")
Back to Journals » International Journal of Nanomedicine » Volume 12
Cyclic RGD peptide-modified liposomal drug delivery system for targeted oral apatinib administration: enhanced cellular uptake and improved therapeutic effects
Authors Song Z , Lin Y, Zhang X, Feng C, Lu Y, Gao Y, Dong C
Received 24 October 2016
Accepted for publication 31 January 2017
Published 10 March 2017 Volume 2017:12 Pages 1941—1958
DOI https://doi.org/10.2147/IJN.S125573
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Zhiwang Song, Yun Lin, Xia Zhang, Chan Feng, Yonglin Lu, Yong Gao, Chunyan Dong
Department of Oncology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China
Abstract: Apatinib is an oral tyrosine kinase inhibitor, which selectively targets vascular endothelial growth factor receptor 2 and has the potential to treat many tumors therapeutically. Cyclic arginylglycylaspartic acid (cRGD)- and polyethylene glycol (PEG)-modified liposomes (cRGD-Lipo-PEG) were constructed to act as a targeted delivery system for the delivery of apatinib to the human colonic cancer cell line, HCT116. These cRGD-modified liposomes specifically recognized integrin αvβ3 and exhibited greater uptake efficiency with respect to delivering liposomes into HCT116 cells when compared to nontargeted liposomes (Lipo-PEG), as well as greater death of tumor cells and apoptosis. The mechanism by which cRGD-Lipo-PEG targets cells was elucidated further with competition assays. To determine the anticancer efficacy in vivo, nude mice were implanted with HCT116 xenografts and treated with apatinib-loaded liposomes or free apatinib intravenously or via intragastric administration. The active and passive targeting of cRGD-Lipo-PEG led to significant tumor treatment targeting ability, better inhibition of tumor growth, and less toxicity when compared with treatments using uncombined apatinib. The results presented strongly support the case for cRGD-Lipo-PEG representing a targeted delivery system for apatinib in the treatment of colonic cancer.
Keywords: integrin αvβ3, cRGD, targeted oral therapy, apatinib, colorectal cancer
Introduction
Colorectal cancer (CRC) is the third most prevalent cancer, with the fourth highest mortality rate over all age groups globally. Almost 694,000 people die from the disease annually. The incidence of CRC in young adults has been rising during recent years.1 Along with the improvements in science, although numerous new diagnosis tools and specific drugs for therapy of CRC emerge continually,2 the 5-year survival rate of patients with late-stage CRC still remains only 12%.3
Vascular endothelial growth factor receptor (VEGFR) has been closely investigated as a regulator of many signaling pathways that control angiogenesis and tumor growth, with the aim of developing drugs with novel molecular targets. Examples are receptor-specific antibodies and low-molecular-weight chemical inhibitors of angiogenic receptor tyrosine kinase inhibitors (TKIs). VEGFR inhibitors have also been recently developed.4
Apatinib is an orally administered TKI, which selectively targets VEGFR-2, similarly to vatalanib, but with a higher binding affinity and greater efficacy than either vatalanib or sorafenib.5,6 It was found to inhibit the efflux function of the multiple ATP-binding cassette transporter.7 Clinical trials have confirmed the efficacy and safety of apatinib for the therapeutic treatment of patients with advanced gastric cancer,8 non-small cell lung cancer,9 breast cancer,10 colon cancer,4 and hepatocellular carcinoma.11 However, apatinib has some unfavorable pharmacokinetic properties: poor aqueous solubility, poor oral bioavailability after oral administration (only 9.24% in male dogs and 15.4% in female dogs), and an extensive tissue distribution, with 92.4% protein binding.12
Currently, nanoparticle drug delivery systems, such as solid lipid nanoparticles, micelles, liposomes, microemulsions, and dendrimers have been the major focus of research aimed at finding drug targets with improved druggability, especially in cancer chemotherapy.13 Among these, liposomes have been studied extensively in clinical trials as potential carriers of chemotherapeutic agents with decreased side effects and enhanced efficacy in treating cancer.14 Many liposomal products (amphotericin B: eg, Abelcet®, Amphotec®, and AmBisome®; doxorubicin: eg, Doxil® and Caelyx®; and daunorubicin: eg, DaunoXome®) have been approved by the US Food and Drug Administration.15 Drug-loaded liposomes typically have a prolonged circulation time in vivo and can be passively accumulated in tumors via effects involving enhanced permeability and retention (EPR).16 However, the liposomal effects can be limited due to their rapid clearance from the body via the reticuloendothelial system. Polyethylene glycol (PEG)-modified liposomes have been routinely used in order to avoid this problem, as these allow enhanced colloidal stability in the systemic circulation.17
Apart from passive accumulation, active targeting can be enhanced by modifying liposomes with various ligands, such as antibodies, peptides, or integrin ligands.18 The tripeptide, arginine–glycine–aspartic acid (RGD), is the most studied of the integrin ligands that can be specifically recognized by integrin αvβ3. Integrin αvβ3 is overexpressed in most tumors and sprouting tumor vessels. It also mediates the progression of tumor angiogenesis, growth, and metastasis. Hence, the modification of liposomes with RGD to achieve targeted drug delivery to tumors and to the tumor neovasculature is considered to be a promising strategy both for the detection of cancers and for cancer therapy.19 In addition, multiple cyclic RGD peptides, such as c(RGDfK), c(RGDyK), RGD4C, and RGD10, have high affinity for integrin αvβ3 receptor antagonists and have been investigated widely as active targeting moieties in cancer therapy (Scheme 1).20,21
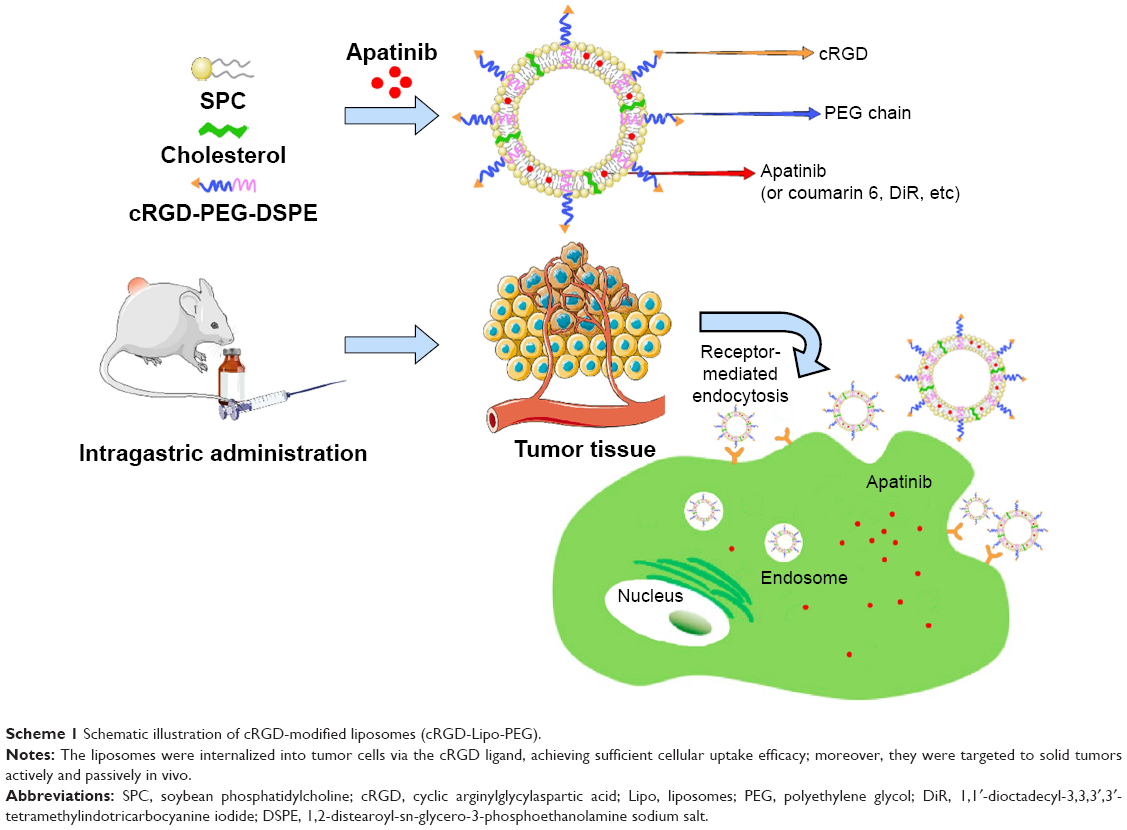 | Scheme 1 Schematic illustration of cRGD-modified liposomes (cRGD-Lipo-PEG). |
Here, we describe tumor-targeted liposomes involving the coupling of a cyclic arginylglycylaspartic acid (cRGD), cyclo(arginine–glycine–aspartic acid-D-Phenylalanine-Cystine [Arg-Gly-Asp-D-Phe-Cys])(cRGDfC), to the ends of N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt (DSPE-PEG2000). The cyclic RGD variant with high affinity for αvβ3 is not only specific but also of high affinity for αvβ3-overexpressing cancer cells. Then, we constructed cRGD-modified liposomes and loaded them with apatinib; these liposomes were termed cRGD-Lipo-PEG/Apa. Their physiological characteristics and cytotoxicity have been described in detail and cRGD-mediated cellular uptake is evaluated in cancerous cells. Finally, this recently developed drug delivery system is also shown to provide enhanced apatinib delivery efficiency in HCT116 tumor cells and greater antitumor efficacy in animal models of colonic carcinoma.
Materials and methods
Materials, cells, and animals
Soybean phosphatidylcholine (SPC) was obtained from Shanghai Advanced Vehicle Technology Pharmaceuticals Ltd (Shanghai, China), DSPE-PEG2000 was obtained from Lipoid GmBH (Ludwigshafen, Germany), DSPE-PEG2000-maleimide (DSPE-PEG2000-Mal) was purchased from Advanced Vehicle Technology Co, Ltd (Shanghai, China). Cholesterol was obtained from Tokyo Chemical Industry Co, Ltd (Tokyo, Japan). Coumarin 6 and Annexin V–FITC/PI apoptosis detection kit were purchased from Beyotime Institute of Biotechnology (Haimen, China). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) was obtained from AAT Bioquest Inc (Sunnyvale, CA, USA). Fetal bovine serum (FBS) and McCoy’s 5A (Modified) medium were purchased from Gibco (Invitrogen, Waltham, MA, USA). Integrin β3 rabbit monoclonal antibodies (mAbs) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Mesylate apatinib was provided by Shanghai Hengrui Pharmaceutical Co Ltd (Shanghai, China). For in vitro assays, the apatinib solution was prepared in dimethyl sulfoxide (DMSO). For in vivo studies, apatinib was diluted in 0.5% (w/v) carboxymethyl cellulose–Na (CMC-Na) solution. cRGDfC was synthesized according to standard solid phase peptide synthesis by SynPeptide Co, Ltd (Shanghai, China) with a purity of 95%, as confirmed by high-performance liquid chromatography (HPLC). All the other chemicals and reagents used in this study were of analytical or HPLC grade and provided by Sinopharm Chemical Reagent Co, Ltd (Shanghai, China).
A human colon cancer cell line (HCT116) and a human embryonic kidney 293T cell line were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in McCoy’s 5A (Modified) medium with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified incubator (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C with 5% CO2.
Female BALB/c nude mice (4 wk old) were supplied by the Shanghai Laboratory Animal Center (Shanghai, China). The animals were maintained on a 12 h light/dark cycle with free access to food and water. This study was approved by the Institutional Animal Care and Use Committee of Tongji University. All the experimental protocols were performed in accordance with the national guidelines of the principles for care and use of laboratory animals.
Synthesis of DSPE-PEG2000-cRGD
The targeted liposomal drug delivery system modified with cyclic RGD, cRGD-Lipo-PEG, was prepared using the thiol-maleimide coupling reaction, by reacting cRGD peptide so that it attached to the distal ends of DSPE-PEG2000-Mal due to the reaction between the thiol and maleimide groups. To synthesize the cRGD-DSPE-PEG2000, DSPE-PEG2000-Mal (1 mmol) was dissolved in 0.1 M PBS (pH 7.2, 0.15 M NaCl, 2 mM EDTA) containing triethylamine (TEA; 2 mmol). Two molar equivalents of cRGD peptides (2 mmol) per maleimide group were added to the DSPE-PEG2000-Mal solution with stirring, and the solution was made to react further in the presence of argon for 12 h at room temperature in the dark. The final solution was dialyzed against ultrapure water using a dialysis membrane (molecular weight cutoff [MWCO] =1,000 Da) to purify the products and to remove any unreacted cRGD peptides. Following dialysis, the final product was lyophilized overnight and stored at −80°C until further use. The product was also confirmed by proton nuclear magnetic resonance (1H-NMR) spectroscopy.
Preparation of liposomes
Liposomes were prepared by the thin-film hydration method.22 Briefly, the liposome compositions of nontargeted liposomes (Lipo-PEG) and cRGD-modified targeted liposomes (cRGD-Lipo-PEG) were SPC/cholesterol/DSPE-PEG2000 5.8:1:4.2 (molar ratio) and SPC/cholesterol/cRGD-DSPE-PEG2000 5.8:1:5.5 (molar ratio). All regents were first dissolved in 4 mL chloroform in a rotary flask. Then, the organic solvent was formed into a transparent thin film by evaporation under vacuum at 37°C, and the film was further dried under vacuum for 8 h to remove traces of organic reagents. The dried thin film was then hydrated in 5% (w/v) glucose solution (pH 7.4) containing apatinib, coumarin 6, or DiR at 40°C and then sonicated for 30 min. The suspension was then dispersed and extruded through a polycarbonate membrane of 200 nm pore size 25 times at 25°C, using an Avanti Polar lipid apparatus, to form smaller and more stable liposomes. The residual free apatinib in liposomal suspensions was removed using column chromatography (Sephadex G-50 column).
Characterization of liposomes and drug loading
The determination of the mean diameters and zeta potentials of the two kinds of liposomes was performed using a Zetasizer analyzer (Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK). Data were determined independently three times for each sample. The apatinib concentration in liposomes was determined by HPLC.
In brief, a C18 column (Diamonsil Plus 5 μm, 250×4.6 mm; flow rate: 1 mL/min; and column temperature; 40°C) was used, and the mobile phase was isocratic with 67% solvent A (1% TEA in distilled water) and 37% solvent B (methyl alcohol). Apatinib concentrations were determined by measuring the absorbance of the samples at 260 nm.
The apatinib loading capacity (LC) was calculated using the following equation:
|
|
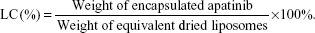
The apatinib encapsulation efficiency (EE) was calculated by the following equation:
|
|
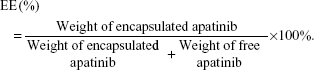
Stability of liposomes in vitro
In order to demonstrate the stability of liposomes, particle sizes and potential charges were evaluated for 12 d when liposomes were maintained at 4°C. Briefly, complexes were prepared according to the method described earlier; then, at different time intervals, 100 μL of sample was diluted to 900 μL with 5% (w/v) glucose solution for determination of particle size and potential charges using a Zetasizer analyzer (Zetasizer Nano ZS90; Malvern Instruments).
Investigation of drug release in vitro
Apatinib release was studied using a dialysis method in vitro. In detail, 0.5 mL of each apatinib-loaded liposome or free apatinib (apatinib concentration: 0.8 μg/mL) was placed into a dialysis device (10K MWCO; Pierce Inc., [Appleton, WI, USA]). Then the device was immersed in 200 mL PBS (pH 5.6 and pH 7.4) containing 0.5% (w/w) Tween 80 as the dissolution medium and incubated in an incubator shaker (THZ-D; Tai Cang Scientific Equipment Plant, [Suzhou City, Jiangsu Province, People’s Republic of China]) at 37°C for 80 h with mild and horizontal oscillation at 60 rpm. At selected time intervals, 1 mL release medium was taken from the bottle and replaced with 1 mL fresh medium. Samples were then diluted with methyl alcohol and the concentration of apatinib was determined by HPLC. Experiments were performed in triplicate.
Receptor expression assay using Western blotting
Western blotting was performed to study the expression of β3 in HCT116 cells and solid tumors. HCT116 cells and tumors were lysed. Released proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and were transferred to nitrocellulose membranes using semidry blotting with 10% (v/v) goat serum as the blocking agent. Primary antibodies (integrin in β3 rabbit mAbs) were used for Western blotting analysis for overnight incubation at 4°C.
Investigation of cellular uptake in vitro
HCT116 cells were plated at a density of 2×105 cells per well in a 12-well plate. After 24 h incubation, coumarin 6-loaded liposomes, termed Lipo-PEG/C6 or cRGD-Lipo-PEG/C6, at the same final coumarin-6 concentration of 1 μg/mL, diluted in culture medium, were applied to each well and incubated for a further 4 h. After treatment with Lipo-PEG/C6 or cRGD-Lipo-PEG/C6, the cells were washed with cold PBS three times, fixed, and imaged using a Zeiss Axio Observer Z1 fluorescence microscope.
The enrichment of liposomes in HCT116 cells was also determined by flow cytometry. Briefly, HCT116 cells were incubated with coumarin 6-loaded liposomes, namely, Lipo-PEG/C6 or cRGD-Lipo-PEG/C6, at the same coumarin-6 concentration of 0.05 μg/mL (diluted in culture medium) for 4 h at 37°C. The cells were then harvested, washed three times with cold PBS (pH 7.4), tested by flow cytometry, and analyzed using FlowJo 7.6 software.
In competitive binding experiments, free cRGD peptides were used in order to measure cRGD ligand-mediated cellular uptake of cRGD-Lipo-PEG. Thirty minutes before cRGD-Lipo-PEG/C6 treatment, a 10-fold excess of free cRGD peptides were added to culture medium, followed by continued coincubation with cRGD-Lipo-PEG for another 4 h at 37°C. Then, the cells were treated as previously described.23 Moreover, 293T cells, which lack β3 receptor expression, were used as a control cell line for cellular uptake study.
Cytotoxicity evaluations in vitro
The cytotoxicity of free apatinib, blank liposomes, and apatinib-loaded cRGD-Lipo-PEG and Lipo-PEG, termed cRGD-Lipo-PEG/Apa and Lipo-PEG/Apa, against HCT116 cells was measured using the MTT assay. Briefly, cells were seeded into a 96-well plate at a density of 6×103 cells per well. After 24 h, the cells were incubated with free apatinib and apatinib-loaded liposomes at the same final concentrations for 48 h. Blank liposomes were also added at different concentrations as controls. Cells were then incubated with 20 μL MTT solution (5 mg/mL in PBS) for 4 h, medium was removed, and cells were dissolved with 200 μL DMSO; the absorbance was read at 570 and 490 nm with a microplate reader (Thermo Fisher Scientific). Cell viability (%) was calculated from the following formula:
|
|
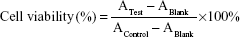
ATest, AControl, and ABlank represent the absorbances of the cells with different treatments, untreated cells, and blank culture media, respectively. Each experiment was performed six times.
Half-lethal concentrations (LC50) were calculated for all the samples by survival analysis using GraphPad Prism 5 software.
Apoptosis assays in vitro
Cell apoptosis was assessed by flow cytometry and Annexin V–FITC/PI staining. HCT116 cells were treated with free apatinib and apatinib-loaded cRGD-Lipo-PEG and Lipo-PEG (the concentration of apatinib was 2 μM) for 24 h. After treatment, cells were washed with binding buffer and stained with 1 μL Annexin V–FITC and 1 μL PI for 15 min in the dark. Acquisition was measured by flow cytometry (BeckmanCoulter, Fullerton, CA, USA). Analysis was performed with FlowJo 7.6 software.
Biodistribution studies
The HCT116-bearing mice were divided randomly into two groups (n=3). The imaging studies started when tumors reached a size of between 100 and 200 mm3. For biodistribution studies, DiR-loaded liposomes were administered via the tail vein at the same dose of 1 mg/kg. Images were taken on an IVIS® Spectrum system (Caliper, Hopkington, MA, USA) at 0.5, 1, 2, 4, 6, 10, and 24 h after intragastric (ig) injection (40 μg per mouse) and intravenous (iv) administration (20 μg per mouse). After the 24 h point, mice were sacrificed, and the tumors and major organs (heart, liver, spleen, lungs, and kidneys) were all excised, washed, weighed, and imaged ex vivo.
Images were unmixed by the vendor-provided software, Living Image. DiR uptake was calculated using Spectrum Living Image 4.0 software.
Antitumor efficacy in vivo
For cancer therapy studies, HCT116 cells (5×106 per mouse) were implanted in the back of each nude mouse. When the initial mean tumor size reached 50–100 mm3, mice were randomly divided into three groups (n=5): a control group (PBS), a group treated with free apatinib, and a group treated with apatinib-loaded liposomes (cRGD-Lipo-PEG/Apa) and then administered the ig treatment at a dose of 50 mg/kg every 2 d for 3 wk. Tumor size and body weight were inspected and recorded every 3 d, and the tumor volume (V) was calculated using the following formula:
|
|
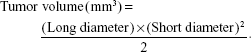
After therapy, mice were sacrificed, and the major organs and tumors were weighed and collected for the preparation of histological sections. Collected tissues were fixed with 4% paraformaldehyde and then embedded in paraffin for hematoxylin and eosin (H–E) staining and subsequent histological examination.
Statistical analysis
Graphing and statistical analysis were routinely performed using Origin 9.0 and GraphPad Prism 5 software. The statistical significance of the results was evaluated using the Student’s t-test in Microsoft Excel. A P-value of <0.05 was considered to be significant and that of <0.01 was considered a statistically significant difference.
Results and discussion
The preparation and characterization of liposomal formulations
cRGD-DSPE-PEG was prepared by the reaction between the cysteine residue of cRGD and the maleimide of DSPE-PEG-Mal. Then, the targeted PEGylated liposomes, namely, cRGD-Lipo-PEG, were prepared by a thin-film dispersion method. Nontargeted PEGylated liposomes, ie, Lipo-PEG, were also prepared by the same method, except with different mass ratios. As shown in Figure 1, the 1H-NMR spectrum showed the characteristic signals of cRGD-DSPE-PEG2000. The peak at 12 ppm (surrounded by a red box) represents the carboxyl group in cRGD; meanwhile, the peaks at 1–2 ppm (surrounded by a green box) represent the –CH– group in DSPE-PEG-Mal.
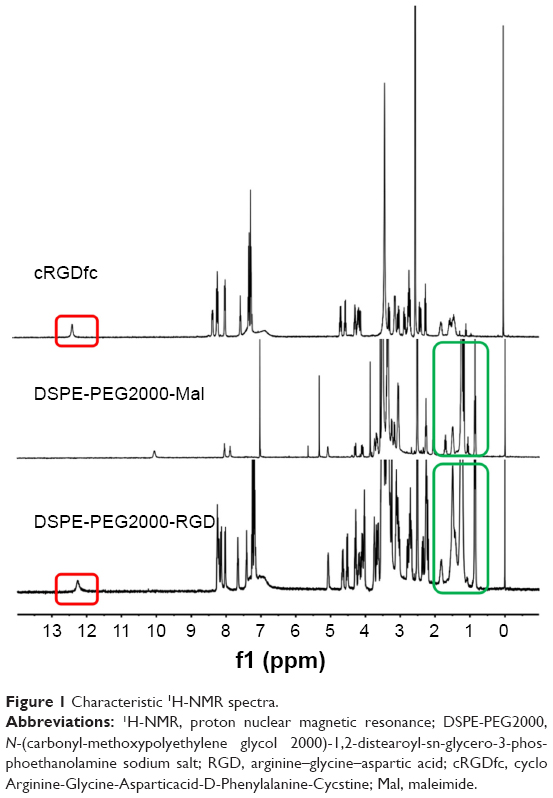 | Figure 1 Characteristic 1H-NMR spectra. |
The apatinib encapsulation efficiencies (AEEs) and drug loading of Lipo-PEG/Apa and cRGD-Lipo-PEG/Apa were measured and are shown in Table 1. For the two liposome types, up to 1.0 mg/mL of liposomal apatinib was achieved after concentration by ultrafiltration, with an entrapment efficiency of more than 90%. These results suggest that the modification of the cRGD peptide did not affect Lipo-PEG micelle AEEs.
 | Table 1 Apatinib-loading capacity and encapsulation efficiency of Lipo-PEG and cRGD-Lipo-PEG liposomes |
The mean diameters of the blank and drug-loaded liposomes are shown in Figure 2A. The blank Lipo-PEG and cRGD-Lipo-PEG liposomes had mean sizes of 151.95 and 149.9 nm, respectively, with small polydispersity index (PDI) values (<0.15). Mean diameters of the two liposome types were not statistically different, indicating that incorporating the cRGD peptide did not change liposome formation. Then, the particle sizes of apatinib-loaded liposomes were measured and compared to the sizes of blank liposomes. The results indicate that the particle sizes of the drug-loaded liposomes, ie, Lipo-PEG/Apa and cRGD-Lipo-PEG/Apa, were 159.15 and 162.3 nm, respectively. This represents a minor increase after apatinib loading. PDI values were also small (<0.1). The size distribution graphs of apatinib-loaded liposomes, viz, Lipo-PEG/Apa and cRGD-Lipo-PEG/Apa, are shown in Figure 2B. Particle size may play an important role in drug-loaded liposome delivery in vivo. Liposomes with a smaller size are of benefit for accumulation in tumors by passive targeting and for effective cellular uptake.24
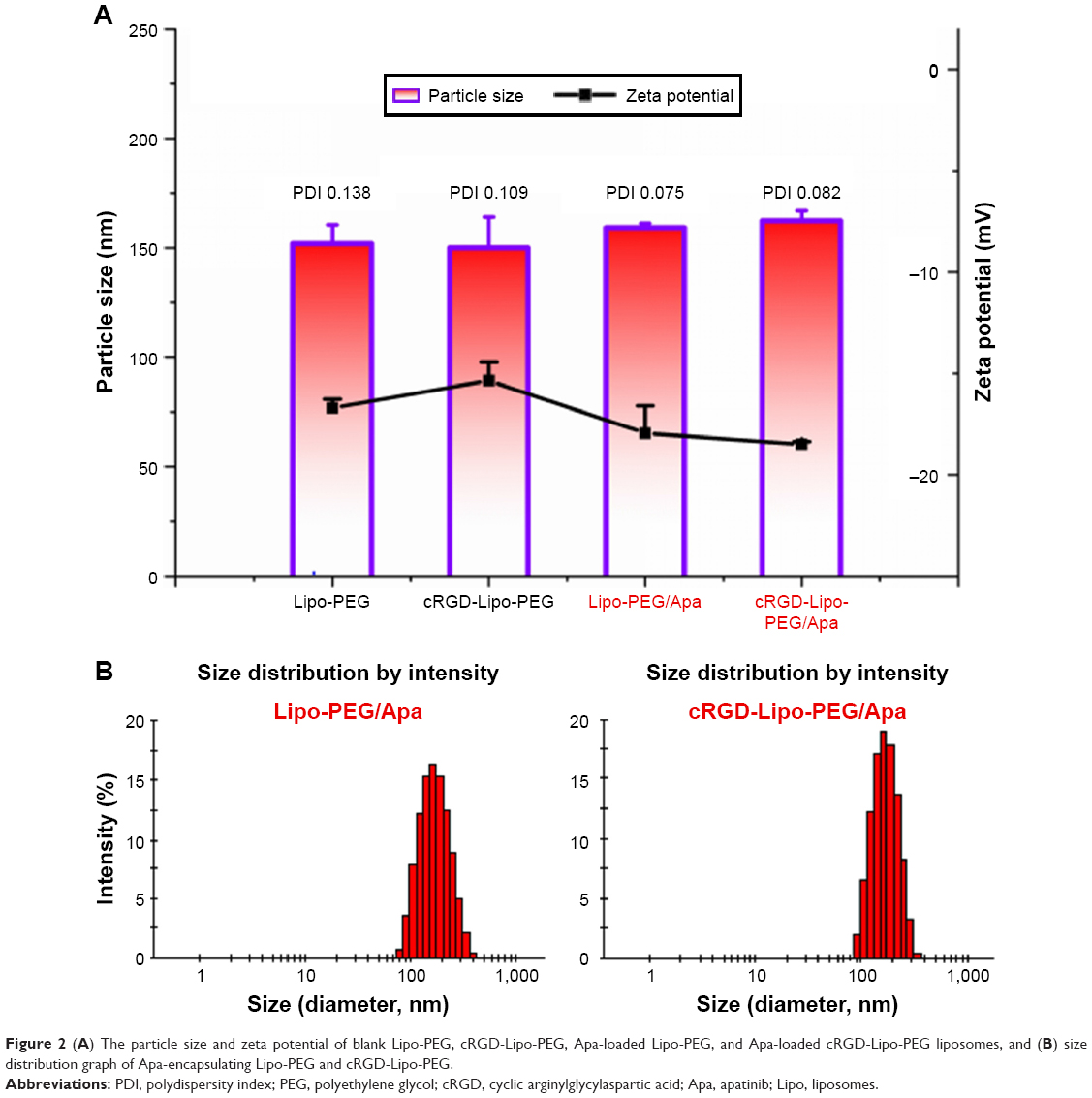 | Figure 2 (A) The particle size and zeta potential of blank Lipo-PEG, cRGD-Lipo-PEG, Apa-loaded Lipo-PEG, and Apa-loaded cRGD-Lipo-PEG liposomes, and (B) size distribution graph of Apa-encapsulating Lipo-PEG and cRGD-Lipo-PEG. |
In addition, both blank and drug-loaded liposomes showed a negative zeta potential of around −17 mV (−16.4 and −16 mV for the blank liposomes and −17.95 and −18.5 mV for apatinib-loaded liposomes). The zeta potentials were reduced after drug loading, possibly due to the changes in size and surface charge density. Resina et al’s25 results showed that anionic particles can easily enter cells favoring energy-independent transport, despite proteoglycans with anionic charges being located on the plasma membrane. Simultaneously, fusion and endocytosis of anionic and neutral lipid membranes with Ehrlich ascites carcinoma (EAC) cells was also studied by the fluorescent method.26 Although liposomes with positive zeta potentials can enter cells more effectively via a temperature-dependent transport, the dose-dependent pulmonary inflammatory and toxic effects of cationic lipids in vivo are limitations restricting the potential applications of cationic liposomal vectors in drug delivery.27 However, anionic lipids are endogenous components of eukaryotic cell membranes with reduced immune response activation, enhanced serum stability, and lower toxicity.28 So, in this study, we prepared the targeted liposomes using anionic lipids with negatively charged groups.
Stability in serum in vitro
To quantify liposomal particle stability for further applications, dynamic light scattering was used to measure particle sizes and zeta potentials of drug-loaded systems at a time point set when preparations had been stored at 4°C for 12 d. After 12 days, liposomes still appeared as a milky white colloidal solution.
As indicated by the data presented in Figure 3, there was no apparent change in the particle sizes and zeta potentials for either apatinib-encapsulating Lipo-PEG/Apa or cRGD-Lipo-PEG/Apa over 12 d, indicating that the liposomes had been maintained in a suitable form with no obvious aggregation. This was likely to be due to PEGylation stabilizing the liposomes. All the results showed that there were no significant changes when cRGD was added to liposomes.
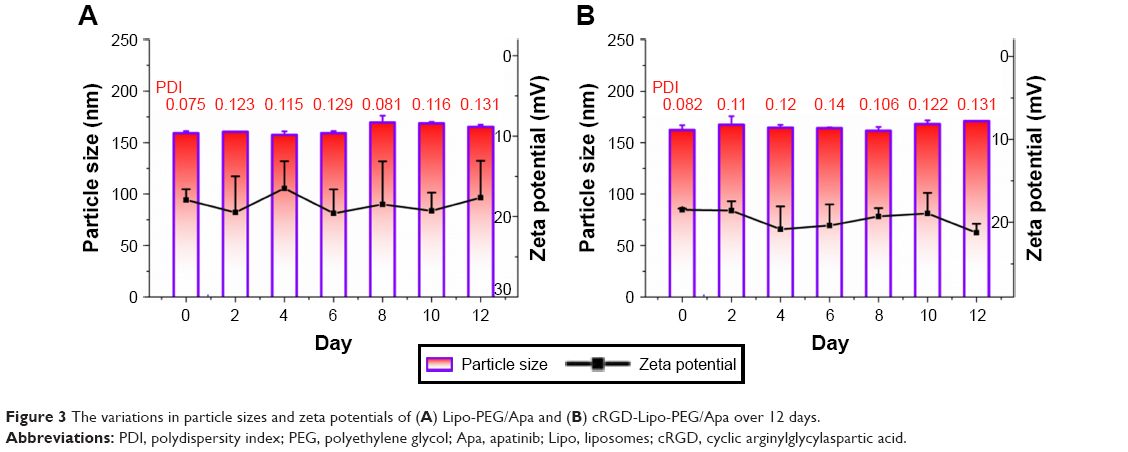 | Figure 3 The variations in particle sizes and zeta potentials of (A) Lipo-PEG/Apa and (B) cRGD-Lipo-PEG/Apa over 12 days. |
Apatinib release behavior in vitro
Apatinib release in vitro was also investigated using the dialysis method in PBS (pH 7.4 and pH 5.6) at 37°C for 72 h to examine the drug release properties of liposomes (Figure 4).
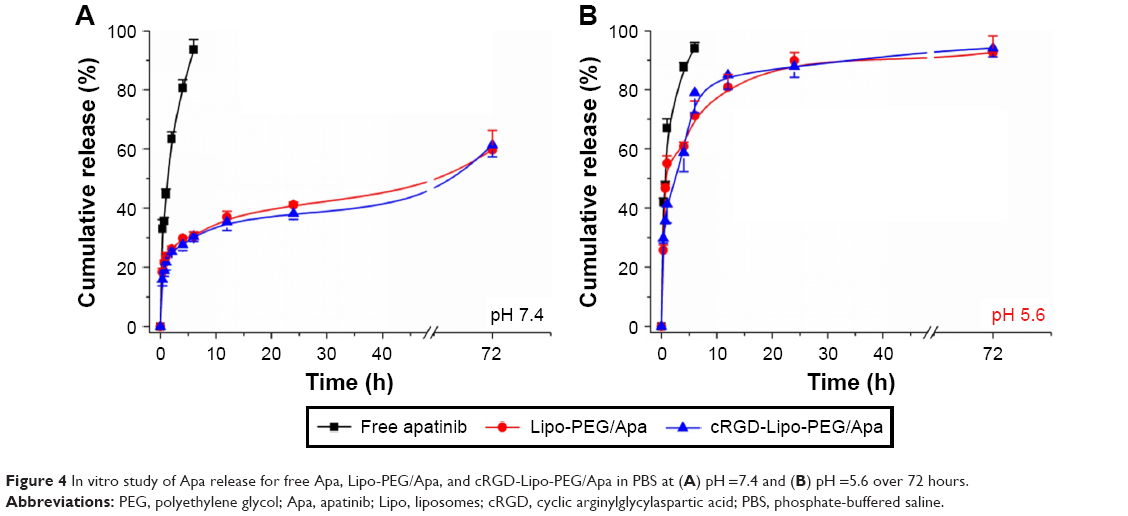 | Figure 4 In vitro study of Apa release for free Apa, Lipo-PEG/Apa, and cRGD-Lipo-PEG/Apa in PBS at (A) pH =7.4 and (B) pH =5.6 over 72 hours. |
Unlike the rapid release of free apatinib, as predicted, both liposome types, Lipo-PEG/Apa and cRGD-Lipo-PEG/Apa, exhibited similar gradual release properties for pH 7.4 (Figure 4A). A more detailed analysis of the release profiles indicates that up to 80.60%±2.76% of the apatinib was released in the first 4 h. In comparison, the release rate of apatinib from Lipo-PEG/Apa and cRGD-Lipo-PEG/Apa liposomes decreased significantly, with only 29.78%±0.11%, and 27.56%±1.96% of apatinib released in the same period. The release rate of apatinib from liposomes was maintained at a sustained rate for at least 72 h. After 6 h, the release rate of free apatinib was more than 90%, but release from liposomes was consistently about 30%.
Interestingly, the apatinib release was pH dependent (eg, 59.72%±6.53% vs 82.68%±5.60% at 72 h at pH 7.4 and 5.6, respectively, for Lipo-PEG/Apa) (Figure 4B). The full name of apatinib that we used for preparation of the liposomes was “mesylate apatinib”. In addition, desalted apatinib would dissolve rapidly upon entering an acidic environment because it is ionized readily in low pH, while displaying slow release in neutral medium. There was also an increase in release for free mesylate apatinib in acidic buffers (35.65%±1.17% and 47.23%±1.77% at pH 7.4 and pH 5.6, respectively, at 40 min for the free drug). Therefore, it could reduce drug leaking in the blood but facilitate drug release in acidic organelles (eg, endosomes, tumors).
These results indicate that Lipo-PEG and cRGD-Lipo-PEG could be used as drug carriers for retarding apatinib release. This would assist in decreasing drug leakage before drug-loaded systems arrive at the targeted tumors and cells.
Western blotting
We analyzed β3 integrin expression using the Western blotting technique. As shown in Figure 5A, HCT116 cells are enriched with β3 integrins, receptors that are rarely expressed in normal 239T cells. In addition, β3 integrins are overexpressed specially in solid tumors rather than in liver and kidneys from HCT116 human colonic cancer xenografts (Figure 5B).29 As a specific recognition site on the ligands for β3, cRGD can be modified on liposomes to produce a HCT116-targeted apatinib delivery system.
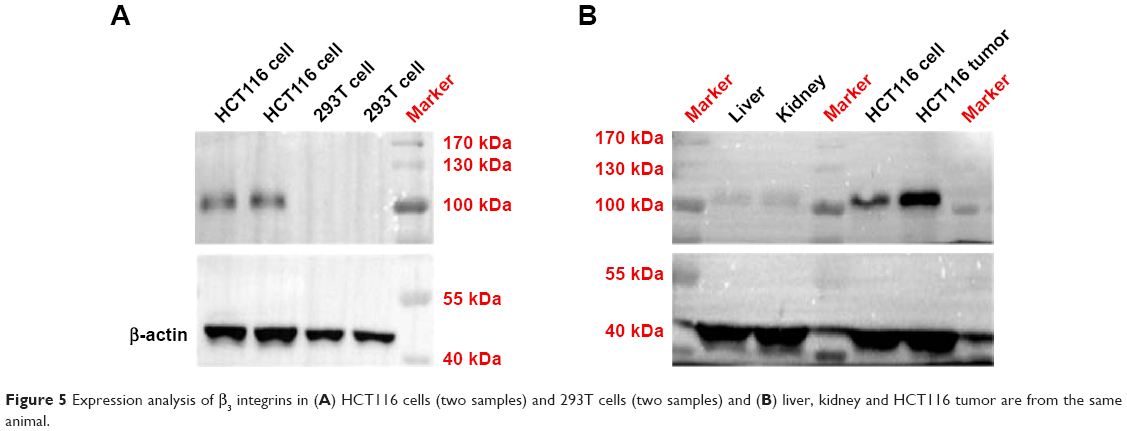 | Figure 5 Expression analysis of β3 integrins in (A) HCT116 cells (two samples) and 293T cells (two samples) and (B) liver, kidney and HCT116 tumor are from the same animal. |
Cellular uptake of liposomes
To assess the liposomal drug delivery capacities into cells in vitro, cellular uptake was measured by coumarin 6-loaded liposomes. Fluorescence microscopy and flow cytometry were used to determine the uptake of drug-loaded liposomes by HCT116 cells.
Fluorescence microscopy images showed the uptake of coumarin 6-loaded liposomes by HCT116 cells in vitro after incubation with the different liposome formulations for 4 h at 37°C. Figure 6A illustrates that the cellular uptake of liposomal coumarin 6 by HCT116 cells was enhanced after the modification of Lipo-PEG liposomes with the cRGD peptide.
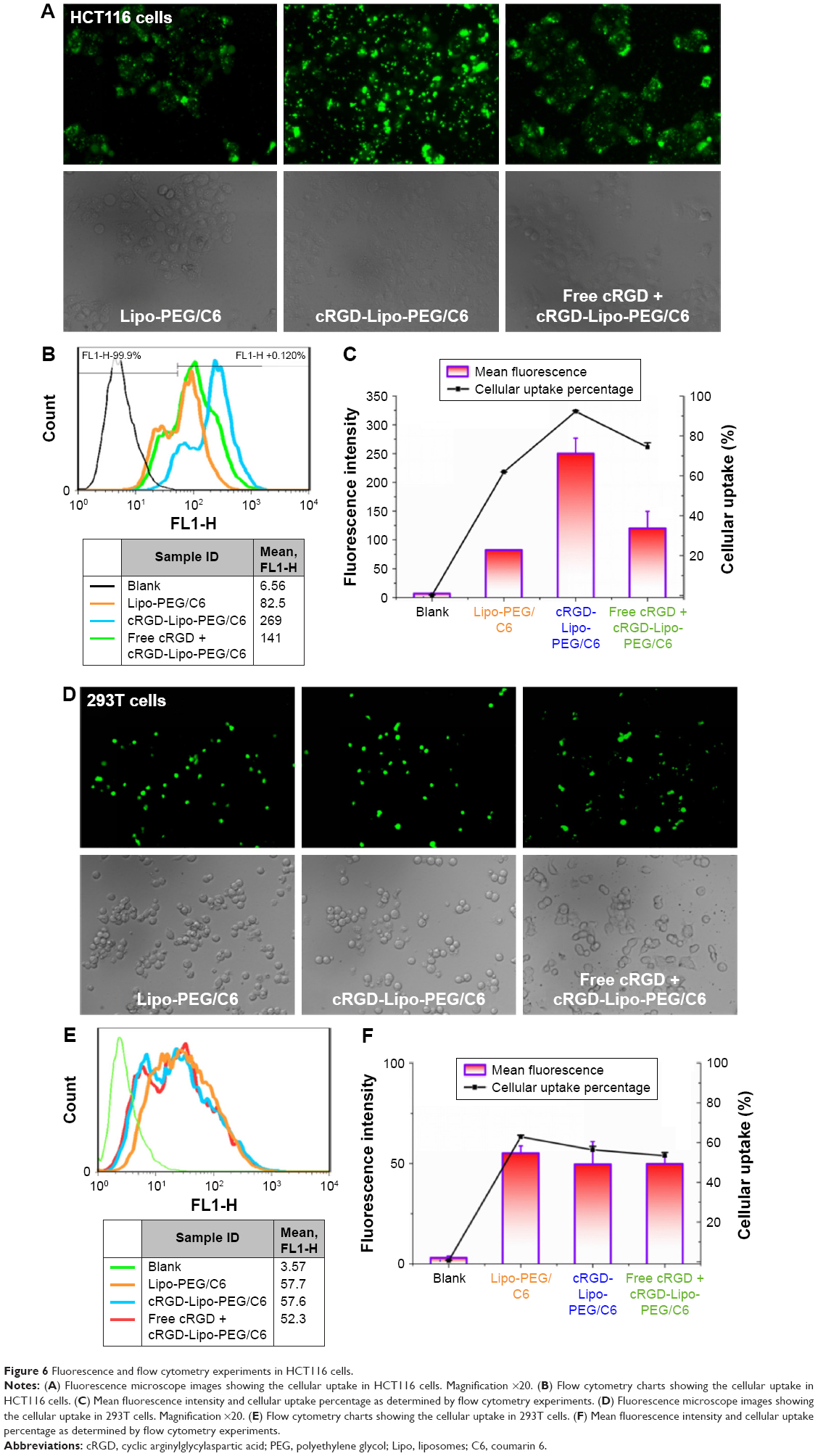 | Figure 6 Fluorescence and flow cytometry experiments in HCT116 cells. |
Flow cytometric data support the results of fluorescence microscopy studies (Figure 6B). After the same incubation time, the coumarin 6 fluorescence intensity in HCT116 cells was enhanced when coumarin 6 was loaded into cRGD-modified liposomes (cRGD-Lipo-PEG/C6) when compared with the intensity produced by nontargeted control liposomes (Lipo-PEG/C6). The cellular coumarin 6 uptake level of cRGD-Lipo-PEG/C6 (92.25%) was greater than that of nontargeted Lipo-PEG/C6 (61.95%). Mean fluorescence intensities (Figure 6C) for cRGD-Lipo-PEG/C6 liposomes were approximately threefold higher than those for Lipo-PEG/C6 liposomes. The uptake of cRGD-Lipo-PEG/C6 potentially involved specific interactions of the cRGD group with its receptor, as β3-positive HCT116 cells accumulated far more of the cRGD-modified liposomes. Hence, the overexpression of integrin receptors (β3) on HCT116 cells could promote cellular entry of liposomes with the RGD modification (Figure 5).
In addition, to confirm the role of cRGD peptide in cellular uptake, a competition assay was performed. Excess free cRGD peptides were added to HCT116 cells 30 min prior to liposomal treatment. After 4 h incubation, cRGD-Lipo-PEG/C6 showed a decrease in fluorescence intensity levels (50%) with free cRGD peptides (Figure 6C). The cRGD-Lipo-PEG/C6 uptake process could be competitively blocked by excess free cRGD, indicating that pretreatment by excess free cRGD peptide inhibited the cellular binding and uptake by specific competitive binding of the peptide to αvβ3 integrins on HCT116 cell membranes.
Meanwhile, a control cell line that lacks the β3 receptor expression, 293T, was set as the negative control. The uptake performances of Lipo-PEG/C6, cRGD-Lipo-PEG/C6, and excess free cRGD peptide with cRGD-Lipo-PEG/C6 were basically the same (Figure 6D–F). These results indicate that cRGD modification enhanced the specific uptake of liposomal drug in integrin-overexpressing HCT116 cells via a cRGD-mediated process.
Cytotoxicity studies and apoptosis assays in vitro
For anticancer effects in vitro, HCT116 cells were treated with either free apatinib or with Lipo-PEG/Apa or cRGD-Lipo-PEG/Apa apatinib-loaded liposomes at 18 different concentrations of drug (0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.25, 2.5, 2.75, 3, 3.25, 3.5, 3.75, 4, 4.25, and 4.5 μM). Cells were then subjected to an MTT assay. As shown in Figure 7C, significant reductions in cell viability were observed in both the Lipo-PEG/Apa-treated group and in the cRGD-Lipo-PEG/Apa-treated group, while the cytotoxicity of cells was clearly lower in the free apatinib-treatment group. In addition, blank control liposomes without apatinib showed no apparent cytotoxicity to HCT116 cells, even at the highest concentration used (2.5 mg/mL) (Figure 7B).
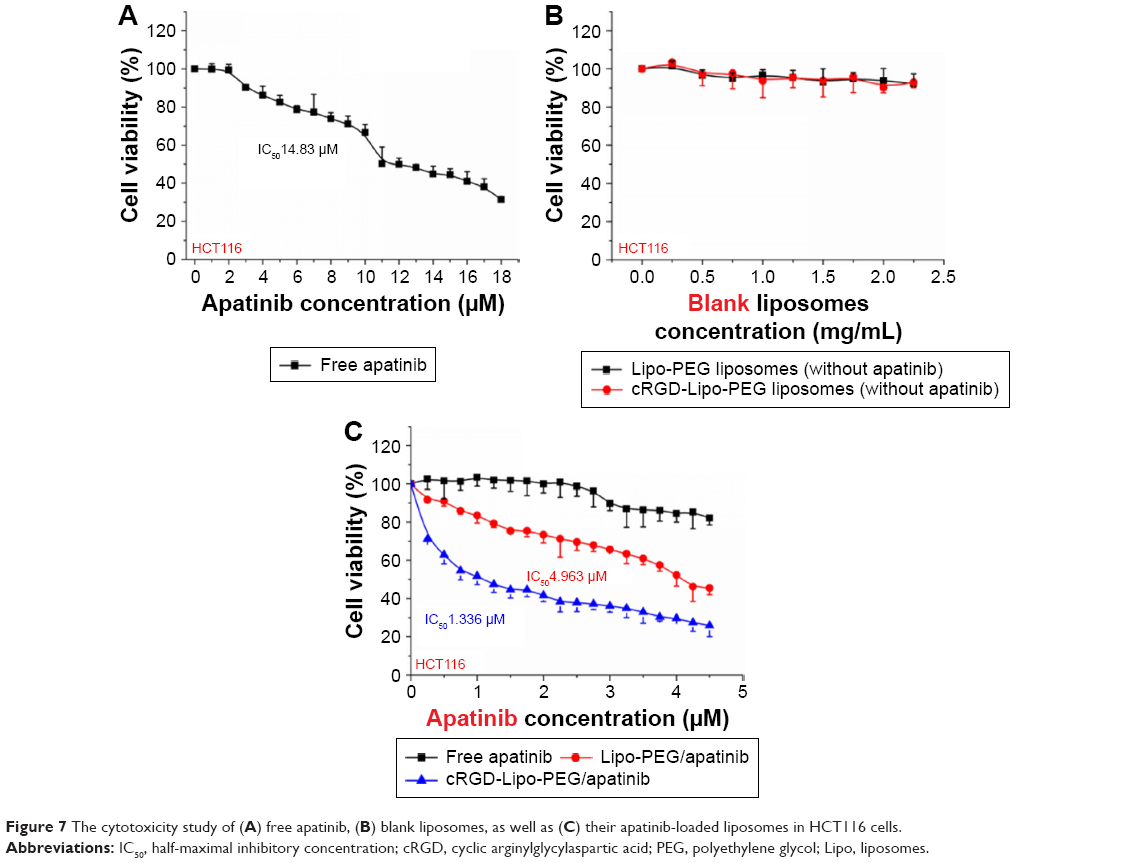 | Figure 7 The cytotoxicity study of (A) free apatinib, (B) blank liposomes, as well as (C) their apatinib-loaded liposomes in HCT116 cells. |
Determination of half-maximal inhibitory concentration (IC50) values was done by using GraphPad Prism 5 (Figure 7A and C). Briefly, the IC50 value of the cRGD-Lipo-PEG/Apa liposome was 1.336 μM compared with a value of 14.83 μM for free apatinib and 4.963 μM for the nontargeted liposome, Lipo-PEG/Apa.
To investigate the apoptosis effect, HCT116 cells were treated with free apatinib, Lipo-PEG/Apa, and cRGD-Lipo-PEG/Apa at the same concentration of apatinib. No significant apoptosis in terms of the apoptotic ratio was found in the control group (3.01%±2.9%; Figure 8) treated with a low concentration (2 μM) of free apatinib. Simultaneously, significant apoptosis was found in the liposome-treated groups: early apoptotic ratios were 6.95%±1.78% and 13.5%±3.96%, while late apoptotic ratios were 4.43%±0.62% and 27.05%±6.01% for Lipo-PEG/Apa and cRGD-Lipo-PEG/Apa, respectively.
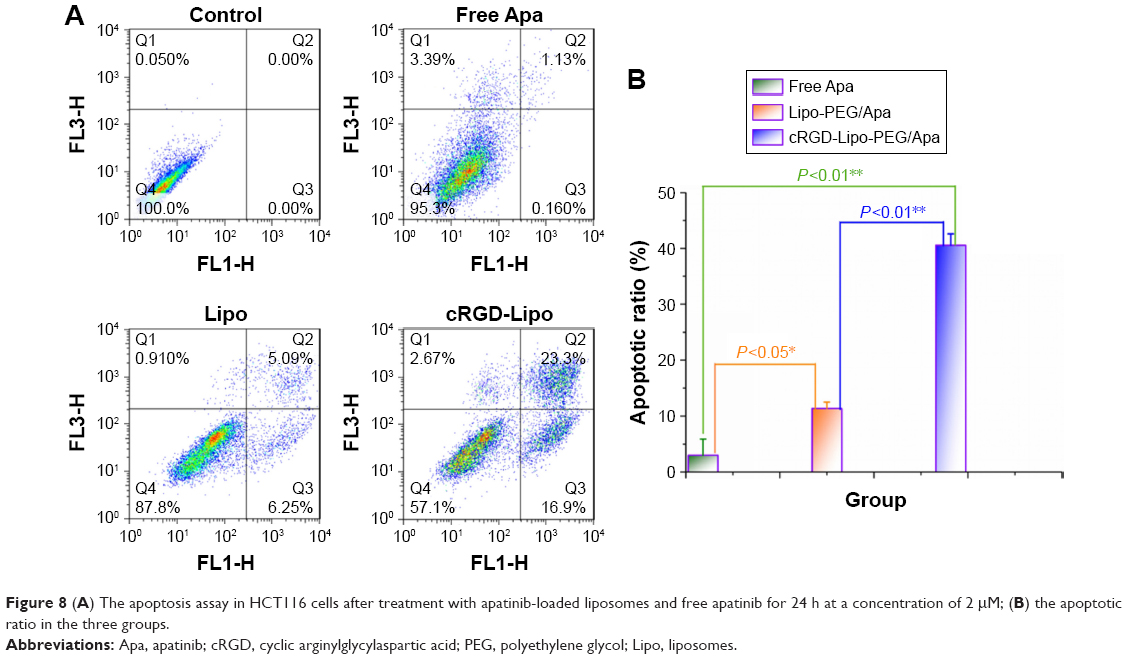 | Figure 8 (A) The apoptosis assay in HCT116 cells after treatment with apatinib-loaded liposomes and free apatinib for 24 h at a concentration of 2 μM; (B) the apoptotic ratio in the three groups. |
The results indicate that 1) the liposomal drug delivery system increased the druggability of poorly soluble apatinib and improved cellular uptake and 2) cRGD modification enabled the cRGD-Lipo-PEG/Apa liposome to target HCT116 cells with overexpressed β3 integrins. The latter can be inferred to indicate that cRGD enhanced the effect of apatinib. It is also important to note here that encapsulation in liposomes enhanced drug efficacy in vitro.
Investigation of the biodistribution of nontargeted and targeted liposomes
Liposomes can protect drugs in physiological environments and at tumor target sites via EPR. Nude mice bearing HCT116 human colonic cancer xenografts were established to estimate the biodistribution and tumor selectivity characteristics of the two types of liposomes.30
DiR was used as the fluorescence probe and model drug. The accumulation of DiR at selected time points after iv injection (20 μg per mouse) and ig administration (40 μg per mouse) with different formations is shown in Figures 9 and 10.
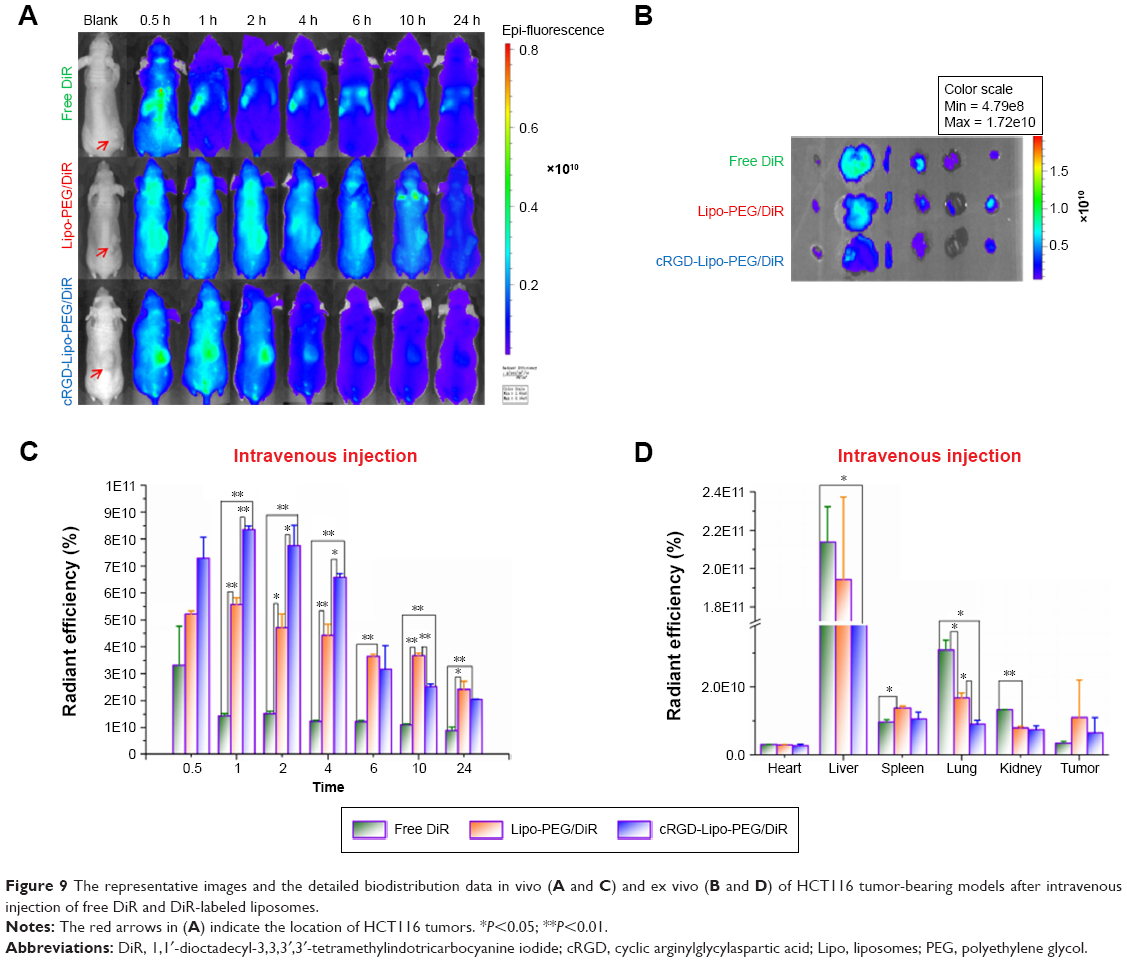 | Figure 9 The representative images and the detailed biodistribution data in vivo (A and C) and ex vivo (B and D) of HCT116 tumor-bearing models after intravenous injection of free DiR and DiR-labeled liposomes. |
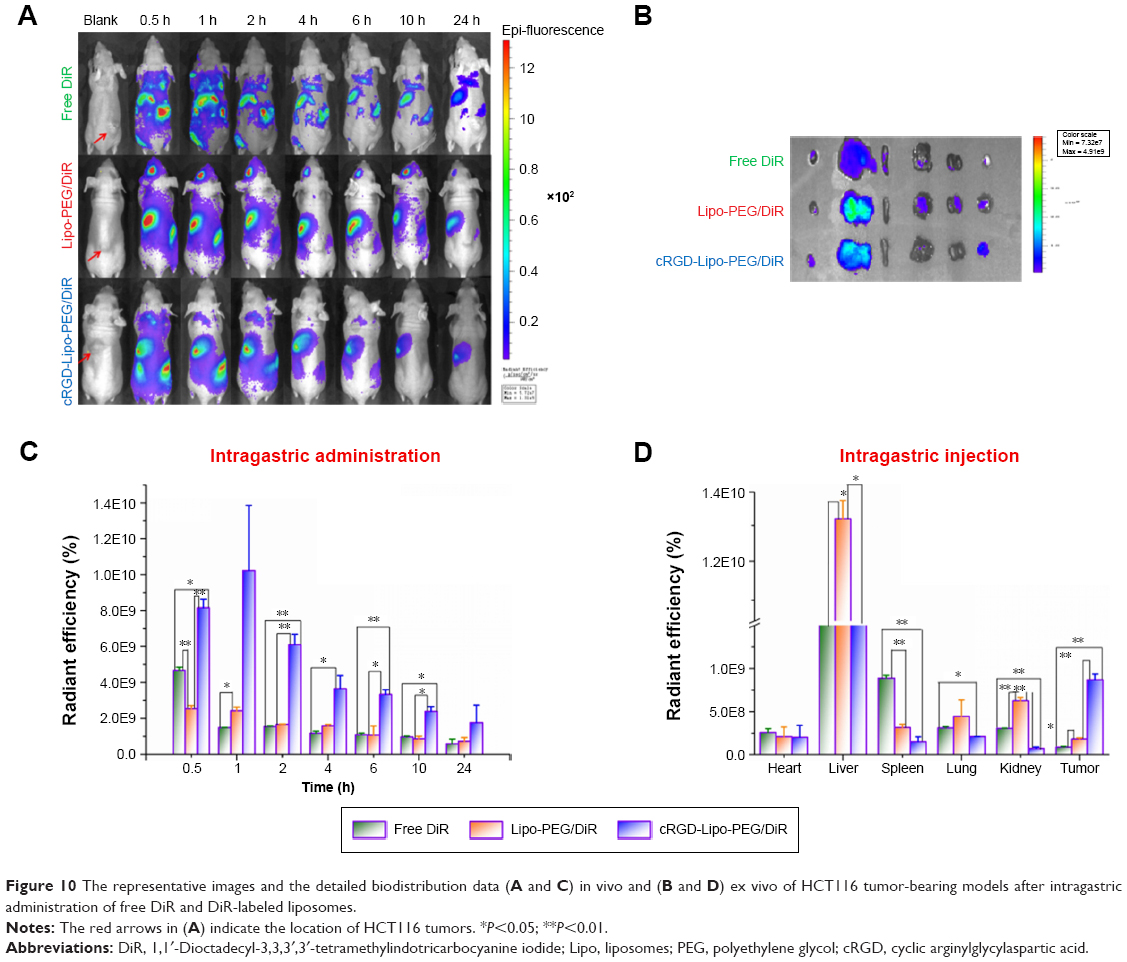 | Figure 10 The representative images and the detailed biodistribution data (A and C) in vivo and (B and D) ex vivo of HCT116 tumor-bearing models after intragastric administration of free DiR and DiR-labeled liposomes. |
Figure 9A and C shows the real-time images of free DiR (as the control), Lipo-PEG/DiR, and cRGD-Lipo-PEG/DiR at varying time points after iv administration in vivo. DiR-loaded targeted liposomes, ie, cRGD-Lipo-PEG/DiR, clearly accumulated in the tumor, and the fluorescence signal at 1 h was highest (8.34×1010±0.14×1010), which was comparable to that of Lipo-PEG/DiR (5.58×1010±0.25×1010). Conversely, tumor uptake of free DiR was only 1.44×1010±0.09×1010 at 1 h postinjection. The significant differences between free DiR and liposomal DiR indicate that PEG-modified liposomes could assist with drug accumulation in tumors and drug stability in the systemic circulation. In addition, the modification of cRGD maintained targeted function and did not alter other properties.
However, the fluorescence signal of cRGD-Lipo-PEG/DiR dropped rapidly after 2 h. The DiR signal of nontargeted Lipo-PEG/DiR gradually became weak with time and was maintained at stronger levels than for targeted liposomes after 6 h. Although tumors in the cRGD-Lipo-PEG/DiR group had lower fluorescence (2.05×1010±0.02×1010) at 24 h compared to those in the nontargeted Lipo-PEG/DiR group (2.42×1010±0.3×1010), the highest fluorescence signal was still observed in this group at 1 h (8.34×1010±0.14×1010). Furthermore, according to recent reports, when cRGD31 and other targeted ligands (folate, transferrin, and others)32,33 are used to modify liposomes, these are usually cleared faster than nontargeted liposomes.
Immediately after imaging at 24 h, animals were sacrificed. Major organs and tumors were harvested, washed, and imaged in an ex vivo imaging study (Figure 9B and D). The delivery of different formations of DiR to the remaining tissues occurred in the following order: liver > lungs > kidney > spleen > heart ~ tumor for free DiR, liver > lungs > spleen > tumor > kidney > heart for Lipo-PEG/DiR, and liver > spleen > lungs > kidney > tumor > heart for cRGD-Lipo-PEG/DiR. As previously reported, the enrichment levels in the spleen via ligand modification of liposomes were 1.5–2 times higher than for nontargeted liposomes.34
As apatinib is an orally administered small molecule receptor drug, we also studied its biodistribution after ig administration (40 μg per mouse). It was found that biodistribution and drug stability were hindered with this administration pathway, most likely due to the conditions in the gastrointestinal tract.
Although all DiR fluorescence was clearly lower than that with iv injections, the cRGD-modified cRGD-Lipo-PEG/DiR group (10.24×109±3.62×109) had a 4.21-fold higher DiR accumulation than the Lipo-PEG/DiR group (2.43×109±0.2×109) and 6.86-fold higher level than the free DiR group (1.49×109±0.01×109) at 1 h (Figure 10A and C).
Detailed biodistribution data in the major organs are shown in Figure 10B and D. We found that the DiR fluorescence signals in liver and tumors were high, being weak in other major organs. For comparative purposes, we evaluated the DiR uptake of the liposomal group. The distribution of cRGD-Lipo-PEG/DiR in liver was lower (10.06×109±0.61×109) than in the liver of the Lipo-PEG/DiR group (13.19×109±0.55×109), and the accumulation in tumors was higher for the targeted liposomes (0.87×109±0.07×109) than for nontargeted liposomes (0.18×109±0.01×109) after 24 h.
The results implied an in vivo enhancement of targeted selective delivery in HCT116 tumors of the cRGD-Lipo-PEG/DiR-treated group compared with the Lipo-PEG/DiR-treated group, with both iv injection treatment and ig administration.
As we have known for several years, radiolabeled (99mTc, 18F, 125I, 64Cu, 68Ga, 90Y, 203Pb, 177Lu, and 111In) cRGD peptides have been used as integrin αvβ3-targeted radiotracers for imaging integrin αvβ3-positive tumors by single photon emission computed tomography (SPECT)/computed tomography (CT)/positron emission tomography (PET), because of their higher integrin αvβ3-binding affinity and radiotracer tumor uptake, with longer tumor retention time than linear RGD.35–38 In addition, tissue sections, biodistribution, and pharmacokinetics of cRGD-peptide-conjugated quantum dots (QDs),39 gold nanoclusters,40 doxorubicin-loaded nanoparticles,41 polymeric micelles,42 and dendrimers43 were imaged or analyzed in mice bearing tumor xenografts, confirming specific tumor targeting.
All the results presented here as well as reports in the literature suggest that cRGD-Lipo-PEG liposomes would result in improved tumor inhibition in mouse models.
Colon tumor inhibition in vivo
To determine the therapeutic effects of the apatinib-loaded liposomes, ie, cRGD-Lipo-PEG/Apa, on colonic cancer in vivo, nude mice were implanted with HCT116 cells and used for therapeutic efficacy evaluation of free apatinib or apatinib liposomes.
Briefly, free apatinib (1 mg/kg 1 time every two days) was injected ig every 2 d for 3 wk (n=5). In the liposomal group, cRGD-Lipo-PEG/Apa was injected at the same dose (n=5). Tumors were found to be more sensitive to cRGD-Lipo-PEG/Apa at the dose used, with a tumor volume of 272.24±168.11 mm3 and tumor mass of 0.18±0.08 g, compared to that of the PBS-treated control group (volume of 1,368.0±522.73 mm3 and mass of 0.98±0.18 g) and the free apatinib-treated group (volume of 975.38±226.33 mm3 and a mass of 0.72±0.14 g). The results represent a significant effect on the rate of reduction of tumor mass of 82.18% for cRGD-Lipo-PEG/Apa, which is a statistically greater effect than that of only 26.39% for free apatinib (Figure 11A–C).
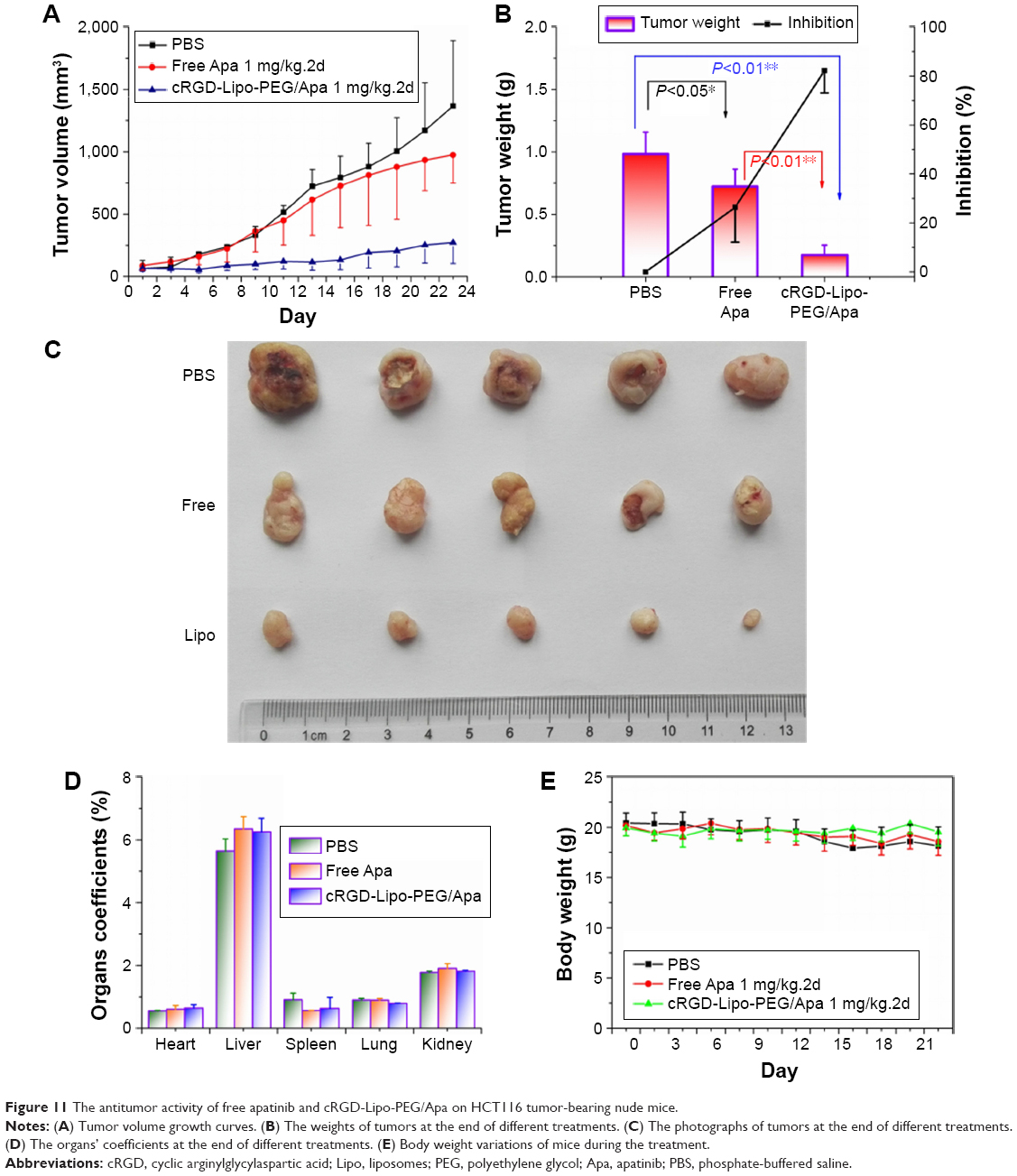 | Figure 11 The antitumor activity of free apatinib and cRGD-Lipo-PEG/Apa on HCT116 tumor-bearing nude mice. |
From Figure 11, which confirms the results we obtained in vivo, it is evident that the liposomal encapsulation drug performed better than free apatinib. The cRGD- and PEG-modified liposomes lead to enhanced accumulation of apatinib in tumors due to the EPR effect and due to integrin-targeted delivery, augmenting the anticancer effect after both ig and iv administration in vivo (Figures 9 and 10). Liposomes allowed sustained release of the entrapped apatinib, resulting in a decrease in the dose administered and in the toxic side effects. In addition, targeted delivery of integrin-mediated liposomes in vivo may be enhanced by modification of PEG by avoiding nonspecific binding.44
Toxicity in normal organs has been one common limiting factor in terms of the use of anticancer drugs in clinical therapy. Therefore, finally, the body weight of mice and survival rates were tested in order to evaluate the toxicity of drug-loaded liposomes. There were no significant weight loss and organ coefficient changes in the groups treated with different formulations of apatinib (free apatinib and cRGD-Lipo-PEG/Apa) (Figure 11D and E). No deaths occurred until the completion of this study.
H–E staining determined whether there was organ damage. The results suggested that there were no visible lesions in the heart, spleen, lungs, and kidneys in any of the four groups, suggesting that there were no significant acute toxic effects caused by apatinib in vivo. The only slight pathological abnormality was inflammatory cell infiltration, which was observed in the liver sample of the free apatinib group, because apatinib still has slightly toxic side effects in terms of acting as a drug (Figure 12).4,11
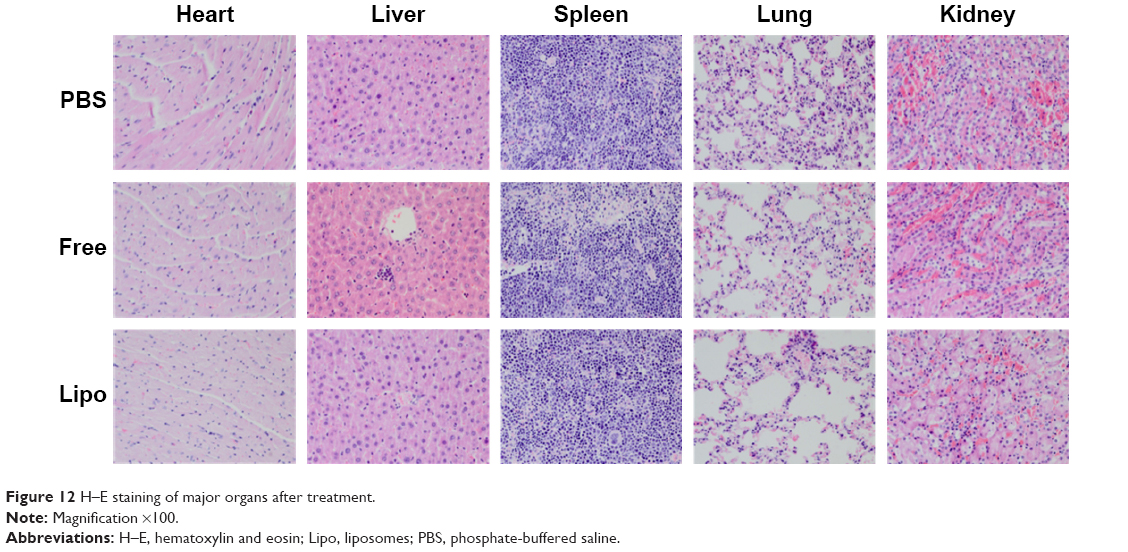 | Figure 12 H–E staining of major organs after treatment. |
In contrast, all the major organs of the cRGD-Lipo-PEG/Apa group showed no toxicity compared with the control group. As shown in Figure 10, cRGD-Lipo-PEG liposomes efficiently targeted HCT116 tumors and decreased nonspecific delivery to major organs, especially the liver, eg, the tumor/liver ratio of cRGD-Lipo-PEG/DiR was 0.0976±0.001 at 24 h, which was much better (P<0.01) than that of free DiR (0.01±0.003 at 24 h), indicating that there may be some minor liver toxicity involved in using cRGD-Lipo-PEG as an anticancer drug delivery system. So, cRGD-Lipo-PEG liposomes may be perceived to be a biologically safe and efficient delivery system for apatinib in the treatment of colonic cancer.
Conclusion
In this study, apatinib-loaded liposomes with cRGD peptides and PEG modification were successfully prepared in order to achieve tumor-specific delivery of the drug both in vitro and in vivo. These liposomes were evaluated for various physicochemical properties, including suitable particle size, negative zeta potential, excellent colloidal stability, retention of drug loading (4.93±0.07), drug encapsulation efficiencies (92.79±0.41), and behavior in terms of sustained drug release. The flow cytometry results indicate a considerably higher cellular uptake for the targeted liposome, cRGD-Lipo-PEG, than for Lipo-PEG in αvβ3 integrin-expressing cancer cells, HCT116. Competition assays with free cRGD peptides showed that there was cRGD-mediated delivery of cRGD-Lipo-PEG. Furthermore, DiR- and apatinib-loaded cRGD-Lipo-PEG represented an improvement in the biodistribution, tumor selectivity characteristics, and cytotoxic/apoptotic activities as compared to Lipo-PEG and free apatinib in both HCT116 cells and solid tumors. The remarkable improvement in activity observed may well be attributable to the increased degree of internalization in HCT116 cells, which is mediated by integrin receptors, and HCT116 tumor-bearing mice.
In summary, the targeted delivery system described for apatinib along with the promising tumor growth control results have potential in terms of tumor-targeted particle enrichment as well as protection of orally administered drug. Therefore, the delivery mechanism described could be a potentially effective approach for improvement in the efficacy of apatinib in the treatment of colon cancer.
Acknowledgments
This research was supported in part by the National Nature Science Foundation of China (81573008), the Fund of Pudong Health Bureau of Shanghai (PWRd2014-01), and the Project of Key Disciplines Group Construction of Pudong Health Bureau of Shanghai (PWZxq2014-04).
Disclosure
The authors report no conflicts of interest in this work.
References
Karanikas M, Esebidis A. Increasing incidence of colon cancer in patients <50 years old: a new entity. Ann Transl Med. 2016;4(9):164–167. | ||
Jiang Y, Lu J, Wang Y, et al. Molecular-dynamics-simulation-driven design of a protease-responsive probe for in-vivo tumor imaging. Adv Mater. 2014;26(48):8174–8178. | ||
Pabla B, Bissonnette M, Konda VJ. Colon cancer and the epidermal growth factor receptor: current treatment paradigms, the importance of diet, and the role of chemoprevention. World J Clin Oncol. 2015;6(5):133–141. | ||
Li J, Zhao X, Chen L, et al. Safety and pharmacokinetics of novel selective vascular endothelial growth factor receptor-2 inhibitor YN968D1 in patients with advanced malignancies. BMC Cancer. 2010;10(1):529–537. | ||
Tian S, Quan H, Xie C, et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011;102(7):1374–1380. | ||
Wilhelm SM, Carter C, Tang LY, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. | ||
Mi Y, Liang Y, Huang H, et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010;70(20):7981–7991. | ||
Qin S. Phase III study of apatinib in advanced gastric cancer: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2014;32(15 suppl):4003. | ||
Ding L, Li QJ, You KY, Jiang ZM, Yao HR. The use of apatinib in treating nonsmall-cell lung cancer: case report and review of literature. Medicine. 2016;95(20):1–4. | ||
Fan M, Zhang J, Wang Z, et al. Phosphorylated VEGFR2 and hypertension: potential biomarkers to indicate VEGF-dependency of advanced breast cancer in anti-angiogenic therapy. Breast Cancer Res Treat. 2014;143(1):141–151. | ||
Zhang H. Apatinib for molecular targeted therapy in tumor. Drug Des Devel Ther. 2015;9:6075–6081. | ||
Ding J, Chen X, Gao Z, et al. Metabolism and pharmacokinetics of novel selective vascular endothelial growth factor receptor-2 inhibitor apatinib in humans. Drug Metab Dispos. 2013;41(6):1195–1210. | ||
Eloy JO, Claro de Souza M, Petrilli R, Barcellos JP, Lee RJ, Marchetti JM. Liposomes as carriers of hydrophilic small molecule drugs: strategies to enhance encapsulation and delivery. Colloids Surf B Biointerfaces. 2014;123:345–363. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Bozzuto G, Molinari A. Liposomes as nanomedical devices. Int J Nanomedicine. 2015;10:975–999. | ||
Stapleton S, Milosevic M, Allen C, et al. A mathematical model of the enhanced permeability and retention effect for liposome transport in solid tumors. PLoS One. 2013;8(12):e81157. | ||
Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297–315. | ||
Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine (Lond). 2013;8(9):1509–1528. | ||
Hosokawa S, Tagawa T, Niki H, Hirakawa Y, Nohga K, Nagaike K. Efficacy of immunoliposomes on cancer models in a cell-surface-antigen-density-dependent manner. Br J Cancer. 2003;89(8):1545–1551. | ||
Park JA, Lee YJ, Lee JW, et al. Cyclic RGD peptides incorporating cycloalkanes: synthesis and evaluation as PET radiotracers for tumor imaging. ACS Med Chem Lett. 2014;5(9):979–982. | ||
Zheng Y, Ji S, Czerwinski A, Valenzuela F, Pennington M, Liu S. FITC-conjugated cyclic RGD peptides as fluorescent probes for staining integrin αvβ3/αvβ5 in tumor tissues. Bioconjug Chem. 2014;25(11):1925–1941. | ||
Gubernator J. Active methods of drug loading into liposomes: recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin Drug Deliv. 2011;8(5):565–580. | ||
Chen W, Jarzyna PA, van Tilborg GA, et al. RGD peptide functionalized and reconstituted high-density lipoprotein nanoparticles as a versatile and multimodal tumor targeting molecular imaging probe. FASEB J. 2010;24(6):1689–1699. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Resina S, Prevot P, Thierry AR. Physico-chemical characteristics of lipoplexes influence cell uptake mechanisms and transfection efficacy. PLoS One. 2009;4(6):e6058. | ||
Goryacheva YA, Vekshina OM, Yashin VA, Kim YA. Fusion and endocytosis of anionic liposomes with Ehrlich ascitic carcinoma cells. Bull Exp Biol Med. 2005;140(6):733–735. | ||
Scheule RK, George JAS, Bagley RG, et al. Basis of pulmonary toxicity associated with cationic lipid-mediated gene transfer to the mammalian lung. Hum Gene Ther. 1997;8(6):689–707. | ||
Patil SD, Rhodes DG, Burgess DJ. Anionic liposomal delivery system for DNA transfection. AAPS J. 2004;6(4):13–22. | ||
Buckel L, Savariar EN, Crisp JL, et al. Tumor radiosensitization by monomethyl auristatin E: mechanism of action and targeted delivery. Cancer Res. 2015;75(7):1376–1387. | ||
Ji S, Czerwinski A, Zhou Y, et al. 99mTc-Galacto-RGD2: a novel 99mTc-labeled cyclic RGD peptide dimer useful for tumor imaging. Mol Pharm. 2013;10(9):3304–3314. | ||
Chen Z, Deng J, Zhao Y, Tao T. Cyclic RGD peptide-modified liposomal drug delivery system: enhanced cellular uptake in vitro and improved pharmacokinetics in rats. Int J Nanomedicine. 2012;7:3803–3811. | ||
Iinuma H, Maruyama K, Okinaga K, et al. Intracellular targeting therapy of cisplatin-encapsulated transferrin-polyethylene glycol liposome on peritoneal dissemination of gastric cancer. Int J Cancer. 2002;99(1):130–137. | ||
Turk MJ, Waters DJ, Low PS. Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett. 2004;213(2):165–172. | ||
Emanuel N, Kedar E, Bolotin EM, Smorodinsky NI, Barenholz Y. Targeted delivery of doxorubicin via sterically stabilized immunoliposomes: pharmacokinetics and biodistribution in tumor-bearing mice. Pharm Res. 1996;13(6):861–868. | ||
Ji S, Zhou Y, Voorbach MJ, et al. Monitoring tumor response to linifanib therapy with SPECT/CT using the integrin αvβ3–targeted radiotracer 99mTc-3P-RGD2. J Pharmacol Exp Ther. 2013;346(2):251–258. | ||
Doss M, Kolb HC, Zhang JJ, et al. Biodistribution and radiation dosimetry of the integrin marker 18F-RGD-K5 determined from whole-body PET/CT in monkeys and humans. J Nucl Med. 2012;53(5):787–795. | ||
Nwe K, Kim YS, Milenic DE, et al. 111In-and 203Pb-labeled cyclic arginine-glycine-aspartic acid peptide conjugate as an αvβ3 integrin-binding radiotracer. J Labelled Comp Radiopharm. 2012;55(11):423–426. | ||
Tateishi U, Oka T, Inoue T. Radiolabeled RGD peptides as integrin alpha(v)beta3-targeted PET Tracers. Curr Med Chem. 2012;19(20):3301–3309. | ||
Yong KT. Biophotonics and biotechnology in pancreatic cancer: cyclic RGD-peptide-conjugated type II quantum dots for in vivo imaging. Pancreatology. 2010;10(5):553–564. | ||
Yin HQ, Bi FL, Gan F. Rapid synthesis of cyclic RGD conjugated gold nanoclusters for targeting and fluorescence imaging of melanoma A375 cells. Bioconjug Chem. 2015;26(2):243–249. | ||
Bibby DC, Talmadge JE, Dalal MK, et al. Pharmacokinetics and biodistribution of RGD-targeted doxorubicin-loaded nanoparticles in tumor-bearing mice. Int J Pharm. 2005;293(1):281–290. | ||
Li AJ, Zheng YH, Liu GD, Liu WS, Cao PC, Bu ZF. Efficient delivery of docetaxel for the treatment of brain tumors by cyclic RGD-tagged polymeric micelles. Mol Med Rep. 2015;11(4):3078–3086. | ||
Dijkgraaf I, Rijnders AY, Soede A, et al. Synthesis of DOTA-conjugated multivalent cyclic-RGD peptide dendrimers via 1, 3-dipolar cycloaddition and their biological evaluation: implications for tumor targeting and tumor imaging purposes. Org Biomol Chem. 2007;5(6):935–944. | ||
Zhang Q, Tang J, Fu L, et al. A pH-responsive α-helical cell penetrating peptide-mediated liposomal delivery system. Biomaterials. 2013;34(32):7980–7993. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.