
11. de Sousa-pereira P, Amado F, Abrantes J, et al. An evolutionary perspective of mammal salivary peptide families: cystatins, histatins, statherin and PRPs. Arch Oral Biol. 2013;58(5):451–458. doi:10.1016/j.archoralbio.2012.12.011
12. Dai DN, Li Y, Chen B, et al. Elevated expression of CST1 promotes breast cancer progression and predicts a poor prognosis. J Mol Med (Berl). 2017;95(8):873–886. doi:10.1007/s00109-017-1537-1
13. de Sousa-pereira P, Abrantes J, Pinheiro A, et al. Evolution of C, D and S-type cystatins in mammals: an extensive gene duplication in primates. PLoS One. 2014;9(10):e109050. doi:10.1371/journal.pone.0109050
14. Cao X, Li Y, Luo RZ, et al. Expression of Cystatin SN significantly correlates with recurrence, metastasis, and survival duration in surgically resected non-small cell lung cancer patients. Sci Rep. 2015;5:8230. doi:10.1038/srep08230
15. Fukuoka A, Matsushita K, Morikawa T, et al. Human cystatin SN is an endogenous protease inhibitor that prevents allergic rhinitis. J Allergy Clin Immunol. 2018. doi:10.1016/j.jaci.2018.06.035
16. Jiang J, Liu HL, Liu ZH, et al. Identification of cystatin SN as a novel biomarker for pancreatic cancer. Tumour Biol. 2015;36(5):3903–3910. doi:10.1007/s13277-014-3033-3
17. Heinrich PC, Behrmann I, Muller-Newen G, et al. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi:10.1042/bj3340297
18. Li T, Xiong Q, Zou Z, et al. Prognostic significance of cystatin SN associated nomograms in patients with colorectal cancer. Oncotarget. 2017;8(70):115153–115163. doi:10.18632/oncotarget.23041
19. Dsamou M, Palicki O, Septier C, et al. Salivary protein profiles and sensitivity to the bitter taste of caffeine. Chem Senses. 2012;37(1):87–95. doi:10.1093/chemse/bjr070
20. Bahreini A, Li Z, Wang P, et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017;19(1):60. doi:10.1186/s13058-017-0851-4
21. Choi EH, Kim JT, Kim JH, et al. Upregulation of the cysteine protease inhibitor, cystatin SN, contributes to cell proliferation and cathepsin inhibition in gastric cancer. Clin Chim Acta. 2009;406(1–2):45–51. doi:10.1016/j.cca.2009.05.008
22. Oh BM, Lee SJ, Cho HJ, et al. Cystatin SN inhibits auranofin-induced cell death by autophagic induction and ROS regulation via glutathione reductase activity in colorectal cancer. Cell Death Dis. 2017;8(9):e3053. doi:10.1038/cddis.2017.446
23. Mullapudi N, Ye B, Suzuki M, et al. Genome wide methylome alterations in lung cancer. PLoS One. 2015;10(12):e0143826. doi:10.1371/journal.pone.0143826
24. Oh SS, Park S, Lee KW, et al. Extracellular cystatin SN and cathepsin B prevent cellular senescence by inhibiting abnormal glycogen accumulation. Cell Death Dis. 2017;8(4):e2729. doi:10.1038/cddis.2017.153
25. Pierobon M, Ramos C, Wong S, et al. Enrichment of PI3K-AKT-mTOR pathway activation in hepatic metastases from breast cancer. Clin Cancer Res. 2017;23(16):4919–4928. doi:10.1158/1078-0432.ccr-16-2656
26. Pascual J, Turner NC. Targeting the PI3-kinase pathway in triple negative breast cancer. Ann Oncol. 2019. doi:10.1093/annonc/mdz133
27. Han F, Li CF, Cai Z, et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat Commun. 2018;9(1):4728. doi:10.1038/s41467-018-07188-9
28. He L, Liu X, Yang J, et al. Imbalance of the reciprocally inhibitory loop between the ubiquitin-specific protease USP43 and EGFR/PI3K/AKT drives breast carcinogenesis. Cell Res. 2018;28(9):934–951. doi:10.1038/s41422-018-0079-6
29. Geyer FC, Li A, Papanastasiou AD, et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat Commun. 2018;9(1):1816. doi:10.1038/s41467-018-04128-5
30. Mendez-Pertuz M, Martinez P, Blanco-Aparicio C, et al. Modulation of telomere protection by the PI3K/AKT pathway. Nat Commun. 2017;8(1):1278. doi:10.1038/s41467-017-01329-2
31. Burris HA
32. Dey N, De P, Leyland-Jones B. PI3K-AKT-mTOR inhibitors in breast cancers: from tumor cell signaling to clinical trials. Pharmacol Ther. 2017;175:91–106. doi:10.1016/j.pharmthera.2017.02.037
33. Sun M, Paciga JE, Feldman RI, et al. Phosphatidylinositol-3-OH kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res. 2001;61(16):5985–5991.
34. Kumar R, Zakharov MN, Khan SH, et al. The dynamic structure of the estrogen receptor. J Amino Acids. 2011;2011:812540. doi:10.4061/2011/812540
35. Bosch A, Li Z, Bergamaschi A, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med. 2015;7(283):283ra51. doi:10.1126/scitranslmed.aaa4442
36. Toska E, Osmanbeyoglu HU, Castel P, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017;355(6331):1324–1330. doi:10.1126/science.aah6893
37. Ding J, Wang X, Zhang Y, et al. Inhibition of BTF3 sensitizes luminal breast cancer cells to PI3Kalpha inhibition through the transcriptional regulation of ERalpha. Cancer Lett. 2019;440–441:54–63. doi:10.1016/j.canlet.2018.09.030
38. Basho RK, Gilcrease M, Murthy RK, et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol. 2017;3(4):509–515. doi:10.1001/jamaoncol.2016.5281
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.